
Abstract
Purpose
In many streams worldwide including those on the south coast of Western Australia (WA), sediments of the > 2-mm fraction often contribute up to 50% of the streambed. However, most analysis and interpretation of sediment chemistry, including phosphorus (P), is conducted on the < 2-mm fraction as this fraction is considered the most chemically reactive. This paper aims to identify the contribution of the > 2-mm fraction to P retention and release in sandy-gravely streams.
Material and methods
Sediment samples were collected from streams in agricultural catchments, and P retention and release by the < 2-mm and > 2-mm (typically lateritic; iron rich) sediment fractions were examined using fluvarium and batch experiments. Phosphorus sorbed by sediment was estimated on a mass (mg P kg−1) and area basis (mg P m−2).
Results and discussion
Phosphorus sorption measurements suggested that mineralogy as well as particle size were important factors influencing P retention by stream sediments. Stream sediments retained approximately 30% of added P. In a desorption phase, approximately 8% of the retained P was released into stream water.
Conclusions
Stream sediments in south western WA appear to be net immobilisers of P, retaining more P than they release, dependent on the stream P concentration. Exclusion of the > 2-mm fraction when determining stream sediment P dynamics may therefore underestimate whole stream sediment P retention and release.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Phosphorus (P) is an essential macro nutrient for agricultural production (Sharpley and Tunney 2000; McDowell et al. 2003). However, repeated P applications can lead to the P fertility of soils exceeding agronomic requirements (Sharpley et al. 1994, 2013; Buczko and Kuchenbuch 2007; Weaver and Wong 2011) and P movement into surface and groundwaters causing eutrophication of receiving waters (Sharpley et al. 2013; Jarvie et al. 2013; Kusmer et al. 2018).
The P transport and transfer pathways by which losses occur, including streams and drains, have been assessed for many agricultural systems (Haygarth and Jarvis 1999; McDowell et al. 2004; Gourley and Weaver 2012). Sediments are an important component of the nutrient cycle in agricultural systems (Sharpley et al. 2007) retaining or releasing P through biological and chemical mechanisms. However, little emphasis has been placed on the relevance of sediment particle size, particle size distribution or mineralogy in stream sediments on P cycling.
A threshold of 2-mm is the standard to distinguish between the notionally reactive fine earth fraction (< 2-mm) and larger, notionally inert fractions in soil science (Rayment and Lyons 2011). Most research on retention and release of P from streambed sediments has focused on the < 2-mm fraction as it is considered the most reactive under natural conditions (Lucci et al. 2010). Additionally, P sorption curves and the EPC0 (Equilibrium Phosphorus Concentration—the concentration at which P is neither sorbed nor desorbed by sediments) have typically used the < 2-mm fraction (Kovar and Pierzynski 2009). However, in some regions of the world, such as southwest Western Australia (WA) and northwestern Arkansas in the USA (Rogers et al. 2011), the > 2-mm fraction can represent a significant component (~ 30–50%) of stream sediments. Given that a large proportion of sedimentary material can be this size (Rogers et al. 2013), the > 2-mm fraction may have significant impacts on P sorption by the whole stream sediment.
The relative amounts and mineral composition of different size fractions may influence the P retention and release of whole sediments, such that:
-
(1)
if the > 2-mm fraction is poorly P retentive relative to the < 2-mm fraction, the > 2-mm fraction will reduce the overall sediment surface area for a given mass diluting the capacity of the whole sediment to retain or release P or
-
(2)
if the > 2-mm fraction of sediment is strongly P retentive (e.g. lateritic gravels), the > 2-mm fraction may provide P sorption sites in addition to those provided by the < 2-mm fraction (Weaver et al. 1992) and contribute to overall P retention or release by the whole sediment.
In addition, it is commonly accepted that P sorption by sediment or soil is evaluated and expressed in terms of mass (i.e. milligrammes of P sorbed per kilogramme (mg P kg−1)) of soil (Rayment and Lyons 2011), since mass is easily determined. However, P adsorbs to the surface of particles irrespective of size. Larger size fractions have a smaller surface area to volume ratio and will likely sorb less P for the same mass as finer sized particles. Expressing P sorption as a function of mass does not account directly for surface area, which may be an important characteristic when comparing different size fractions with the same or different mineralogy. Therefore, expressing P sorbed on an areal basis as well as a mass basis may provide insights not possible when using mass-based measures alone. When sediment mineralogy is disparate across different size fractions, mineralogy may become more important than surface area alone.
Weaver et al. (1992) examined P sorption of lateritic gravel fractions of the same mineralogy, but of varying size (2–2.8-mm, 2.8–4-mm, 4–5.6-mm, 5.6–8-mm and > 8-mm) with different proportions representing a toposequence found in southwest WA. Lateritic gravel with the largest proportion of smaller size fractions adsorbed the most P over a 5-week period.
When reframed and normalised by surface area (Walling 2005; Ballantine et al. 2009) rather than mass, the P sorption data of Weaver et al. (1992) can be described by a single curve independent of particle size, rather than separate curves for different size distributions. For this case, it could be concluded that surface area was the dominant factor influencing P sorption rather than differences in mineralogy or surface microtopography of the > 2-mm fraction.
The interaction between mineralogy and surface area on P transport is important in regions with highly-weathered soils, such as those on the south coast of WA where lateritic gravels can be abundant in otherwise coarse-textured sandy soils (Schoknecht and Pathan 2013). Stream sediment in these regions can be derived from a variety of sources including stream banks with exposed subsoils, which can include > 2-mm lateritic gravels, as well as erosion of fine particulate matter from surface soils (Weaver and Summers 2014). Stream sediments on the south coast of WA are mainly exposed to soluble P transported via subsurface pathways from adjacent agricultural land (McKergow et al. 2003; Weaver and Summers 2014). In addition, catchment-based modelling for southwest WA suggests that 42% of surplus P in agriculture is lost to streams, with 87% estimated to be retained by stream sediments (Keipert et al. 2008).
This paper aims to explore the potential for the < 2-mm and > 2-mm fractions of stream sediment to retain and release soluble P in agricultural-dominated catchments on the south coast of WA using both batch and fluvarium experiments.
2 Materials and methods
2.1 Site description
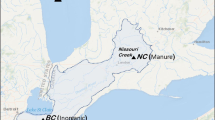
The Wilson Inlet Catchment (WIC) drains into the Wilson Inlet (Brearly 2005) and is located on the south coast of WA to the west of the city of Albany (35.0° S 117.8° E). Average annual rainfall ranges from 710 mm in the north of the catchment to 940 mm in the south (Master 2009) (Fig. 1). The catchment is dominated by Podosols and Chromosol soil orders (Australian Soil Classification) (Isbell et al. 1997) with sandy surface soils (52%) including deep sands and sandy duplex soils (Moore 1998; Master 2009). The Oyster Harbour Catchment (OHC) is dominated by Podosol and Sodosol soil orders (Australian Soil Classification) (Isbell et al. 1997) with sandy surface soils (59%) including deep sands and sandy duplex soil classifications (Master 2008). The OHC drains to Oyster Harbour (Hillman et al. 1990) via the Kalgan and King Rivers (Brearly 2005) with rainfall varying from 500 mm per annum in the north to 950 mm per annum in the south (Master 2008) (Fig. 1).

Left map: south west Western Australia and the Wilson Inlet and Oyster Harbour catchments. Right map: catchment boundaries (thick solid lines), annual rainfall isohyets (thin dashed line), stream network (grey lines – thin lines are low Strahler stream order and thick lines high stream order), labelled study sites (filled circles) and sub-catchment boundaries (thick dashed lines) for sites A and B where sediment was collected for fluvarium experiments. Sites A–J where sediment was collected for preliminary experiments (Online resource 1)
Two sites were selected for sediment sampling, one in WIC and one in OHC (Fig. 1). Both sediments were from ephemeral streams which undergo annual wetting and drying due to the mediterannean climate. Site A in WIC site was 3 km upstream from the entrance of Cuppup Creek to the Wilson Inlet. Site B is located in the OHC approximately 3.6 km from the headwater of an intermittent stream on an unnamed tributary (8 km in length) that feeds into the upper Kalgan River. Both grazing and cropping are undertaken in this sub-catchment. Sediments from both sites were classified as ‘sand’ based on the USDA soil texture triangle with 22–30% > 2-mm material (w/w) making up the sediment (Table 1).
2.2 Sample collection and processing
For batch experiments, a composite sample from the top 3 cm of the streambed sediment was collected using a 80-mm PVC sample collecting tube. For fluvarium (simulated stream apparatus as described in McDowell and Sharpley (2003)) experiments, a composite sample was prepared by collecting the top 3 cm of streambed sediment with a shovel at a minimum of five random points along a 75-m transect. The composite sample was laid out on plastic sheets to air dry.
An air-dried subsample was passed through a 2-mm sieve to determine the percentage of > 2-mm and < 2-mm fractions by mass. The remaining air-dried sediment was passed through a 2-mm sieve and the < 2-mm fraction and > 2-mm fraction separated. The > 2-mm fraction was thoroughly washed with water to remove traces of fine sediment adhering to the surface. Where necessary, sediments were artificially recombined from the > 2-mm and < 2-mm fractions to create ‘whole’ sediment samples with the original proportions of < 2-mm and > 2-mm fractions. This resulted in a > 2-mm fraction, < 2-mm fraction and whole sediment subsample for each site (Table 1).
2.2.1 Phosphorus sorption and EPC0 estimation
It has been reported (Simpson et al. 2018) that drying of sediment can alter its EPC0 and P dynamics. Preliminary experiments on sediments from these catchments (sites A–J, Fig. 1) compared EPC0 from moist and air-dried sediment and did not find significant differences (Online resource 1, Electronic Supplementary Material). Therefore, in triplicate, each sediment (Table 1) was left in contact with 0, 0.5, 1, 2, 5 and 10 mg P L−1 in a potassium chloride (KCl) solution at a sediment solution ratio of 1:20 for 24 h at room temperature (approximately 21 °C) prior to a subsample of the solution being taken for P analysis. Potassium chloride was used as the supporting electrolyte since calcium chloride can lead to the precipitation of calcium phosphates (Allen and Jeffery 1990). A 0.0005 M KCl solution was used for site A and a 0.02 M KCl solution for site B. This increase in molarity of KCl for site B was made to enhance laboratory processes such as filtration (< 0.45 μm) after centrifugation. Preliminary P desorption experiments from ten different streambed sediments in the presence of de-ionised water, 0.02 M KCl and 0.0005 M KCl showed no statistically significant difference (ANOVA, P > 0.05) between P adsorption for different KCl concentrations and de-ionised water.
Samples were agitated gently by hand for 30 s after 1, 3 and 23 h to avoid abrasion of surfaces of the > 2-mm fraction (Barrow and Shaw 1979; Stone and Murdoch 1989). This minimised the risk of increased P retention due to surface abrasion. Subsamples were collected after 24 h, filtered (0.45 μm) and stored below 3 °C prior to analysis for Filterable Reactive P (FRP) using the modified molybdenum blue method of Murphy and Riley (1962).
Equilibrium Phosphorus Concentration (EPC0) (Taylor and Kunishi 1971; Froelich 1988) was determined for each of the sediments using the lowest points on the sorption isotherm where the sorption isotherm is linear (Lucci et al. 2010). The three to four P concentrations below 2 mg L−1 (0, 0.25, 0.5, 1 mg L−1) were used to calculate the EPC0 using a plot of initial FRP in solution vs. P sorbed (mg kg−1) (Eq. (1)).
Where S = P sorbed or desorbed (mg kg−1), m = slope, c = y-intercept, Ci = initial P concentration.
The Freundlich equation, S = acbwas used to describe the P sorption curves, where S indicates sorption (mg kg−1), c the solution concentration (mg L−1) and a and b are coefficients (Barrow 2008). DataDesk 6.1 (Data Description. 2011, New York) was used to determine significant differences between the P sorbed and EPC0 of each sediment (P < 0.05) using ANOVA.
2.2.2 Phosphorus sorption by ground sediment components
The potential of different sediment components to adsorb P was examined by separating them by colour and size. Sediment components were selected visually and included < 2-mm and > 2-mm fractions consisting of colours indicative of iron (Fe) (Torrent et al. 1983; Schwertmann 1993) from both sites A and B. The non-red material < 2-mm at sites A and B was very pale brown (10YR 7/4). The > 2-mm non-red material at site A was pale yellow (2.5Y 7/3) in colour and site B had yellow (10YR 7/6) material. The > 2-mm red coloured material at site A was yellowish red to strong brown (7.5YR 4/6) and site B material was strong brown. Using a grinding process to reduce particle size, each fraction was ground to a fine powder in a puck and ring mill (Rocklabs Benchtop Ring Mill). Triplicate ground samples were shaken end-over-end for 20 h at a soil solution ratio of 1:20 with P concentrations of 0, 0.25, 0.5, 1, 2, 5, 10 and 100 mg L−1, centrifuged, filtered (0.45 μm) and stored at 3 °C prior to analysis for FRP as above.
2.2.3 Phosphorus retention and release using simulated stream conditions in a fluvarium
The ability of whole sediments, < 2-mm fraction and > 2-mm fraction (Table 1) to retain and release P under simulated stream conditions was examined using a fluvarium. Four fluvarium channels (9.8-m long, 0.2-m wide), based on the design by McDowell and Sharpley (2003), were filled to 3-cm depth with various sedimentary materials, one with > 2-mm, one with < 2-mm and two with whole sediments from each site. Sediments were gently compacted, and the slopes of the fluvarium troughs were set to 1% to simulate the slope at each sample site.
There were two phases (uptake and release) in this experiment that were repeated multiple times for each sediment. During the uptake phase, KH2PO4 was added to 396 L of rainwater in the fluvarium sump to provide a starting concentration of approximately 2.5 mg P L−1 and this solution was cycled over the sediment for 48 h. During the release phase (48 h), 396 L of rainwater (median FRP of 0.06 mg L−1) was cycled over sediment that had sorbed P in previous uptake phases. At the end of each 48 h run, the starting 396 L of water was removed, and replenished with water of the appropriate P concentration to represent the starting conditions of the uptake or release phase. The site A sediments were exposed to ten consecutive uptake phases, followed by four release phases, a further single uptake phase and finally a release phase. The sediments from site B were exposed to five consecutive uptake phases, two release phases then a single uptake phase. During each phase, samples were automatically collected using an ISCO 6712 portable autosampler (Teledyne ISCO, Lincoln, NE, USA) at 0, 5, 10, 15, 45 min and 1, 4, 8, 10, 12, 20, 24, 32, 36 and 48 h. The flow rate for the experiments was approximately 1 L s−1, measured on each fluvarium channel by 0.5-ft H-flumes. Samples were filtered (< 0.45 μm) and analysed for FRP as above.
The surface area of different sediments was estimated based on mass in a known volume, particle size distribution (50, 31.5, 20, 4, 2.8 and 2-mm sieves) and median particle size within a size range. Surface area of the fractions of gravel or sand was then estimated based on the following equation (Eq. (2)), where Af = surface area (m2) of the size fraction:
where Wsize = total mass (g) of counted particles for a size group, n = number of particles counted for a size group and %size = the % (by mass) of a size group of the whole sediment. To calculate the total surface area of the combined fractions in the fluvarium channel, Afluv = Sum of Af from each size fraction.
2.2.4 Mineral determination of sediment fractions
Semi-quantitative analysis using X-ray diffraction of samples > 2-mm was carried out using a Panalytical Xpert Pro with a PIXCEL 1D detector, angular range of 5 to 110° with ½° and 1° fixed slits. Radiation used was Co kα 1.788965 with a 45 Kv 40 Ma generator.
3 Results
3.1 Phosphorus sorption and EPC0 estimation
Phosphorus adsorption experiments showed that the purely < 2-mm fraction had the highest P retention (up to 65 mg P kg−1), and purely > 2-mm fraction had the lowest P retention (up to 30 mg P kg−1) at both sites (Fig. 2a, b). Linear regressions between initial P concentration in solution and P sorbed (r2 = 0.88–0.99) provided a strong basis for calculating EPC0 for these sediments. There were no statistically significant differences between EPC0 values with varying gravel proportions at sites A or B (P > 0.05). However, the EPC0 at site A, increased from 0.03 to 0.07 mg P L−1 as the proportion of gravel in the sediment increased (Table 2). Site B was dissimilar, with the purely > 2-mm sample having an EPC0 of 0.15 mg P L−1, 75% gravel and EPC0 of 0.12 mg P L−1, and 30% gravel and EPC0 of 0.17 mg P L−1 (Table 2). The application of the Freundlich equation (S = acb) to P sorption values (Fig. 2a, b and Table 2) identified that a, an indicator of P energy of adsoprtion (Pant and Reddy 2001) was negatively correlated to the amount of gravel in the sample at both sites.

Freundlich P adsorption curves for a site A and b site B. < 2-mm fraction (filled circle, short dash), whole sediment with 22% > 2-mm fraction or 30% > 2-mm fraction (filled square, long and short dash), whole sediment with 75% > 2-mm fraction (filled triangle, medium dash), 100% >2-mm fraction (filled diamond, solid line). Error bars indicate the 95% confidence interval of the medians
3.2 Phosphorus sorption by ground sediment components
Red-coloured material, indicative of the presence of Fe, dominated the > 2.8-mm size fraction. Below 2.8 mm in size, the material was predominantly non-red coloured, suggesting that the > 2.8-mm material had Fe minerals present that were more conducive to retaining P. Ground red-coloured > 2-mm fraction from site A sorbed 1073 mg P kg−1 at an initial concentration of 100 mg P L−1, 4–10 times more than the ground < 2-mm fraction and the yellow > 2-mm fraction. The ground red-coloured > 2-mm fraction adsorbed 1229 mg P kg−1 at an initial concentration of 100 mg P L−1, approximately twice as much as the ground pale yellow > 2-mm fraction and the ground < 2-mm fraction at site B. Exposing the interior of the > 2-mm fraction by grinding increased the sorption approximately ten times more compared to the intact > 2-mm material.
The X-ray diffraction analysis of a mixed (red- and yellow-coloured material) > 2-mm sample from each site identified their mineralogy and relative proportions of each of the minerals (Online resource 2, Electronic Supplementary Material). The major P-sorbing minerals present at both sites were the iron oxides haematite (α-Fe2O3) and goethite (α-FeO(OH)).
3.3 Phosphorus retention and release under simulated stream conditions in a fluvarium
During the uptake phases, each run was typified by a decay in FRP in solution over 48 h, and diminished cumulative P retained compared to the previous run. These within-run trends of diminished cumulative P retention were also evident within the uptake phase for each of the sediments examined. In addition, P was released at a slower rate than P uptake, showing some hysteresis. During the uptake phase, the % P retained diminished between each 48 h run (Fig. 3a, b). The uptake rate declined with repeated additions of P (Fig. 4). During sequential release phases, 36% of the P sorbed at site A and 9–15% of the P sorbed at site B (Table 3) was released. For all sediments, P retention diminished with consecutive additions of P at sites A and B (Fig. 3a, b). Site A sediment retained up to 83% of the P added in the first run, but only 18–30% of the added P after the tenth run (Fig. 3a). Site B retained less P after one addition of P compared to site A, with a maximum of 50% of added P being retained. After five consecutive additions of P, sorption of P by sediments from site B was reduced to 15–30% of added P (Fig. 3b). After two sequential P release phases, the capacity to retain P at site B increased.

a % P retained by sediment for site A for each run. The closed symbols represent P retained after the addition of P after four sequential release phases of P. b % P retained by sediment for site B for each run. The closed symbols represent P retained after the addition of P after two sequential release phases of P

Cumulative P sorbed (mg kg−1) by sediment at sites A and B
During the first uptake phase, site A removed 71–84% of the added P whereas site B removed 29–46% of the added P (Fig. 4). The > 2-mm fraction from site B required four uptake phases of approximately 2.5 mg P L−1 to sorb the same amount of P sorbed during the first uptake phase at site A.
4 Discussion
This study identified the importance of the > 2-mm fraction in the regulation of soluble P by stream sediments. Previous studies ignore the > 2-mm fraction on the assumption that it is inert or contributes very little to P dynamics (Miller and Guthrie 1984; Stone and English 1993; Webster et al. 2001; van der Perk et al. 2007; Ryan et al. 2007; Zhuan-xi et al. 2009). The > 2-mm fraction in these studies was not inert and does not simply occupy volume and acts as a diluent. All the sediments tested in this study behaved like soils in that repeated applications of P diminished the sediments capacity to adsorb more P. Both mineralogy and surface area of various size fractions are important determinants of P retention characteristics for the whole sediment.
The > 2-mm fraction, its mineralogical composition and possible porosity (Tiessen et al. 1991, 1993) can influence P retention and release. The types of minerals that comprise the > 2-mm fraction will influence the ability of this fraction to sorb P. For the sediments studied here, the > 2-mm fraction contained P-binding minerals including goethite (α-FeO (OH)), kaolinite (Al2Si2O5(OH)4) and gibbsite (Al(OH)3 which is consistent with previous analysis of iron oxides in soils of southwest WA (Singh and Gilkes 1992). Iron is considered important in the sorption of P (Murphy 1939; Borggaard 1983; Haggard et al. 1999; Kröger and Moore 2011). This is reflected in the mineralogical differences between the two sites and the potential for the different size fractions to sorb P. Despite having a smaller surface area, the capacity of the > 2-mm fraction to sorb P was similar to the < 2-mm fraction because of the low proportion of Fe in the < 2-mm fraction.
Specific surface area measurements of the > 2-mm fractions of soils and sediments in the literature suggest that even relatively inert materials in the 2–6-mm size range can have surface areas as large as primary minerals such as kaolinite (Tokunga et al. 2003). Additionally Drizo et al. (1999), reported a surface area of 6.8 m2 g−1 for bauxite (a similar material to the > 2-mm red coloured fraction in the sediments studied here) which was 97% by weight > 6.8 mm in size. This is comparable to the specific surface area of kaolinite at 8 m2 g−1. The much greater P sorption by stream sediment that had been through the grinding process than unground stream sediment in our study reinforces that P is sorbed predominantly to the exterior surface of the > 2-mm fraction (Weaver et al. 1992); however, some lateritic gravels are known to be porous (Tiessen et al. 1991, 1993). The > 2-mm fraction adsorbed considerably more P per unit area than either the < 2-mm fraction or the whole sediment. This may be due to our estimation of surface area rather than direct measurement (Alymore et al. 1970). Our estimate of P sorbed by gravel may be conservative, as not all the material was red coloured with an estimated 20% of the gravel non-red in colour. For the red coloured lateritic > 2-mm fraction, a specific surface area of 0.0011 m2 g−1 was estimated for a 2-mm-sized gravel. This is 3 to 4 orders of magnitude less than specific surface area of similar bauxitic (lateritic) material reported by Drizo et al. (1999). Therefore, it is likely that the > 2-mm fraction has considerably higher surface area than we estimated. The > 2-mm fraction should not be excluded from the assessment of P retention by sediments, particularly those with reactive gravels.
In the fluvarium, the rate of P uptake by sediment did not remain constant with sequential P additions. This was likely due to displacement of singly coordinated hydroxyl groups by the phosphate ion (Barrow 1983a; Froelich 1988; Stone et al. 1995; Lair et al. 2009), which reduces the binding sites available for the next addition of P (Barrow 1974). Another explanation, as presented by Barrow et al. (1998) identified that the increased presence of phosphate ions which are negatively charged provides a negative charge on the particle surface. The presence of increased negative charge decreases the electric potential of the sediment and thus reduces the capacity of the sediment to hold more P. Desorption of lesser amounts of P in the release phase suggests that the majority of P was strongly retained by the sediment (Barrow 1974, 2008). This is consistent with catchment scale P budgets in a WA coastal catchment that suggested significant P retention by stream sediment (Keipert et al. 2008). Barrow (1989) further identified that desorption processes, whilst much slower than diffusive adsorption processes in soils, offer potential for more P to be released over time. However, other factors influencing desorption, such as stream water concentration and sediment EPC0, also need to be taken into account.
Rogers et al. (2013) identified that the sediment in their study removed 85–96% of the P. Sharpley et al. (2007) obtained similar results; however, this previous research included only one retention phase in a fluvarium. In the first run of the first uptake phase conducted here, the percentage of P removed by site A was similar to that of Sharpley et al. (2007) and Rogers et al. (2013). However, the percentage of P sorbed decreased with sequential additions of P, likely due to a decrease in the P buffering capacity of the sediment (Barrow 1974; Barrow et al. 1998). Likewise, more P was desorbed by sediment in the early release phases than subsequent release phases, most likely due to loosely bound P (Barrow 1983b). Thus, basing sediment P-release characteristics on a single desorption curve or release phase may overestimate P desorption, dependent on the sequence of events that occur in a stream environment. This finding is important to both future research on sediments and instream P transfer modelling, as it identified that stream sediment P uptake is dynamic. A single uptake curve will not adequately describe temporal P dynamics and may overestimate sediment P retention capacity, hence modelling must consider temporal decreases in sediment P-buffering.
The < 2-mm fraction adsorbed the most P in the batch experiments whereas the same fraction in the fluvarium experiment adsorbed the least. Lower P sorption in the fluvarium may be due to reduced contact between the <2-mm fraction and P, and reduced opportunity for P diffusion. Once the pore space of the < 2-mm fraction is saturated with water, the surface area of the sediment matrix that is in direct contact with flowing water is reduced because water flows over the top of the sediment rather than through it. The > 2-mm fraction has larger pore spaces due to its particle size, allowing P-rich water to enter and interact with much of the surface area of the gravel. Similarly, different soil:solution ratios will affect the adsorption of P in laboratory experiments (White 1966; Hope and Syers 1976; Barrow and Shaw 1979). A soil:solution ratio of 1:20 was used in batch experiments compared with a 1:4 ratio in the fluvarium. Phosphorus sorbed after 24 h for the same initial P concentration at a soil:solution ratio of 1:4 was between 20 and 80% less than at a soil:solution ratio of 1:20. This is because for a soil:solution ratio of 1:20, there will be more P available to be adsorbed than at 1:4 ratio.
Sorbed P varied for sediment components within a site, as well as between sites. The within- and between-site variability may be explained by a range of factors, including the greater abundance of the iron oxide (haematite) at site B. Haematite has a lower affinity and slower uptake of P (Torrent et al. 1994) compared to goethite, which was in higher proportion at site A. We found no consistent increase in the EPC0 values with increasing gravel proportions as demonstrated by Haggard and Sharpley (2007) and McDowell et al. (2002) for increasing sand proportions. This suggests that the mineralogy of the > 2-mm fraction may be as important as particle size in regulating stream P concentrations. On a finer fraction of sediment (< 0.5-mm), Stone and Murdoch (1989) suggested that the key factors controlling P adsorption onto sediment were chemical composition and mineralogy, which were sometimes more important than particle size.
Our results suggest that sediments at both sites were net immobilisers of P. The ‘whole’ sediments generally retained more P than either the 100% < 2-mm or > 2-mm sediments. This may be due to a combination of factors such as (a) the high P-sorbing mineralogy of the > 2-mm fraction and (b) the higher surface area of the low P-sorbing < 2-mm fraction which occupied voids between the > 2-mm particles and provided high surface area for P adsorption. The combination of < 2-mm and > 2-mm fractions may also offer the greatest opportunities for surface contact between P in flowing water and particles.
5 Conclusions
The stream sediments examined here from south western WA are net immobilisers of P, retaining more P than they release, depending on the stream P concentration.
The > 2-mm fraction can be an important component of stream sediments as it influences both the retention and release of P. The > 2-mm fraction can retain P if it has iron-rich mineralogy and should be considered when estimating sediment characteristics such as EPC0. Measuring only the < 2-mm fraction of a sediment that also contains reactive fractions > 2-mm may underestimate the sediment’s capacity to adsorb P, particularly when much of the < 2-mm fraction is P-inert. Similarly, if a sediment has a P-inert > 2-mm fraction, measuring the uptake and release of only the < 2-mm fraction may overestimate sediment P retention capacity, because the inert > 2-mm fraction occupies volume, which may not be accounted for if measuring only the < 2-mm fraction.
Similar to soils, P sorption by sediment diminishes with repeated P additions. Experiments with single uptake and release phases do not adequately describe the long-term potential of sediment to retain or release P as sorbed P accumulates. Expressing P sorbed per unit area may be a useful adjunct to traditional mass-based P-sorption metrics, and may assist to better understand the potential for different sedimentary components to retain P.
References
Allen D, Jeffery R (1990) Methods of analysis of phosphorus in Western Australian soils. Report of investigation no. 37. East Perth. http://www.chemcentre.wa.gov.au/Files/pdf/publication-pdf/Method-for-Analysis-of-Phosphorus-in-WA-Soils.aspx. Accessed January 2019
Alymore LAG, Sills ID, Quirk J (1970) Surface area of homoionic illite and montmorillonite clay minerals as measured by the sorption of nitrogen and carobon dioxide. Clay Clay Miner 18:91–96
Ballantine DJ, Walling DE, Collins AL, Leeks GJL (2009) The content and storage of phosphorus in fine-grained channel bed sediment in contrasting lowland agricultural catchments in the UK. Geoderma 151:141–149
Barrow N (1974) Effect of previous additions phosphate on phosphate adsorption by soils. Soil Sci 118:82–89
Barrow NJ (1983a) On the reversibility of phosphate sorption by soils. J Soil Sci 34:751–758
Barrow NJ (1983b) A mechanistic model for describing the sorption and desorption of phosphate by soil. J Soil Sci 34:733–750
Barrow NJ (1989) The reaction of plant nutrients and pollutants with soil. Aust J Soil Res 27:475–492
Barrow NJ (2008) The description of sorption curves. Eur J Soil Sci 59:900–910
Barrow NJ, Shaw TC (1979) Effects of solution: soil ratio and vigour of shaking on the rate of phosphate adsorption by soil. Eur J Soil Sci 30:67–76
Barrow N, Bolland M, Allen D (1998) Effect of previous additions of superphosphate on sorption of phosphate. Aust J Soil Res 36:359–372
Borggaard OK (1983) The influence of iron oxides on phosphate adsorption by soil. J Soil Sci 34:333–341
Brearly A (2005) Ernest Hodgkin’s Swanland: estuaries and coastal lagoons of southwestern Australia, 1st edn. University of Western Australia
Buczko U, Kuchenbuch RO (2007) Phosphorus indices as risk-assessment tools in the U.S.A. and Europe—a review. J Plant Nutr Soil Sci 170:445–460
Drizo A, Frost CA, Grace J, Smith KA (1999) Physico-chemical screening of phosphate-removing substrates for use in constructed wetland systems. Water Res 33:3595–3602
Froelich PN (1988) Kinetic control of dissolved phosphate in natural rivers and estuaries: a primer on the phosphate buffer mechanism. Limnol Oceanogr 33:649–668
Gourley CJP, Weaver DM (2012) Nutrient surpluses in Australian grazing systems: management practices, policy approaches, and difficult choices to improve water quality. Crop Pasture Sci 63:805–818
Haggard B, Sharpley A (2007) Phosphorus transport in streams: processes and modeling considerations. In: Radcliffe D, Cabrera M (eds) Modeling phosphorus in the environment. CRC. USA, Boca Raton, pp 105–130
Haggard BE, Stanley EH, Hyler R (1999) Sediment-phosphorus relationships in three northcentral Oklahoma streams. Trans ASAE 42:1709–1714
Haygarth PM, Jarvis SC (1999) Transfer of phosphorus from agricultural soils. Adv Agron 66:195–249
Hillman K, Lukatelich RJ, Bastyan G, McComb AJ (1990) Distribution and biomass of sea grasses and algae, and nutrient pools in water, sediments and plants in Princess Royal Harbour and Oyster Harbour. Environmental Protection Authority Technical Series 40, November 1990
Hope G, Syers J (1976) Effects of solution: soil ratio on phosphate sorption by soils. J Soil Sci 27:301–306
Isbell R, McDonald W, Ashton L (1997) Concepts and rationale of the Australian soil classification. Australian Collaborative Land Evaluation Program, Canberra, ACT
Jarvie HP, Sharpley A, Spears B, Buda A, May L, Kleinman PJA (2013) Water quality remediation faces unprecedented challenges from “legacy phosphorus”. Environ Sci Technol 47:8991–8998
Keipert N, Weaver D, Summers R, Clarke M, Neville S (2008) Guiding BMP adoption to improve water quality in various estuarine ecosystems in Western Australia. Water Sci Technol 57:1749–1756
Kovar JL, Pierzynski GM (2009) Methods of phosphorus analysis for soils, sediments , residuals, and waters. South Coop Ser Bull 408:1–131
Kröger R, Moore MT (2011) Phosphorus dynamics within agricultural drainage ditches in the lower Mississippi alluvial valley. Ecol Eng 37:1905–1909
Kusmer AS, Goyette JO, MacDonald GK, Bennett EM, Maranger R, Withers PJA (2018) Watershed buffering of legacy phosphorus pressure at a regional scale: a comparison across space and time. Ecosystems. https://doi.org/10.1007/s10021-018-0255-z
Lair GJ, Zehetner F, Khan ZH, Gerzabek MH (2009) Phosphorus sorption – desorption in alluvial soils of a young weathering sequence at the Danube River. Geoderma 149:39–44
Lucci GM, McDowell RW, Condron LM (2010) Evaluation of base solutions to determine equilibrium phosphorus concentrations (EPC0) in stream sediments. Int Agrophys 24:157–163
Master R (2008) Oyster Harbour catchment appraisal - Resource management technical report 320. Albany, Western Australia. https://researchlibrary.agric.wa.gov.au/rmtr/302/. Accessed January 2019
Master R (2009) Wilson Inlet catchment appraisal 2007 - Resource management technical report 329 https://researchlibrary.agric.wa.gov.au/rmtr/311/. Accessed January 2019
McDowell RW, Sharpley AN (2003) Uptake and release of phosphorus from overland flow in a stream environment. J Environ Qual 32:937–948
McDowell RW, Sharpley AN, Chalmers AT (2002) Land use and flow regime effects on phosphorus chemical dynamics in the fluvial sediment of the Winooski River, Vermont. Ecol Eng 18:477–487
McDowell RW, Sharpley AN, Folmar G (2003) Modification of phosphorus export from an eastern USA catchment by fluvial sediment and phosphorus inputs. Agric Ecosyst Environ 99:187–199
McDowell RW, Biggs BJF, Sharpley AN, Nguyen L (2004) Connecting phosphorus loss from agricultural landscapes to surface water quality. Chem Ecol 20:1–40
McKergow LA, Weaver DM, Prosser IP, Grayson RB, Reed AEG (2003) Before and after riparian management : sediment and nutrient exports from a small agricultural catchment, Western Australia. J Hydrol 270:253–272
Miller FT, Guthrie RL (1984) Classification and distribution of soils containing rock fragments in the United States. In 'SSSA Special Publication, Erosion and Productivity of Soils Containing Rock Fragments, 13:1–6.' (Soil Science, Society of America, 677 South Segoe Road, Madison, WI 53711. Erosion and Productivity of Soils Containing Rock Fragments
Moore G (1998) Soilguide. A handbook for understanding and managing agricultural soils. Agriculture Western Australia Bulletin No. 4343, Perth, Western Australia
Murphy HF (1939) Clay minerals and phosphate availability: 1. Adsoprtion of phosphate ions by clay minerals. Proc Oklahoma Acad Sci 1939:79–81
Murphy J, Riley JP (1962) A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27:31–36
Pant HK, Reddy KR (2001) Phosphorus sorption characteristics of estuarine sediments under different redox conditions. J Environ Qual 30:1474–80
Rayment GE, Lyons DJ (2011) Soil chemical methods - Australasia. CSIRO Publishing
Rogers CW, Sharpley AN, Haggard BE, Scott JT, Drake BM (2011) Physicochemical characterization of sediment in northwest Arkansas streams. J Environ Prot 2:629–638
Rogers CW, Sharpley AN, Haggard BE, Scott JT (2013) Phosphorus uptake and release from submerged sediments in a simulated stream channel inundated with a poultry litter source. Water Air Soil Pollut 224:1361
Ryan RJ, Packman AI, Kilham SS (2007) Relating phosphorus uptake to changes in transient storage and streambed sediment characteristics in headwater tributaries of Valley Creek, an urbanizing watershed. J Hydrol 336:444–457
Schoknecht N, Pathan S (2013) Soil groups of Western Australia: a simple guide to the main soils of Western Australia (4th ed.) https://researchlibrary.agric.wa.gov.au/rmtr/348/. Accessed January 2019
Schwertmann U (1993) Relation between iron oxides, soil color, and soil formation. Soil Sci Soc Am J 31:51–69
Sharpley A, Tunney H (2000) Phosphorus research strategies to meet agricultural and environmental challenges of the 21st century. J Environ Qual 29:176–181
Sharpley AN, Chapra SC, Wedepohl R, Sims JT, Daniel TC, Reddy KR (1994) Managing agricultural phosphrous for protection of surface waters: issue and options. J Environ Qual 23:437–451
Sharpley A, Krogstad T, Kleinman P et al (2007) Managing natural processes in drainage ditches for nonpoint source phosphorus control. J Soil Water Conserv 62:197–206
Sharpley A, Jarvie HP, Buda A, May L, Spears B, Kleinman P (2013) Phosphorus legacy: overcoming the effects of past management practices to mitigate future water quality impairment. J Environ Qual 42:1308–1326
Simpson ZP, McDowell RW, Condron LM (2018) The error in stream sediment phosphorus fractionation and sorption properties effected by drying pretreatments. J Soils Sediments. https://doi.org/10.1007/s11368-018-2180-3
Singh B, Gilkes RJ (1992) Properties and distribution of iron oxides and their association with minor elements in the soils of south-estern Australia. J Soil Sci 43:77–98
Stone M, English MC (1993) Geochemical composition, phosphorus speciation and mass transport of fine-grained sediment in two Lake Erie tributaries. Hydrobiologia 253:17–29
Stone M, Murdoch A (1989) The effect of particle size, chemistry and mineralogy of river sediments on phosphate sorption. Environ Technol Lett 10:501–510
Stone M, Mulamoottil G, Logan L (1995) Grain size distribution effects on phosphate sorption by fluvial sediment: implications for modelling sediment- phosphate transport. Hydrol Sci 40:67–81
Taylor AW, Kunishi HM (1971) Phosphate equlibria on stream sediment and soil in a watershed draining an agricultural region. J Agric Food Chem 19:827–831
Tiessen H, Frossard E, Mermut AR, Nyamekye AL (1991) Phosphorus sorption and properties of ferruginous nodules from semiarid soils from Ghana and Brazil. Geoderma 48:373–389
Tiessen H, Abekoe MK, Salcedo IH, Owusu-Bennoah E (1993) Reversibility of phosphorus sorption by ferruginous nodules. Plant Soil 153:113–124
Tokunga TK, Olson KR, Wan J (2003) Moisture characteristics of Hanford gravels: bulk, grain-surface, and intragranular components. Vadose Zone J 2:322–329
Torrent J, Schwertmann U, Fechter H, Alferez F (1983) Quantitative relationships between soil color and hematite content. Soil Sci 136:354–358
Torrent J, Schwertmann U, Barrón V (1994) Phosphate sorption by natural hematites. Eur J Soil Sci 45(1):45–51
van der Perk M, Owens PN, Deeks LK, Rawlins BG, Haygarth PM, Beven KJ (2007) Controls on catchment-scale patterns of phosphorus in soil, streambed sediment, and stream water. J Environ Qual 36:694–708
Walling DE (2005) Tracing suspended sediment sources in catchments and river systems. Sci Total Environ 344:159–184
Weaver DM, Summers RN (2014) Fit-for-purpose phosphorus management: do riparian buffers qualify in catchments with sandy soils? Environ Monit Assess 186:2867–2884
Weaver DM, Wong MTF (2011) Scope to improve phosphorus (P) management and balance efficiency of crop and pasture soils with contrasting P status and buffering indices. Plant Soil 349:37–54
Weaver DM, Ritchie GSP, Gilkes RJ (1992) Phosphorus sorption by gravels in lateritic soils. Aust J Soil Res 30:319–330
Webster IT, Ford PW, Hancock G (2001) Phosphorus dynamics in Australian lowland rivers. Mar Freshw Res 52:127–137
White RE (1966) Studies on the phosphate potential of soils IV. The mechanism of the “soil/solution ratio effect”. Aust J Mar Freshwat Res 4:77–85
Zhuan-xi L, Bo Z, Jia-liang T, Tao W (2009) Phosphorus retention capacity of agricultural headwater ditch sediments under alkaline condition in purple soils area, China. Ecol Eng 35:57–64
Acknowledgements
We thank the landowners who kindly allowed us to access their properties for sample collection.
Funding
This work was supported with research funding from the WA State Centre of Excellence for Ecohydrology, UWA and the Department of Primary Industries and Regional Dvelopment and received financial support from the University of Western Australia through University Postgraduate Award and Top-up scholarships.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Brian Kronvang
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Clarendon, S.D.V., Weaver, D.M., Davies, P.M. et al. The influence of particle size and mineralogy on both phosphorus retention and release by streambed sediments. J Soils Sediments 19, 2624–2633 (2019). https://doi.org/10.1007/s11368-019-02267-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-019-02267-w