
Abstract
Although age-related macular degeneration (AMD) is a multifactorial disorder with angiogenic, immune, and inflammatory components, the most common clinical treatment strategies are antiangiogenic therapies. However, these strategies are only applicable to neovascular AMD, which accounts for less than 20% of all AMD cases, and there are no FDA-approved drugs for the treatment of dry AMD, which accounts for ~ 80% of AMD cases. Here, we report that the elimination of senescent cells is a potential novel therapeutic approach for the treatment of all types of AMD. We identified senescent retinal pigment epithelium (RPE) cells in animal models of AMD and determined their contributions to retinal degeneration. We further confirmed that the clearance of senescent RPE cells with the MDM2-p53 inhibitor Nutlin-3a ameliorated retinal degeneration. These findings provide new insights into the use of senescent cells as a therapeutic target for the treatment of AMD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Age-related macular degeneration (AMD) is a progressive neurodegenerative disorder of the retinal pigment epithelium (RPE), retina, and choriocapillaris and the leading cause of vision loss in developed countries [1, 2]. AMD is divided into early, intermediate, and advanced forms. Early and intermediate AMD are characterized by extracellular deposits called drusen, focal hyper- or hypopigmentation in the retina, and the thickening of Bruch’s membrane [3]. Advanced forms of AMD include neovascular AMD (nAMD) with choroidal neovascularization (CNV) or geographic atrophy (GA), which involves atrophy or loss of RPE, photoreceptors and underlying choriocapillaris [4] and causes blindness. No FDA-approved treatment is available for early and intermediate AMD and GA (these three types are classified as “dry” AMD), which account for 80–90% of AMD cases. nAMD accounts for 10–20% of AMD cases, and currently, vision loss attributable to nAMD has been reduced by 50–70% in some countries due to the use of anti-vascular endothelial growth factor (anti-VEGF) treatments targeting the most dominant angiogenic mediator, VEGF, in the eye [5,6,7]. However, repetitive intravitreal injections of anti-VEGF are needed for many years to control disease activity in the majority of patients with nAMD [8], and GA frequently develops within 5 years of starting nAMD therapy [9]. Moreover, up to 30% of patients with nAMD do not respond to anti-VEGF drugs [7, 10, 11].
Although the exact mechanism of AMD is not completely understood, environmental conditions such as aging and oxidative stress and evidence of inflammatory and immune system disturbances have accumulated in the eye over time. Analyses of single nucleotide polymorphisms have also revealed that intronic and common variants in genes, including the complement factor H gene, increase the risk of AMD [12,13,14]. In addition to antiangiogenic therapeutics, drugs currently being investigated in clinical trials for AMD mostly target complement regulation and/or inflammation. However, most of these attempts have failed [15], which is not surprising because AMD is caused by multiple factors, which are still incompletely understood. Thus, comprehensive therapeutic approaches to the development of a new AMD drug are needed.
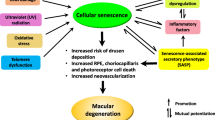
Cellular senescence, an irreversible type of cell growth arrest, protects against cancer but also contributes to tissue deterioration underlying aging and age-related pathologies [16]. Senescent cells accumulate in tissues upon aging and adopt a senescence-associated secretory phenotype (SASP), which involves the secretion of proinflammatory cytokines, chemokines, and growth factors and is associated with the pathophysiology of age-related diseases, including osteoarthritis, atherosclerosis, chronic obstructive pulmonary disease, and age-related brain diseases [17]. Aging-accelerated OXYS rats show AMD-like retinopathy, including RPE degeneration and loss of photoreceptors [18]. In addition, senescent cells have been detected in the retinas and RPEs of older humans [19] and primates [20]. Although the roles of senescent RPE cells in AMD pathogenesis and development/progression in vivo remain unknown, several of the proposed causes of AMD, including oxidative stress, DNA damage, and mitochondrial reactive oxygen species, are known to induce and be linked to cellular senescence [21,22,23,24,25,26]. Therefore, strategies targeting senescent RPE cells might be new therapeutic approaches for treating AMD.
Currently, two major strategies for targeting senescent cells have been developed. First, chronic inflammation is reduced by suppressing the release of SASP from senescent cells, ameliorating the “bystander effect” or paracrine senescence [27, 28]. However, in the context of AMD treatment, the strategies for reducing SASP, although they appear rather safe, are insufficient because drugs targeting complement and inflammatory dysregulation have been proven to be unsuccessful [15, 29]. A more fundamental and groundbreaking strategy is needed. Second, the elimination of the source of chronic inflammation by removing senescent cells from tissues has been reported to improve tissue homeostasis and ameliorate several disease conditions [30, 31]. The application of these “senolytics” as a therapy for AMD has never been reported. However, because RPE atrophy or death is progressive and deteriorates due to the centrifugal progressive nature at the boundary of GA [32], the progression of GA might conceivably be related to the SASP of neighboring senescent RPE cells, affecting the healthy RPE cells around them. Here, we hypothesize that the cellular senescence of the RPE may lead to retinal degeneration and the key pathological phenotypes of AMD. We initially developed a chemically induced mouse model of RPE cell senescence and established measurable parameters of RPE cell senescence in vitro and in vivo to address this crucial question. Then, we investigated whether the clearance of senescent RPE cells prevented retinal degeneration in this model, mouse models of AMD (laser-induced CNV models and a GA model), and old mice [33,34,35].
Methods
Human RPE cell line and induction of senescence and drug treatment in cell culture
ARPE-19 cells were obtained from American Type Culture Collection (ATCC, CRL-2302, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (WellGene, LM 002–04, Gyeongsangbuk-do, Republic of Korea) supplemented with 10% fetal bovine serum (Invitrogen 16,000, Waltham, USA) and penicillin–streptomycin (pen/strep, WellGene, LS 202–02) at 37 °C in a humidified incubator with 5% CO2. Cells were used after reaching approximately 70–80% confluence and at passages 15–25.
ARPE-19 cells were seeded into 24- or 6-well plates at a density of 25,000 or 100,000 cells per well prior to the induction of cellular senescence with doxorubicin (Dox; Tocris Bioscience #2252, Bristol, UK). After 24 h, cells were treated with 250 nM Dox for 3 days. After 3 days, the cells were placed in Dox-free medium, which was changed daily. At 10 days, Dox-treated senescent cells (SnCs) and nonsenescent cells (non-SnCs; only the culture medium was changed) were seeded in 24- or 6-well plates for further experiments. After 24 h of incubation, the cells were treated with vehicle (0.2% DMSO) or various concentrations of Nutlin-3a (Sigma, SML-0580, St. Louis, MO, USA) for 24 h.
Mitochondrial ROS for live cell imaging
MitoSox (Invitrogen, M36008, CA, USA) was used to visualize mitochondrial ROS production in ARPE-19 cells. ARPE-19 cells were treated with Dox (250 nM or 1 μM) for 24 h at 37 °C in a humidified incubator with 5% CO2. After incubation, ARPE-19 cells were treated with 5 µM MitoSox solution for 10 min at 37 °C in a humidified incubator with 5% CO2. Mitochondrial ROS in live cells were observed with an inverted microscope (LSM 900, Carl Zeiss, Oberkochen, Germany).
Cell cycle distribution analysis
For cell cycle distribution analysis, the FUCCI-ARPE19 cell line was established using lentivirus. To package lentivirus, HEK293T cells were seeded at ~ 80% confluence the day before transfection. HEK293T cells in 100-mm culture dishes were transfected with 10 μg of pBOB-EF1-FastFUCCI-Puro, 8 μg of psPAX2, and 3 μg of pMD2.G using Lipofectamine 3000 reagent (Life Technologies, Gaithersburg, MD, USA). Two days posttransfection, the cell culture supernatant was filtered through a 0.45-μm cellulose acetate filter (Sartorius) and treated with 8 μg/ml polybrene. After lentiviral infection, FUCCI-ARPE19 cells were selected with 5 μg/ml puromycin, and fluorescence was detected by inverted fluorescence microscopy (Axio200, Carl Zeiss).
Cell viability assay
The toxicity to Nutlin-3a on ARPE-19 cells was assessed using Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). ARPE-19 cells were treated with Nutlin-3a at various concentrations (0, 1, 5, 10, 20, 50, and 100 μM) for 24 h at 37 °C. After 24 h, 10 μl CCK-8 solution with 90 μl of medium was added to the well, and the cells were incubated for 3 h at 37 °C in a humidified incubator with 5% CO2. Cell viability was measured with an ELISA reader (SpectraMAX 190 Microplate Reader, Molecular Devices, CA, USA) at 450 nm.
RNA isolation and real-time PCR
Total RNA was isolated from ARPE-19 cells cultured in 6-well plates or mouse RPE under a microscope using TRIzol reagent (Invitrogen, 15,596,026). After RNA isolation, mRNAs were transcribed to cDNA using reverse transcriptase (Thermo Fisher Scientific, 4,368,813). Human and mouse mRNAs were quantified with specific primers in 40 cycles using a CFX Connect Real-Time system (BIO-RAD, CA, USA).
PCR products were subjected to melting curve analysis. Relative mRNA expression was calculated using the 2−ΔΔCt method. GAPDH served as the internal reference gene. Mean Ct values were normalized to GAPDH. Real-time PCR was performed at least in triplicate for each group. See Table S1 for the list of primers.
Multiplex ELISA
Conditioned medium (CM) from nonsenescent ARPE-19 cells, senescent ARPE-19 cells, and senescent ARPE-19 cells treated with Nutlin-3a was collected, filtered, and stored at − 80 °C. The levels of IL-6, IL-8, MMP-1, MMP-2, MMP-3, and VEGF in CM were measured with a high-sensitivity bead-based multiplex ELISA using a Luminex analysis system from a cytokine service (KOMA Biotechnology, Seoul, Korea).
Experimental animals
Mice were maintained in accordance with the guidelines established by the Konkuk University Institutional Animal Care and Use Committee (IACUC) and housed in a controlled barrier facility within the Konkuk University Laboratory Animal Research Center. All experimental procedures and animal care were conducted according to guidelines approved by the Konkuk University IACUC (KU IACUC; approval No. KU18080). C57BL/6 J male mice (aged 8 weeks) were purchased from Charles River Laboratories Japan Inc. (Yokohama, Japan) and maintained until an age of 24 months. Male C57BL/6 J or C57BL/6 N mice (aged 8 weeks) were purchased from Charles River Laboratories Japan Inc. or Orient Bio (Seongnam, Korea) and were allowed to acclimate to the facility for 1 week before the experiments. For all experiments, the mice were randomly assigned to control or treatment groups. All animal experiments were conducted in a blinded manner. The mice were anesthetized with a mixture of Zoletil (Carros, France) and xylazine (Leverkusen, Germany) (4:1, diluted with normal saline), and pupils were dilated with topical Tropherine eye drops (single use, phenylephrine hydrochloride (5 mg/ml) and tropicamide (5 mg/ml), Hanmi Pharm, Seoul, Korea). An antibiotic ophthalmic ointment (Tarivid, Santen, Osaka, Japan) was applied to all eyes after the procedures.
Synthesis of in vitro transcribed Alu RNA
pT7Alu (T7 promoter-containing Alu expression plasmid, Goodrich laboratory, Boston, USA) was used to synthesize the Alu RNA as previously described [35, 36]. Briefly, the plasmid was incubated with DraI in 10 × FastCut buffer at 37 °C for 1 h to linearize the plasmid template. If the linearized DNA contained T7Alu, bands of 19, 692, 1131, and 1146 bp were observed after electrophoresis on a 1% agarose gel in 1 × TAE buffer. The linearized DNA was purified with phenol:chloroform:isoamyl alcohol solution. Alu RNA was generated from linearized DNA with an Epicentre AmpliScribe T7 Flash transcription kit (#ASF3507, Epicentre, Madison, WI, USA) according to the manufacturer’s instructions. RNA was purified with a NucleoSpin RNA clean-up kit (Macherey–Nagel, #740,948.50, Düren, Germany).
Subretinal and intravitreal injection
Under an optical microscope (Olympus SZ51, Tokyo, Japan), a small hole was created at the limbus with a 30-gauge sterile needle (BD Science, San Jose, USA). Then, a blunt 35-gauge Hamilton microsyringe (Hamilton Company, NV, USA) was inserted slowly through the hole. One microliter of 100 ng/µl Dox or 1.4 µg/µl Alu RNA was injected into the subretinal space of C57BL/6 N or C57BL/6 J mice, respectively. In some cases, 20 ng/µl Nutlin-3a (#SML0580, St. Louis, USA) or vehicle (0.4% DMSO in normal saline) was intravitreally injected with a blunt 35-gauge Hamilton microsyringe, followed by a subretinal injection.
Mouse model of laser-induced CNV and two-stage laser-induced mouse model
C57BL/6 N mice at the age of 9 weeks were anesthetized, and topical 0.5% proparacaine and Tropherine were dropped into their eyes for local anesthesia and pupil dilation, respectively. Laser photocoagulation was applied to the mice as described in previous studies [37, 38] with some modifications. A slit-lamp delivery system with a laser photocoagulator (532-nm laser, Carl Zeiss AG, Oberkochen, Germany) was used with the following settings: 200 mW, a 0.1-s duration, and a 50-μm spot size. Three laser spots were generated around the optic nerve head in each lubricated (with hypromellose) eye with a cover slip measuring 12 mm in diameter used as a contact lens. Gaseous bubbles confirmed the disruption of Bruch’s membrane, and only laser spots whose production produced bubbling were included in the study. Intravitreal injection of vehicle, Nutlin-3a (20 ng/µl), or aflibercept (Eylea, 2 µg/µl) was performed immediately after laser treatment. Intravitreal injection of vehicle or Nutlin-3a was additionally performed at 3 days; evaluations were conducted at 7 days.
To verify the treatment effect over a longer period of time and to simulate a more chronic stage of nAMD, a two-stage laser-induced mouse model was generated according to a recent paper by Little et al. [39]. Briefly, 7 days after the first laser photocoagulation, a second laser was applied. Three laser spots were generated again at the CNV lesion created at day 0. The laser settings were the same as those of the first laser photocoagulation. Nutlin-3a (20 ng/µl) was intravitreally injected at 0, 3, 7, and 10 days. The results were obtained twice, at 14 and 28 days.
Cryosectioning and hematoxylin and eosin (H&E) staining
The eye cup was enucleated and fixed in 4% paraformaldehyde for 12 h at 4 °C. After fixation, the eye cup was transferred to 15% sucrose for 12 h at 4 °C and transferred to 30% sucrose for at least 3 h until the eye cup settled to the bottom of the tube. Then, the eye cup was embedded in frozen section compound (FSC 22 Clear Frozen Section Compound; Biosystems Inc., Wetzlar, Germany). Frozen blocks were then cryosectioned (Leica CM1860, Leica Biosystems Inc.) at 10-µm thickness and stained with hematoxylin and eosin (H&E). Outer nuclear layer (ONL) thickness was measured in the stained sections by using ImageJ software (NIH, USA). ONL thickness averages were calculated for at least three different sections close to the optic nerve.
Assessment of RPE senescence: SA-β-gal assay performed on RPE/choroid flat mounts, retinal flat mounts, retinal sections, and cultured cells
Mice were deeply anesthetized with CO2 gas, and their eyes were immediately enucleated. First, the anterior segment was removed, and retinas were carefully removed from the RPE/choroid/sclera under a microscope. After washing with cold PBS, retinas, RPE/choroid/scleral complex tissues, or sectioned retinas were immediately fixed with the fixative solution provided with the SA-β-galactosidase (SA-β-gal) staining kit for 20 min at room temperature and stained with the provided staining solution mix according to the manufacturer’s protocol (BioVision, #K320, CA, USA). After overnight SA-β-gal staining at 37 °C, heavy melanin pigment in the RPE/choroid was bleached to reveal SA-β-gal staining obscured by the melanin pigment in these tissues. For depigmentation of the RPE/choroid, RPE/choroid/scleral complex tissues were immersed in 30% H2O2, incubated for 45 min on a 55 °C heat block, rinsed with PBS, and then flat mounted with fine-point forceps and a surgical blade (World Precision Instruments, FL, USA) under an optical microscope (Olympus SZ51). Images of stained retinal flat mounts, RPE/choroid flat mounts or sectioned retinas were captured using an inverted microscope (Leica Microsystems #DMi1, Wetzlar, Germany).
For SA-β-gal staining of ARPE-19 cells, cells were grown on 24-well plates, and senescence was induced. The cells were fixed with fixative solution for 15 min at room temperature, washed with PBS, and stained with the staining solution for 6 h. Images were captured using an inverted microscope (Carl Zeiss Axio Scope A1, Gottingen, Germany).
TUNEL assay
An in situ Cell Death Detection Kit (Roche, 11684795910) was used to detect cellular apoptosis following the manufacturer’s instructions. Retinal flat mounts and RPE/choroid flat mounts were fixed with freshly prepared 4% PFA for 1 h. The tissues were washed three times with PBS and then incubated in permeabilization solution for 2 min on ice. The fixed and permeabilized tissues were incubated in 50 μl of TUNEL reaction mixture for 1 h at 37 °C in a humidified atmosphere in the dark. The incubated tissues were washed three times with PBS and incubated with Hoechst 33342 (1:3000, Thermo Fisher Scientific, H3570) for 15 min at room temperature. After washing with PBS, tissues were mounted with mounting medium (Polysciences, 18606–20, PA, USA). The stained tissues were observed with an inverted microscope (Carl Zeiss, LSM 900, Oberkochen, Germany).
Immunofluorescence of RPE/choroid flat mounts, retinal flat mounts, sectioned retinas, and cultured cells and cell proliferation assay (BrdU immunolabeling)
Bromodeoxyuridine (BrdU, Sigma-Aldrich, B5002, MO, USA) solution (200 µl, 5 mg/ml) was injected into mice intraperitoneally using a sterile 30-G needle attached to a 1-ml syringe 4 days prior to enucleation. ARPE-19 cells were treated with 10 µM BrdU for 4 h at 37 °C in a humidified incubator with 5% CO2. Tissues or cells were fixed with 4% PFA for 20 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 min. For BrdU immunolabeling, tissues or cells were incubated for 30 min at 37 °C in 2 N HCl and washed with 0.1 M borate buffer (pH 8.5) for 5 min twice.
After blocking with 1% BSA in PBS for 1 h, fixed tissues or cells were incubated overnight at 4 °C with primary antibodies against BrdU (1:1000, Invitrogen, MA3-071, CA, USA), p53 (1:50, Santa Cruz Biotechnology, TX, USA), p21 (1:50, Santa Cruz Biotechnology), p16 (1:100, BD Bioscience), Ki67 (1:1000, Abcam, Cambridge, UK), HMGB1 (1:1000, Abcam, ab18256), H3K9me3 (1:1000, Abcam, ab8898), γ-H2AX (1:1000, Abcam, ab2893), ZO-1 (1:250, Invitrogen, 61–7300, 33–9100), APOE (1:50, Santa Cruz Biotechnology), RPE65 (1:250, Novus Biologicals, Centennial, CO, USA, NB100-355), GS-IB4 (1:300, Vector Laboratories, Burlingame, CA, USA, FL1201), or CD34 (1:200, Invitrogen, MA1-10202). The stained tissues or cells were washed with PBS for 5 min twice and incubated for 2 h at room temperature with the following Alexa Fluor-conjugated secondary antibodies: Alexa Fluor 555-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, A-11029, Waltham, USA), Alexa Fluor 488-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, A-21424), Alexa Fluor 555-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific, A-21424), or Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific, A-11034). All secondary antibodies were used at a dilution of 1:1000.
After incubation with secondary antibodies, tissues or cells were stained with the nuclear dye DAPI (1:4000, Thermo Fisher Scientific, 62248) or Hoechst 33342 (1:3000, Thermo Fisher Scientific, H3570) in PBS for 15 min at room temperature. Afterward, tissues were mounted with mounting medium (Polysciences, 18606–20, PA, USA). The stained cells were observed with an inverted microscope (, Oberkochen, Germany).
Western blotting
Mouse RPE cells or ARPE-19 cells grown on 6-well plates were lysed in RIPA buffer (Thermo Fisher Scientific, 89901) supplemented with a protease inhibitor cocktail (Roche, 11697498001, Basel, Switzerland). The protein concentration was quantified using a BCA assay (Pierce, 23227, Waltham, USA). Equal amounts of proteins were separated by SDS-PAGE on 10–12% polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked with 5% nonfat dry milk in buffer for 1 h and incubated overnight at 4 °C with anti-p53 (1:500, Santa Cruz Biotechnology, SC-126), anti-phospho-p53 (1:2000, Cell Signaling, 9284S), anti-p21 (1:500, Santa Cruz Biotechnology, SC-6246), anti-p16 (1:1000, Abcam, ab189034), anti-caspase-3 (1:2000, Cell Signaling, 9664S), and anti-β-actin (1:2000, Abcam, ab8226) antibodies, followed by incubation with horseradish peroxidase-conjugated anti-mouse IgG (1:5000, Cell Signaling, 7076S) antibodies. For HMGB1 immunoblotting, incubation with the anti-HMGB1 (1:1000, Abcam, ab79823) antibody was followed by incubation with horseradish peroxidase-conjugated anti-rabbit IgG (1:5000, Cell Signaling, 7074S). Immunoreactive proteins were visualized using a chemiluminescence substrate (Amersham, RDN2232, Little Chalfont, UK) and quantified by densitometry using ImageJ software (NIH, USA). All experiments were performed at least in triplicate.
Color fundus photography and fundus autofluorescence imaging
Color fundus photographs and fundus autofluorescence images of mouse eyes were captured after dilation with a TRC-50 IX camera (Topcon, Tokyo, Japan) linked to a digital imaging system (Nikon, Japan) and HRA2 (Heidelberg Engineering, Heidelberg, Germany), respectively.
Transmission electron microscopy
Eyeballs from mice were fixed with a solution containing 2% glutaraldehyde and 3.75% sucrose in 0.1 M sodium phosphate buffer (pH 7.0) for 30 min and washed with 0.1 M PBS containing 3.75% sucrose three times over 30 min to prepare samples for TEM. The eyeballs were postfixed with a solution containing 0.1% osmium tetroxide and 5% sucrose in a 0.05 M sodium phosphate buffer (pH 7.0) for 1 h and rinsed with distilled water three times. Samples were then dehydrated in a graded series of acetone solutions (20% steps) and embedded in epoxy resin. The resin was polymerized at 70 °C for 12 h. Ultrathin Sects. (100 nm) were obtained with an RMC CR-X ultramicrotome and then imaged under a JEOL JEM-1400 electron microscope. The thickness of Bruch’s membrane was measured using TEM. The average thickness of Bruch’s membrane was determined by measuring the thickness in 9 images from each group.
Full-field electroretinogram (Ganzfeld ffERG)
A dark-adapted scotopic ffERG (Ganzfeld ffERG) was recorded to examine overall retinal function and visual defects using a Ganzfeld ERG and the quick setup guide according to the manufacturer’s instructions (Phoenix Research Labs, Pleasanton, USA). Mice were dark adapted for more than 12 h for scotopic experiments and prepared for ERG experiments under deep red illumination. After the pupils were dilated with topical Tropherine eye drops, the mice were anesthetized using a mixture of zoletil and xylazine and placed on a heated stand to measure ERG signals. The eyes were lubricated with 2% hypromellose and contacted the corneal electrode. The results produced a sensitivity curve from ERG a- and b-wave amplitudes. Following the ERG measurement, ointment was applied to both eyes of the mice, and the mice were returned to the animal room after completely recovering on a warm heating mat.
Fluorescein angiography (FA) and quantitation of CNV in a mouse model of laser-induced CNV and a two-stage laser-induced mouse model
FA images were captured 7, 14, or 28 days after laser treatment with HRA2 (Heidelberg Engineering, Heidelberg, Germany). Five minutes after injecting 10% fluorescein sodium (Alcon, Geneva, Switzerland) into the peritoneal cavity (5 ml/kg), late-phase FA images were captured. CNV lesions were visualized as described in previous studies [37, 38] with some modifications. FITC-dextran (5 mg/kg) was intravenously injected for fluorescence labeling. After 3 min, the mice were sacrificed, and the eyes were extracted. The anterior segment of the eyeball and neural retina were immediately detached from the RPE/choroid tissue. The RPE/choroid tissue was fixed with 4% PFA for 30 min and washed three times with PBS. RPE/choroid tissues were cut with four relaxing radial incisions and flat mounted in Aqua Poly/Mount (#18,606–20, Polysciences, Inc., Warrington, PA, USA). Z-section images were captured using a confocal laser scanning microscope (FluoView 1000; Olympus, Tokyo, Japan). The z-section images were reconstructed and quantified using image analysis software (Metamorph; Molecular Devices, Sunnyvale, CA, USA).
Statistics
All data are presented as the mean ± S.D. Statistical significance (P values) was determined using unpaired two-tailed Student’s t-tests, and multiple datasets were compared using one-way ANOVA followed by Fisher’s least significant difference post hoc test or Tukey’s multiple comparisons test. All analyses were performed with GraphPad Prism 5 software (San Diego, CA, USA). Statistical significance was defined as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Results
Doxorubicin-induced cellular senescence in the RPE drives retinal degeneration
We established an in vitro model of RPE senescence using Dox, which induces the formation of superoxide anions and the DNA damage response and has been used as a cellular senescence-inducing chemical [40,41,42]. We exposed ARPE-19 cells, a human RPE cell line widely used in research on senescence [22, 23], to different doses of Dox. Apoptosis was induced at 1 µM; however, at 250 nM, apoptosis did not occur despite increased mitochondrial ROS (mtROS) levels (Supplemental Fig. 1A and B). To create an in vitro RPE senescence model in which > 95% of all cells were senescent, ARPE-19 cells were treated with 250 nM Dox for 3 days; then, the culture medium was refreshed, and the cells were cultured for another 4 days (Supplemental Fig. 1C). Cell cycle analysis revealed a significant increase in the proportion of cells in G1 phase, while the proportion of cells in the S, G2, and M phase was decreased during senescence induction, resulting in the accumulation cells in G1 phase at 4–11 days using a fluorescence ubiquitination cell cycle indicator (FUCCI) reporter system (Supplemental Fig. 1D) [43]. This in vitro model was likely stress-induced premature senescence (SIPS) caused by subcytotoxic concentrations of the DNA-damaging agent Dox and/or oxidative stress [44].
Along with cell cycle arrest at the G1 phase, the senescence phenotype induced by a low concentration of Dox was confirmed by elevated senescence-associated β-gal (SA-β-gal) staining and transcriptional activation of genes encoding senescence and SASP factors, including p53, p21, p16, IL-1β, IL-6, and Mmp-3 (Supplemental Fig. 2A, B, and C). Elevated expression of markers regulating key senescent pathways, such as p53, p21, and p16, was also identified using Western blot analyses (Supplemental Fig. 2D). Dox-induced senescent ARPE-19 cells (SnCs) also showed nuclear morphological abnormalities, formation of DNA double-strand breaks (γ-H2AX), loss of heterochromatin marker (H3K9me3), and a significant decline in cell proliferation, as determined by Ki67 immunofluorescence and a BrdU assay (Supplemental Fig. 2E). SnCs exhibited reduced levels of the nuclear high-mobility-group box 1 (HMGB1) protein, an extracellular alarmin secreted before the development of the SASP in senescent cells [41] (Supplemental Fig. 2F). The in vitro RPE senescence model was established by treating ARPE-19 cells with a low dose of Dox, which led to characteristics of SIPS, whose induction and expansion seem to be related to AMD pathogenesis [45].
Then, we developed a mouse model in which Dox led to rapid senescence in RPE cells to investigate the role of senescent RPE cells in the pathogenesis of retinal degeneration, including AMD [40, 44]. The systemic injection of 10 mg/kg/10 ml Dox caused no acute toxicity but induced therapy-induced senescence in the study by Demaria et al. [40]. Dox (100 ng/µl) was subretinally injected into mice, and cellular senescence and apoptosis in the RPE and retina were monitored by staining RPE/choroid flat mounts and retinal whole mounts for SA-β-gal and TUNEL at 3, 7, and 14 days (Supplemental Fig. 3 and Supplemental Fig. 4A, B, and C). At 3 days, no significant SA-β-gal or TUNEL staining was observed in either RPE/choroid flat mounts or retinal whole mounts from the eyes of Dox-injected mice compared to those from the eyes of controls. The level of SA-β-gal staining in RPE/choroid flat mounts peaked at 7 days after Dox injection and was decreased significantly at 14 days. However, no significant SA-β-gal staining was observed in retinal whole mounts until 14 days, suggesting that the cells that became senescent were RPE cells. No significant cellular apoptosis on RPE/choroid or retinal flat mounts was observed until 14 days. In addition to elevated levels of SA-β-gal staining (Supplemental Fig. 4D and E), RPE senescence was demonstrated by elevated levels of p53 and p21 (Supplemental Fig. 4F). Immunofluorescence analysis of retinal sections confirmed that p53 expression was primarily localized to the RPE/choroid layer (Supplemental Fig. 4G). Loss of nuclear HMGB1 was also shown in eyes exposed to Dox (Supplemental Fig. 4H).
A distinguishing characteristic of mice subretinally injected with Dox was the development of aggregates, which are one of the hallmarks of cellular aging and consist of damaged macromolecules, such as proteins and lipids, similar to drusen observed in patients with AMD. Increased expression of APOE, a well-known component of drusen in patients with AMD [46], was observed in mice subretinally injected with Dox (Supplemental Fig. 4I). Collectively, this mouse model was validated as a model of RPE senescence, which is transient and may be naturally cleared in young adult mice. The subretinal injection of Dox induces cellular senescence in the RPE, which contributes to retinal degeneration. The AMD-like phenotype demonstrated the clinical relevance of this model.
Elimination of senescent RPE cells using senolytic Nutlin-3a alleviates retinal degeneration
Then, we investigated whether the removal of Dox-induced senescent RPE cells would reduce the progression of retinal degeneration. We utilized a senolytic drug that selectively induces apoptosis in senescent cells by targeting proteins associated with the antiapoptotic pathway, including the BCL2 family, p53, PI3K, HSP90, and HIF1α [47]. Based on our observation of the increased p53 level in senescent cells compared to nonsenescent cells (Supplemental Fig. 2C and D, Supplemental Fig. 4F and G), we hypothesized that an MDM2 antagonist would disrupt the MDM2/p53 protein interaction, increase p53 activity, and trigger p53-dependent apoptosis in senescent cells [48]. The effects of Nutlin-3a, a potent small-molecule MDM2/p53 inhibitor, were assessed in Dox-induced senescent ARPE-19 cells to test this hypothesis. SnCs and nonsenescent ARPE-19 cells (non-SnCs) were treated with Nutlin-3a. Nutlin-3a (20 μM) killed 50.3% of the SnCs, whereas no significant cell death was observed in non-SnCs treated with 20 μM Nutlin-3a (Fig. 1A, B, and C). Decreased numbers of p53- and p21-expressing SnCs were confirmed after treatment with Nutlin-3a by immunofluorescence (Fig. 1D). SnC removal decreased the levels of inflammatory cytokine proteins (IL-6, IL-8, MMP-1, MMP-2, MMP-3, and VEGF), as determined using enzyme-linked immunosorbent assays (ELISAs) of the CM (Fig. 1E). Based on these results, the MDM2 antagonist Nutlin-3a selectively induces cell death in SnCs, and SnC removal in turn diminishes features of the SASP [30].

Nutlin-3a selectively eliminates doxorubicin (Dox)-induced senescent RPE cells. A Schematic of the ARPE-19 cell experiments depicted in B–E. A senescent ARPE-19 cell (SnC) monolayer was formed by treatment with 250 nM doxorubicin (Dox) for 3 days and then maintained for 7 days with daily medium changes. After 10 days, the cells were seeded onto 24-well plates at a density of 25,000 cells/cm2. One day after seeding (after 11 days), the cells were treated with Nutlin-3a (1, 5, 10, 20, 50, or 100 μM) for 24 h. B ARPE-19 cells (non-SnCs) were incubated with Nutlin-3a at the indicated concentrations for 24 h. After treatment, cell viability was determined by a Cell Counting Kit-8. No significant cytotoxicity was observed at concentrations lower than 50 μM. C SnCs were incubated with Nutlin-3a at the indicated concentrations for 24 h and subsequently stained for SA-β-gal. The number of SA-β-gal-positive cells in four randomly selected fields from each sample was counted. A total of 50.3% of SnCs were eliminated by 20 μM Nutlin-3a treatment (n = 3). Decreased expression of senescence markers and SASP components in RPE cells (D, E). D Fluorescence photomicrographs of senescent markers in non-SnCs and SnCs treated with 20 μM Nultin-3a for 24 h. Cells were immunostained with an anti-p53 (top panel), anti-p21 (middle panel), or anti-p16 (bottom panel) antibody. The nuclei were stained with Hoechst (blue). E Attenuation of the SASP in SnCs by Nutlin-3a. Analysis of the levels of cytokines (IL-6, IL-8, VEGF, MMP-1, MMP-2, and MMP-3) secreted into the conditioned media from non-SnCs, SnCs, and SnCs treated with 20 μM Nultin-3a by multiplex ELISA (n = 3). One-way ANOVA followed by Tukey’s multiple comparisons test was used for statistical analysis of the data shown in B, C, and E. The data are presented as the mean ± S.D.s. *P < 0.05 and ***P < 0.001 by t-test. The scale bar in C represents 50 µm, and the scale bars in D represent 20 µm
As Nutlin-3a efficiently eliminates senescent RPE cells in vitro, we next investigated the senolytic ability of Nutlin-3a in vivo in a mouse model of RPE senescence (Fig. 2A). Intravitreally, 11.63 ng/µl Nutlin-3a corresponded to approximately 20 µM in vitro. Nutlin-3a was administered at a dose of 20 ng/µl once (at 0 day or 3 days) or twice (at 0 day and 3 days) via intravitreal injection, and its ability to eliminate senescent RPE cells in vivo was evaluated at 7 days. Quantitative analysis of SA-β-gal staining in RPE/choroid flat mounts showed that the administration of multiple Nutlin-3a doses reduced SA-β-gal activity by up to 75% compared to a single dose of Nutlin-3a (44.5% reduction) (Supplemental Fig. 5, Fig. 2B). SA-β-gal staining, conducted on RPE/choroid flat mounts and retinal sections, confirmed that senescent cells in the eyes induced by subretinal injection of Dox were primarily RPE cells, and this effect was significantly reduced with Nutlin-3a treatment. Immunofluorescence analysis of retinal sections confirmed that p53 expression was primarily localized to the RPE/choroid layer in eyes exposed to Dox, while no significant immunofluorescence was identified in the neuroretina (Supplementary Fig. 6A) No significant changes in SA-β-gal activity were observed in the RPE and neurosensory retina from control mice treated with Nutlin-3a, confirming that 20 ng/µl Nutlin-3a delivered intravitreally in vivo did not induce RPE or retinal senescence per se (Fig. 2B). No gross changes in the thickness of the neuronal cell layers of the Nutlin-3a-treated eyes were observed by H&E staining (Fig. 2C), and apparent toxic effects on retinal vasculature in GS-IB4 lectin-stained retinal flat mounts were not observed with two doses of intravitreal Nutlin-3a compared to vehicle injection (Supplementary Fig. 6B), suggesting that 20 ng/µl Nutlin-3 delivered intravitreally is not grossly toxic to the neurosensory retina and does not cause vascular damage to retinal vessels. No particular adverse effects of Nutlin-3a were exhibited in the choriocapillaris/choroid layer (Supplementary Fig. 6A).

Clearance of senescent RPE cells by Nutlin-3a reduces the senescence phenotype induced by Dox. A Schematic of the time course of the experiments shown in B–G. Male C57BL/6 N mice (9 weeks old) received 1 µl of vehicle (0.67% DMSO/saline) or Dox (100 ng/µl in 0.67% DMSO/saline) through subretinal injection. Vehicle (0.4% DMSO in normal saline) or Nutlin-3a (20 ng/µl) was intravitreally injected two times (on days 0 and 3), and eyeballs were harvested on day 7. B Representative images of SA-β-gal staining in RPE/choroid flat mounts (upper panel) and sectioned retinas (lower panel) from the four groups (group 1, mice subretinally injected with vehicle; group 2, mice subretinally injected with vehicle and intravitreally injected with Nutlin-3a; group 3, mice subretinally injected with Dox and intravitreally injected with vehicle; group 4, mice subretinally injected with Dox and intravitreally injected with Nutlin-3a). Quantification of the RPE area by SA-β-gal staining (group 1, n = 14 eyes; group 2, n = 14 eyes; group 3, n = 14; group 4, n = 14 eyes). The relative SA-β-gal areas in the representative images from groups 1, 2, 3, and 4 were 0%, 4.5%, 119.7%, and 38.5%, respectively (the mean value for group 3 was set to 100%). C Hematoxylin and eosin (H&E)-stained images of retinal sections from 4 groups of mice. Quantification of the thickness of the outer nuclear layer (ONL) showed that senescent RPE induced by Dox caused ONL thinning and disorganization of retinal layered structures, and anatomic recovery of the retina was achieved after treatment with Nutlin-3a (n = 5). D Quantification of mRNA expression by qPCR. E, F Western blot analysis and immunofluorescence of p53 and p21 levels in the RPE from the three groups of mice (group 1, subretinal vehicle-injected mice; group 2, subretinal Dox-injected and intravitreal vehicle-injected mice; group 3, subretinal Dox-injected and intravitreal Nutlin-3a-injected mice). p53 and p21 expression was increased in the RPE from group 2 compared to that from group 1 and decreased in the RPE from group 3. The graph depicts the levels of both proteins determined by densitometry quantification relative to β-actin (n = 3). G Representative images of high-mobility-group box 1 (HMGB1) immunofluorescence staining in RPE/choroid flat mounts from the three groups. The RPE from group 2 showed a significant loss of nuclear HMGB1 compared to the control (group 1), but increased nuclear HMGB1 was observed in group 3 (group 1, n = 6 eyes; group 2, n = 6 eyes; group 3, n = 6 eyes). One-way ANOVA followed by Tukey’s multiple comparisons test was used for statistical analysis of the data shown in B, C, D, and E. The data are presented as the mean ± S.D.s. *P < 0.05, **P < 0.01, and ***P < 0.001 by t-test. The scale bars in B represent 200 µm; that in C represents 100 µm; and those in F and G represent 20 µm
Transcriptional activation of the major senescence markers p53, p21, and p16 and the SASP factors IL-1 and VEGF were significantly reduced in RPE cells from Nutlin-3a-treated eyes compared to vehicle-treated eyes (Fig. 2D). The levels of the p53 and p21 proteins were also significantly reduced (Fig. 2E and F), and increased immunostaining for nuclear HMGB1 was observed with intravitreal Nutlin-3a (Fig. 2G). Moreover, the proliferative potential of RPE cells was recovered, as shown by increased Ki67 expression and BrdU incorporation in the RPE of eyes treated with Nutlin-3a (Fig. 3A). Collectively, an increase in Ki67 staining and BrdU incorporation and an overall decrease in the levels of the p53 and p21 proteins indicated an increased number of nonsenescent RPE cells and facilitation of RPE proliferation due to the clearance of senescent RPE cells. Along with the alleviation of the senescence characteristics in the RPE, the subsequent amelioration of retinal degeneration by treatment with Nutlin-3a was confirmed as described below. The areas of atrophic patches and the accumulation of subretinal debris, which are typically observed in retinas from patients with dry AMD, observed using color fundus photography (CFP), were reduced (Fig. 3B, upper panel). The number of autofluorescent dots corresponding to areas in which extracellular debris accumulated was also verified to be decreased in autofluorescence images of the in vivo fundus (Fig. 3B, upper middle panel). Occasional empty spaces and enlarged and irregularly shaped cells at the RPE junction were detected using tight junction protein immunostaining, suggesting that the loss of individual cells in the monolayer in RPE/choroid flat mounts was reversed with the restoration of a homogenous RPE monolayer (Fig. 3B, lower middle panel). The increased expression of APOE, a well-known component of drusen in patients with AMD, was also decreased (Fig. 3B, bottom panel). Transmission electron microscopy further confirmed that sub-RPE deposits and loss of pigmented granules were restored in Dox-injected mice treated with Nutlin-3a (Fig. 3C). Prominent thickening of Bruch’s membrane, one of the most frequent age-related changes in the RPE and/or Bruch’s membrane in humans and mice, was also observed in Dox-injected mice [49, 50], which was reversed upon Nutlin-3a administration.

Nutlin-3a ameliorates retinal degeneration induced by Dox and facilitates the proliferation of RPE cells. A BrdU assay and immunofluorescence staining for Ki67. Ki67 was not expressed in the RPE from normal young adult mice or Dox-treated mice (groups 1 and 2: group 1, mice subretinally injected with vehicle; group 2, mice subretinally injected with Dox and intravitreally injected with vehicle), while in Nutlin-3a-treated mice, RPE clearly expressed Ki67. The number of proliferative RPE cells that incorporated BrdU was dramatically increased with Nutlin-3a-treated mouse RPE (group 3, mice subretinally injected with Dox and intravitreally injected with Nutlin-3a), while BrdU staining was not seen in the RPE from groups 1 and 2 (group 1, n = 6 eyes; group 2, n = 6 eyes; group 3, n = 6 eyes). Nuclei were stained with Hoechst (blue). B Color fundus images (upper panel), autofluorescence images of the fundus (upper middle panel), and immunofluorescence staining of RPE/choroid flat mounts for ZO-1 (lower middle panel) and APOE (bottom panel). Degenerated areas in color images are outlined by blue arrowheads. C TEM images of each group show decreased sub-RPE deposition and Bruch’s membrane thickening in group 2 compared to group 1. In group 3, the morphology of both the RPE and Bruch’s membrane was restored. The graph depicts the quantified thickness of Bruch’s membrane in the three groups (group 1, n = 9 eyes; group 2, n = 9 eyes; group 3, n = 9 eyes). D ERG analysis of mice from the four groups (group 1, mice subretinally injected with vehicle; group 2, mice subretinally injected with vehicle and intravitreally injected with Nutlin-3a; group 3, mice subretinally injected with Dox and intravitreally injected with vehicle; group 4, mice subretinally injected with Dox and intravitreally injected with Nutlin-3a). (group 1, n = 6 eyes; group 2, n = 6 eyes; group 3, n = 6 eyes; group 4, n = 6 eyes). One-way ANOVA followed by Tukey’s multiple comparisons test was used for statistical analysis of the data shown in C and D. The data are presented as the mean ± S.D. *P < 0.05, and ***P < 0.001 by t-test. The scale bars in A and B (lower middle and bottom panels) represent 20 µm, the scale bars in B (upper and upper middle panels) represent 500 µm, and the scale bar in C represents 50 µm
Furthermore, visual function was evaluated by recording electroretinograms (ERGs), which showed significant decreases in a- and b-wave amplitudes in Dox-injected mice compared to control mice, indicating that RPE senescence was associated with visual function impairment through subsequent degeneration of the overlying retina. The a- and b-wave amplitudes were restored after treatment with Nutlin-3a (Fig. 3D). Thus, senescent RPE cells play a role in the development of retinal degeneration, and the intravitreal injection of Nutlin-3a resulted in the amelioration of both anatomical and functional damage in the retina following the elimination of senescent RPE cells.
Nutlin-3a promotes retinal regeneration in AMD mouse models by selectively removing senescent RPE cells
We next evaluated the relevance of senescent RPE cells and their clearance in AMD using AMD mouse models. AMD is a chronic disease that begins in adults over 50 and progresses slowly over a long period of time. No animal model completely recapitulates the pathology of AMD patients. Moreover, the pathology of AMD reflected in animal models occurs within a short period of time. Nevertheless, some animal models are regarded as useful for AMD preclinical research [51].
First, a laser-induced CNV mouse model, the most widely used model for studying nAMD pathogenesis and assessing the efficacy of therapeutic agents, including antiangiogenic agents [37], was exploited (Fig. 4A). At 7 days after laser application, CNV induction by neovascularization was confirmed by FA and through a 3D analysis of FITC-dextran-labeled flat-mounted RPE/choroid (Fig. 4B), and the presence of senescent RPE cells localized mainly near the laser spots was validated by SA-β-gal staining (Fig. 4C). Then, Nutlin-3a (20 ng/µl) was administered by intravitreal injections at 0 day and 3 days, after which SA-β-gal activity was significantly decreased (Fig. 4C), and BrdU incorporation and Ki67 staining were markedly increased, compared to those in the vehicle group or aflibercept (Eylea)-treated group (Fig. 4D, upper). Moreover, dual staining for Ki67 and RPE65 was detected in eyes treated with Nutlin-3a, indicating the elimination of senescent RPE cells and proliferation of RPE cells (Fig. 4D, lower). The 3D analysis of the FITC-dextran-labeled flat-mounted RPE/choroid further showed a 64.8% decrease in the CNV volume of the treatment group compared to the vehicle group (Fig. 4B). This antiangiogenic activity of Nutlin-3a is consistent with the observations of other researchers [34, 52], but our findings further suggest that the elimination of senescent RPE cells hinders pathological angiogenesis because the CNV model is based on the wound healing reaction, and the SASP adopted by senescent RPE cells contributes to reparative vessel growth [53]. mRNA of senescent markers and SASP components, including IL-1, IL-6, and VEGF, using qPCR showed decreased expression in RPE cells from Nutlin-3a-treated eyes (Fig. 4E). Based on these results, the clearance of senescent RPE cells is likely to have therapeutic benefits for nAMD. Remarkable inhibition of CNV formation was observed in eyes treated with Nultin-3a and Eylea, one of the current standard anti-VEGF drugs used to treat nAMD, and no significant differences were observed between the groups (Fig. 4B). However, the decreased SA-β-gal staining (57.8 and 32.2% decrease in eyes treated with Nultin-3a and eyes treated with Eylea, respectively, compared to eyes treated with vehicle) and increased BrdU incorporation and colocalization of Ki67 and RPE65 immunofluorescence staining in eyes treated with Nultin-3a compared to eyes treated with Eylea (Fig. 4C and D) suggest that anti-VEGF treatment has a more limited ability to mitigate cellular senescence in the RPE and restore proliferative capacity than Nutlin-3a. Retinal histology showed that the ONL thickness increased with decreased disorganization of retinal layers in eyes treated with Nutlin-3a compared to eyes treated with vehicle (Fig. 4F). Improvement of retinal function after Nutlin-3a was evaluated by recording ERGs. Both a- and b-wave amplitudes on ERGs were restored in eyes treated with Nutlin-3a (Fig. 4G).

Clearance of senescent RPE cells by Nutlin-3a reduces retinal degeneration and facilitates retinal regeneration in a mouse model of neovascular AMD. Male C57BL/6 N mice (9 weeks old) were used to develop a nAMD model. A Schematic of the time course for the development of the mouse model of nAMD and laser-induced choroidal neovascularization (CNV) analyzed in B–G. B Nutlin-3a reduced the development of CNV in a mouse model of nAMD. (Top and middle panels) At 7 days after laser application, color fundus images and fluorescein angiography (FA) images show a decreased CNV area and decreased fluorescence leakage in mice intravitreally injected with Nutlin-3a or Eylea compared to mice injected with vehicle. (Bottom panel) CNV areas from mice intravitreally injected with vehicle, Nutlin-3a, or Eylea were measured in FITC-dextran-labeled RPE/choroidal flat mounts, and 3D volumes of dextran fluorescence were quantified (the number of spots in eyes administered vehicle = 23; the number of spots in eyes administered Nutlin-3a = 25; the number of spots in eyes administered Eylea = 29). C Representative images of SA-β-gal staining in RPE/choroid flat mounts (upper panel) and sectioned retinas (lower panel) from mice intravitreally injected with vehicle, Nutlin-3a, or Eylea. Senescent RPE areas were measured by calculating the SA-β-gal-stained areas in RPE/choroid flat mounts (vehicle, n = 29 spots; Nutlin-3a, n = 26 spots; Eylea, n = 28 spots). Quantification of RPE areas with SA-β-gal staining (the mean value for the vehicle group was set to 100%, and the values shown for the eyes of treatment groups are relative to this value) showed a mean decrease in the SA-β-gal-positive area by 57.8% in Nutlin-3a-treated eyes and 32.2% in Eylea-treated eyes. D Images of immunofluorescence staining for Ki67 and the BrdU-incorporation assay in RPE/choroid flat mounts from mice intravitreally injected with vehicle, Nutlin-3a, or Eylea. A considerably increased number of Ki67-positive and BrdU-positive cells were observed following Nutlin-3a treatment, while a substantially lower number of cells had proliferative potential following Eylea treatment. Importantly, most Ki67-positive cells were coimmunofluorescence for RPE65, indicating RPE proliferation. (vehicle, n = 6; Nutlin-3a, n = 6; Eylea, n = 6). E Expression of mRNAs encoding p53, p21, p16, IL-1α, IL-6, MMP-13, TNF-α, and VEGF, as determined by qPCR. F H&E-stained images of mouse retinas intravitreally injected with vehicle, Nutlin-3a, or Eylea. The thickness of the ONL increased, and anatomic recovery of retinal layered structures was observed in Nutlin-3a- or Eylea-treated mouse retinas (vehicle, n = 6; Nutlin-3a, n = 6; Eylea, n = 6). G ERG analysis of nAMD model mice treated with vehicle, Nutlin-3a, or Eylea (vehicle, n = 4; Nutlin-3a, n = 4; Eylea, n = 4). The data are presented as the mean ± S.D.s. *P < 0.05, **P < 0.01, and ***P < 0.001 by t-test. The scale bars in B represent 500 µm (top and middle panels) and 50 µm (bottom panel); those in C represent 200 µm, those in D represent 20 µm, and that in F represents 100 µm
To further verify Nutlin-3a’s long-term effect on the nAMD mouse model, CNV with fibrovascular membrane or subretinal fibrosis was induced according to Little et al. [39] (Supplemental Fig. 7A). In this “two-stage” laser-induced mouse model, CNV with fibrovascular proliferation is maintained for more than 4 weeks, making it suitable for longer-term therapeutic effects. Nutlin-3a (20 ng/µl) was administered by intravitreal injections at 0, 3, 7, and 10 days. On 14 days and 28 days, 3D analysis of FITC-dextran-labeled flat-mounted RPE/choroid further showed a 40.7 and 38.1% decrease in the CNV volume of the treatment group compared to the vehicle group along with markedly decreased fluorescent leakage in FA (Supplemental Fig. 7B). Colocalization of Ki67 and RPE65 was detected in Nutlin-3a-treated eyes, indicating RPE proliferation (Supplemental Fig. 7C). A-wave amplitudes on ERGs were restored in eyes treated with Nutlin-3a (Supplemental Fig. 7D).
Second, the relevance of senescent RPE cells and their clearance was investigated in a dry AMD mouse model. A mouse model of GA, an advanced form of dry AMD, was established through the subretinal injection of Alu RNA (1.4 µg/µl) [35]. The occurrence of GA was validated by CFP and immunostaining for ZO-1 (Fig. 5A and B), as well as the thinning of ONL thickness in H&E-stained images of the retinas (Fig. 5C). Cellular senescence was confirmed by elevated levels of SA-β-gal staining (Fig. 5D) and p53 and p21 expression (Fig. 5E). Intravitreal injections of Nutlin-3a (20 ng/µl) at 0 day and 3 days reduced the areas of patchy atrophy and RPE degeneration, as shown by CFP and ZO-1 immunofluorescence staining, respectively (Fig. 5B); this effect was further shown by attenuated SA-β-gal staining (Fig. 5D), expression of p53, p21 (Fig. 5E), and APOE (Fig. 5F), and increased Ki67 staining (Fig. 5G). All of the above findings indicate that senescent RPE cells are a novel therapeutic target for the treatment of dry AMD. mRNA of p53 and p21 and SASP factors in RPE cells from Nutlin-3a-treated eyes also showed decreased expression compared to that in RPE cells from vehicle-treated eyes (Fig. 5H). Furthermore, visual function evaluated using ERG showed a significant recovery of a- and b-wave amplitudes after treatment with Nutlin-3a (Fig. 5I). Collectively, the elimination of senescent RPE cells by Nultin-3a was associated with the attenuation of the AMD-related pathological phenotypes of both neovascular and dry AMD and promoted RPE proliferation and retinal regeneration.

Clearance of senescent RPE cells by Nutlin-3a alleviates retinal degeneration and enhances retinal regeneration in a mouse model of dry AMD. Male C57BL/6 J mice (9 weeks old) were used to develop a mouse model of geographic atrophy (GA). A Schematic of the time course for the development of a mouse model of GA with the subretinal injection of Alu RNA analyzed in B–I. B (Top panel) Color fundus images of mouse eyes subretinally injected with Alu RNA and intravitreally injected with vehicle (n = 6 eyes) or Nutlin-3a (n = 6 eyes). (Bottom panel) Confocal micrographs of immunofluorescence staining for ZO-1 in RPE/choroid flat mounts of the same eyes shown in the top panel. C H&E staining images of mouse retinal sections. The thickness of the ONL increased markedly with Nutlin-3a treatment control, n = 6 eyes; vehicle, n = 6 eyes; Nutlin-3a, n = 6 eyes, **P < 0.01). D Representative images of SA-β-gal staining in RPE/choroid flat mounts (upper panel) and sectioned retinas (lower panel) showing areas of RPE senescence (control, n = 6 eyes; vehicle, n = 6 eyes; Nutlin-3a, n = 6 eyes). E–G Confocal micrographs of immunofluorescence staining for senescent markers (p53 and p21) (E), APOE (F), and Ki67 (G) (vehicle, n = 4 eyes; Nutlin-3a, n = 4 eyes). H Expression of mRNAs encoding p53, p21, IL-1α, IL-1β, IL-6, MMP-13, and TNF-α using qPCR. I Visual function was evaluated by recording ERGs (control, n = 10 eyes; Alu + vehicle, n = 7 eyes; Alu + Nutlin-3a, n = 10 eyes). One-way ANOVA with Tukey’s multiple-comparisons test was used for statistical analysis. The data are presented as the mean ± S.D.s. *P < 0.05, **P < 0.01, and ***P < 0.001 by t-test. The scale bars in B (bottom panel), E, F, and G represent 20 µm, that in B (top panel) represents 500 µm, and those in C and D represent 100 µm
Clearance of senescent RPE cells by Nutlin-3a improves retinal function in aged mice
Our final two tests were conducted to determine whether Nutlin-3a eliminated naturally occurring senescent RPE cells in old mice and reversed retinal degeneration associated with aging. In the first of those tests, the increased presence of senescent RPE cells in wild-type 24-month-old mice compared to 3-month-old mice was confirmed by the significantly increased SA-β-gal activity in the RPE/choroid flat mounts and cryosectioned retinas (Fig. 6A and B). Moreover, the corresponding retinal degeneration was validated by CFP and fundus autofluorescence (Fig. 6C). SA-β-gal activity in mouse eye sections demonstrated that aging retinas express senescence markers primarily in the RPE/choroid layer, not the neurosensory retinas. However, ONL thickness was decreased in 24-month-old mice compared to 3-month-old mice, suggesting retinal degeneration along with RPE senescence in 24-month-old mice (Fig. 6D). Nutlin-3a was administered to 24-month-old mice via intravitreal injections (20 ng/µl, day 0 and day 3), after which SA-β-gal activity in the RPE layer, autofluorescent puncta, and the expression of p21 were attenuated on 7 days (Fig. 6B and C). mRNA expression of the senescent markers p53 and p21 and SASP components in the RPE decreased after Nutlin-3a treatment in 24-month-old mice (Fig. 6E). Although ONL thickness was not recovered significantly in the 24-month-old mice treated with Nutlin-3a compared to mice treated with the vehicle (Fig. 6D), visual function evaluated using ERG showed a recovery of b-wave amplitudes after treatment with Nutlin-3a in the 24-month-old mice treated with Nutlin-3a compared to the 24-month-old mice treated with the vehicle (Fig. 6F).

Clearance of senescent RPE cells by Nutlin-3a promotes retinal regeneration in old mice. Male C57BL/6 J mice at the age of 24 months were used as old mice. A Schematic of the time course for the experiments shown in B–F. B Representative images of SA-β-gal staining in RPE/choroid flat mounts and sectioned retinas from 3-month-old mice intravitreally injected with vehicle (left panel, n = 4 eyes), 24-month-old mice intravitreally injected with vehicle (middle panel, n = 4 eyes), and 24-month-old mice intravitreally injected Nutlin-3a twice (right, n = 4 eyes). C Color fundus photographs and fundus autofluorescence images (AF) (top and middle panels) and representative confocal micrographs showing immunofluorescence staining for p21 (bottom panel). Nuclei were stained with Hoechst (blue). D Analysis of retinal thickness. Representative images of H&E staining in retinal sections of 3-month-old mice intravitreally injected with vehicle (left panel, n = 6 eyes), 24-month-old mice intravitreally injected with vehicle (middle panel, n = 6 eyes), and 24-month-old mice intravitreally injected with Nutlin-3a twice (right, n = 6 eyes). Quantification of the thickness of the ONL. E mRNA encoding p53, p21, IL-1α, IL-1β, IL-6, and MMP-13 using qPCR. F Visual function was evaluated by recording ERGs (3 months old, n = 6 eyes; 24 months old with vehicle, n = 5 eyes; 24 months old with Nutlin-3a, n = 10 eyes). One-way ANOVA with Tukey’s multiple-comparisons test was used for statistical analysis. Data are presented as means ± SD. ***P < 0.001, *P < 0.05 as indicated by the t-test. The scale bars in B and D represent 100 µm and C represents 500 µm (top and middle panels) and 20 µm (bottom panel)
In the second of those tests, we investigated the effects of Nutlin-3a over a month starting at the age of 23 months. An intravitreal injection was given once per week for 4 weeks (for a total of four injections) (Supplemental Fig. 8A). SA-β-gal staining and the immunofluorescence of the p53 and p21 proteins in the RPE/choroid flat mounts were decreased in 24-month-old mice treated with Nutlin-3a four times compared to mice treated with vehicle (Supplemental Fig. 8B). Immunostaining for CD34 and the senescent marker p53 in retinal sections showed that p53 expression was primarily localized in the RPE layer, which was markedly decreased with Nutlin-3a treatment, indicating a reduction in senescent RPE cells (Supplemental Fig. 8C). The presence of Ki67 immunofluorescence confirmed the proliferative activity of RPE cells in 24-month-old mice treated with Nutlin-3a, whereas no immunostaining of Ki67 in the RPE layer was detectable in 24-month-old mice treated with vehicle (Supplemental Fig. 8C). In addition, choriocapillaris and large choroidal vessels that are positive for CD34 immunofluorescence were markedly increased in 24-month-old mice treated with Nutlin-3a compared to 24-month-old mice treated with vehicle. The functional improvement was remarkable, and anatomical restoration, as shown by increased ONL thickness, was also observed in Nutlin-3a-treated 24-month-old mice (Supplemental Fig. 8D and E). Based on these findings, the administration of Nutlin-3a can eliminate naturally occurring senescent RPE cells in old mice, resulting in significant functional improvement in the retina.
Discussion
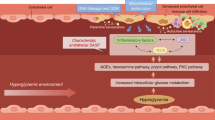
Currently, a curative treatment option is not available for AMD. Therefore, novel treatment strategies and targets for AMD are urgently needed. A recent pioneering study by Oubaha et al. [53] showed the senescence of cells in the ischemic zones of the retina, such as retinal vascular endothelial cells and retinal ganglion cells, and the inhibition of the SASP with metformin ameliorated the spread of senescence and decreased pathological neovascularization in a mouse model of oxygen-induced retinopathy. The key pathological cells in the retina that are responsible for AMD development and progression are RPE cells. RPE cells are postmitotic cells, and aged RPE cells show lysosomal dysfunction with lipofuscin accumulation. Although cellular senescence was initially identified as a mechanism used by mitotic cells to prevent unrestrained cell division, recent studies have suggested that postmitotic cells such as neurons can enter a state of cellular senescence [54,55,56]. In the present study, we hypothesized that cellular senescence in the RPE might represent a new treatment target for AMD. We first developed a mouse model of RPE senescence that exhibited general features of cellular senescence, including increased SA-β-gal expression, enlargement of cell size, HMGB1 relocalization, activation of the SASP, and elevated expression of p53, p21, and p16, to confirm this hypothesis. The phenotypes observed in RPE cellular senescence overlapped with several pathological features of the retina from patients with AMD, such as increases in subretinal deposits, autofluorescence, and the thickening of Bruch’s membrane. Furthermore, in the mouse models of AMD, increased activity of SA-β-gal and increased p53 and p21 expression were observed, indicating the role of RPE cellular senescence in AMD. Then, the selective elimination of senescent RPE cells with Nutlin-3a ameliorated retinal degeneration in an in vivo model of RPE senescence and two mouse models of AMD. Interestingly, the age-dependent degeneration of the retina in old mice was reversed by Nutlin-3a.
The SASP released from senescent cells produces a distinctive proinflammatory microenvironment and aggravates senescence via both autocrine and paracrine mechanisms [57]. Senomorphic drugs that inhibit the SASP of senescent cells without killing these cells might be safer and more easily applicable to AMD. Several drugs, such as rapamycin and metformin, which improve mitochondrial function and decrease inflammation [27, 28], or drugs that target the ERK, p38 MAPK, or JAK/STAT pathways have been investigated [58,59,60]. However, most of these agents exert complex biological effects and are related to multiple signaling pathways other than decreasing the SASP; thus, their clinical value in treating neurodegenerative diseases such as AMD remains uncertain [61, 62]. Indeed, many drugs under clinical investigation, such as antioxidants and complement inhibitors, have proven ineffective in treating AMD [15].
Regarding the possible mechanisms underlying the amelioration of retinal degeneration by the application of a senolytic, first, removing senescent RPE cells with senolytics might increase the proliferative capacity or mitotic competence of the remaining nonsenescent RPE cells, thereby increasing the overall capacity of tissue regeneration and homeostasis of the retina. Senescence has been suggested to decrease the regenerative potential of the tissue by inducing permanent cell cycle arrest in proliferating cell populations [63]. Researchers are still debating whether RPE cells, which might remain nonproliferative throughout life in humans after their complete differentiation early in development, are capable of proliferating under certain circumstances. According to Salero et al., adult human RPE cells are activated to become self-renewing cells under a variety of culture conditions [64]. In addition, RPE cell division has been reported in in vivo models of RPE wound healing [65, 66]. In the present study, the RPE proliferated in vivo after treatment with Nutlin-3a, facilitating retinal regeneration, which might be a key effect of senolytic treatment for AMD. Second, the senolytic-induced death of a significant fraction of senescent RPE cells might reduce harmful paracrine signals sent to neighboring nonsenescent RPE cells by the SASP. SASP factors are known to incite immune cell infiltration, tissue destruction, stem cell exhaustion, and the dissemination of cellular senescence in neighboring cells [63], all of which are relevant to AMD development and progression. Therefore, the effective removal of damaged RPE cells by senolysis will be important for the maintenance of retinal homeostasis. When GA initially occurs, it begins outside the macula, with the area increasing by a median of 1.78 mm2 each year [32], invades the center of the macula with a centrifugal progressive nature, and results in vision loss. In the autofluorescence images of the retina reflecting the intracellular lipofuscin content of the RPE, autofluorescence of the marginal zone surrounding the GA usually increases, suggesting that the cells surrounding GA might be senescent RPE cells. Because atrophy progresses and deteriorates in a centrifugal nature from the boundary of the GA, the healthy RPE cells outside of the margin of the GA are gradually damaged due to the paracrine effect of SASP secreted from the senescent RPE cells at the border of the GA. Thus, the persistence of senescent RPE cells might be responsible for the progression of GA.
The elimination of senescent RPE cells by Nultin-3a mitigates two representative phenotypes of AMD. First, drusen, which represents the major phenotype of early and intermediate AMD, are focal extracellular deposits located between the RPE and Bruch’s membrane and are closely associated with the development and progression of AMD [67, 68]. Increased SA-β-gal activity in senescent RPE cells is consistent with increased autofluorescent deposits observed in in vivo autofluorescence images of a subretinal Dox-injected mouse model and old mice. This autofluorescence in the retina is mainly derived from lipofuscin in the RPE, a nondegradable aggregate of oxidized proteins and lipids that reflects dysfunctional lysosomal processing of phagocytosed photoreceptor outer segments in RPE cells, resulting in drusen formation. Nultin-3a treatment decreased subretinal and autofluorescent deposits in in vivo images, the levels of a drusen marker protein were confirmed using immunofluorescence staining, and extracellular drusen-like deposits and thickened Bruch’s membrane were confirmed using TEM. Furthermore, in the Alu-induced GA model and old mice, Nultin-3a treatment reduced subretinal and autofluorescent deposits in in vivo images. Second, senescent RPE cells might also be responsible for the initiation and progression of CNV and the poor treatment response. The disruption of the integrity of the RPE layer with weakened barrier function by increased heterogeneity in cell size and morphology and decreased adhesion of RPE cells favors the formation of CNV [69]. As shown in the nAMD model analyzed in this study, Nutlin-3a caused the proliferation of RPE cells and decreased the formation of CNV, as evidenced by increased Ki67 expression and decreased CNV volume. In contrast, anti-VEGF treatment did not induce substantial cell proliferation in the nAMD model. The proliferation of RPE cells during anti-VEGF treatments for nAMD might halt CNV progression if neovascular tissue is enveloped by RPE, as rarely observed in patients with pure type 2 CNV [70]. Failure of the current standard anti-VEGF treatment for nAMD in patients, including resistance to anti-VEGF therapy and the development of GA or subretinal fibrosis after multiple anti-VEGF treatments, might at least partly be associated with the persistence of senescent RPE cells. Therefore, the application of a senolytic agent in combination with anti-VEGF represents a potentially promising treatment for nAMD and early and intermediate AMD and GA.
The current study has some limitations. First, extensive data on morphological changes, including senescent cells in the choriocapillaris/choroid in mouse models and the effects of senolysis, could not be analyzed, although the elimination of senescent cells in some parts of the choriocapillaris of the Dox-induced senescent RPE model and the restoration of the choriocapillaris layer in old mice were observed. AMD pathogenesis is associated with early changes and degeneration in both the RPE and the choriocapillaris. Recent single-cell transcriptomics demonstrated inflammatory and degenerative effects in the choriocapillaris/choroid from aged and AMD donors [71, 72]. In addition, optical coherence tomography angiography analyses confirmed age-dependent decreases in macular choriocapillaris flow deficits and choriocapillaris flow voids in AMD patients [73]. Therefore, in-depth investigations on the effects of senolytics on the choriocapillaris/choroid will be needed in future studies.
Second, although we used one chronic model (old mice) that might better recapitulate the long-term chronic progression of AMD in patients to prove the efficacy of the senolytic, short-term treatments did not fairly counteract structural damage in the retina. One week or month of treatment could not completely reverse retinal degeneration in 24- or 23-month-old mice, which had already presented with severe RPE senescence as well as neuroretinal degeneration, even though senescent RPE cells were effectively removed. Therefore, starting periodic treatment at an age where no obvious evidence of senescence and retinal degeneration have been found will have greater therapeutic effects structurally as well as functionally.
In summary, Nutlin-3a eliminates irreparably compromised senescent RPE cells and thereby curtails retinal degeneration, possibly by decreasing the detrimental effects of these cells on neighboring healthy and nonsenescent RPE cells and the overlying retina in AMD mouse models. A cure for nAMD and an approved treatment for dry AMD are not available. Most of the drugs developed to date may temporarily impede tissue degeneration by reducing inflammation or complement dysregulation using anti-inflammatory, antioxidant, or antiangiogenic agents; however, recent clinical trials in patients with dry AMD have not obtained positive results. Our results suggest that cellular senescence of the RPE might be a key contributor to AMD and thus may be a new therapeutic target for AMD. Furthermore, this study provides the first evidence of the use of a senolytic compound as a promising approach for the treatment of AMD. Therefore, further studies are warranted to develop new senolytics for AMD with periodic dosing and longer follow-up and to investigate their therapeutic mechanisms as treatments for AMD.
Data availability (data transparency)
All data generated or analyzed during this study are included in this article (and its supplementary information files) and are available from the corresponding author on reasonable request.
Code availability (software application or custom code)
Not applicable
Change history
16 March 2022
A Correction to this paper has been published: https://doi.org/10.1007/s11357-022-00523-5
References
la Cour M, Kiilgaard JF, Nissen MH. Age-related macular degeneration: epidemiology and optimal treatment. Drugs Aging. 2002;19(2):101–33. https://doi.org/10.2165/00002512-200219020-00003.
Coleman HR, Chan CC, Ferris FL 3rd, Chew EY. Age-related macular degeneration. Lancet. 2008;372(9652):1835–45. https://doi.org/10.1016/S0140-6736(08)61759-6.
Biesemeier A, Taubitz T, Julien S, Yoeruek E, Schraermeyer U. Choriocapillaris breakdown precedes retinal degeneration in age-related macular degeneration. Neurobiol Aging. 2014;35(11):2562–73. https://doi.org/10.1016/j.neurobiolaging.2014.05.003.
Spaide RF. Improving the age-related macular degeneration construct: a new classification system. Retina. 2018;38(5):891–9. https://doi.org/10.1097/IAE.0000000000001732.
Mitchell P, Bressler N, Doan QV, Dolan C, Ferreira A, Osborne A, et al. Estimated cases of blindness and visual impairment from neovascular age-related macular degeneration avoided in Australia by ranibizumab treatment. PLoS One. 2014;9(6):e101072. https://doi.org/10.1371/journal.pone.0101072.
Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann Med. 2006;38(7):450–71. https://doi.org/10.1080/07853890600946724.
Prasad PS, Schwartz SD, Hubschman JP. Age-related macular degeneration: current and novel therapies. Maturitas. 2010;66(1):46–50. https://doi.org/10.1016/j.maturitas.2010.02.006.
Falavarjani KG, Nguyen QD. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye (Lond). 2013;27(7):787–94. https://doi.org/10.1038/eye.2013.107.
Grunwald JE, Pistilli M, Daniel E, Ying GS, Pan W, Jaffe GJ, et al. Incidence and growth of geographic atrophy during 5 years of comparison of age-related macular degeneration treatments trials. Ophthalmology. 2017;124(1):97–104. https://doi.org/10.1016/j.ophtha.2016.09.012.
Lux A, Llacer H, Heussen FM, Joussen AM. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br J Ophthalmol. 2007;91(10):1318–22. https://doi.org/10.1136/bjo.2006.113902.
Tozer K, Roller AB, Chong LP, Sadda S, Folk JC, Mahajan VB, et al. Combination therapy for neovascular age-related macular degeneration refractory to anti-vascular endothelial growth factor agents. Ophthalmology. 2013;120(10):2029–34. https://doi.org/10.1016/j.ophtha.2013.03.016.
Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–4. https://doi.org/10.1126/science.1110189.
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227–32. https://doi.org/10.1073/pnas.0501536102.
Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–21. https://doi.org/10.1126/science.1110359.
Holz FG, Sadda SR, Busbee B, Chew EY, Mitchell P, Tufail A, et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: Chroma and Spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018;136(6):666–77. https://doi.org/10.1001/jamaophthalmol.2018.1544.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35. https://doi.org/10.1038/nm.4000.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. https://doi.org/10.1146/annurev-physiol-030212-183653.
Markovets AM, Saprunova VB, Zhdankina AA, Fursova A, Bakeeva LE, Kolosova NG. Alterations of retinal pigment epithelium cause AMD-like retinopathy in senescence-accelerated OXYS rats. Aging. 2011;3(1):44–54. https://doi.org/10.18632/aging.100243.
Lopez-Luppo M, Catita J, Ramos D, Navarro M, Carretero A, Mendes-Jorge L, et al. Cellular senescence is associated with human retinal microaneurysm formation during aging. Invest Ophthalmol Vis Sci. 2017;58(7):2832–42. https://doi.org/10.1167/iovs.16-20312.
Mishima K, Handa JT, Aotaki-Keen A, Lutty GA, Morse LS, Hjelmeland LM. Senescence-associated beta-galactosidase histochemistry for the primate eye. Invest Ophthalmol Vis Sci. 1999;40(7):1590–3.
Baek A, Yoon S, Kim J, Baek YM, Park H, Lim D, et al. Autophagy and KRT8/keratin 8 protect degeneration of retinal pigment epithelium under oxidative stress. Autophagy. 2017;13(2):248–63. https://doi.org/10.1080/15548627.2016.1256932.
Glotin AL, Debacq-Chainiaux F, Brossas JY, Faussat AM, Treton J, Zubielewicz A, et al. Prematurely senescent ARPE-19 cells display features of age-related macular degeneration. Free Radic Biol Med. 2008;44(7):1348–61. https://doi.org/10.1016/j.freeradbiomed.2007.12.023.
Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. 2016;7:78–87. https://doi.org/10.1016/j.redox.2015.11.011.
Yu B, Ma J, Li J, Wang D, Wang Z, Wang S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat Commun. 2020;11(1):2549. https://doi.org/10.1038/s41467-020-16312-7.
Blasiak J, Pawlowska E, Sobczuk A, Szczepanska J, Kaarniranta K. The aging stress response and its implication for AMD pathogenesis. Int J Mol Sci. 2020;21(22):8840. https://doi.org/10.3390/ijms21228840.
Sreekumar PG, Hinton DR, Kannan R. The emerging role of senescence in ocular disease. Oxid Med Cell Longev. 2020;2020:2583601. https://doi.org/10.1155/2020/2583601.
Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564–74. https://doi.org/10.1111/acel.12587.
Moiseeva O, Deschenes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell. 2013;12(3):489–98. https://doi.org/10.1111/acel.12075.
Gensler G, Clemons TE, Domalpally A, Danis RP, Blodi B, Wells J 3rd, et al. Treatment of geographic atrophy with intravitreal sirolimus: the Age-Related Eye Disease Study 2 Ancillary Study. Ophthalmol Retina. 2018;2(5):441–50. https://doi.org/10.1016/j.oret.2017.08.015.
Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81. https://doi.org/10.1038/nm.4324.
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562(7728):578–82. https://doi.org/10.1038/s41586-018-0543-y.
Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, et al. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology. 2018;125(3):369–90. https://doi.org/10.1016/j.ophtha.2017.08.038.
Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–59. https://doi.org/10.1016/j.cell.2012.03.036.
Chavala SH, Kim Y, Tudisco L, Cicatiello V, Milde T, Kerur N, et al. Retinal angiogenesis suppression through small molecule activation of p53. J Clin Invest. 2013;123(10):4170–81. https://doi.org/10.1172/JCI67315.
Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471(7338):325–30. https://doi.org/10.1038/nature09830.
Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24(1):50–61. https://doi.org/10.1038/nm.4450.
Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez ML, Struman I, et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc. 2013;8(11):2197–211. https://doi.org/10.1038/nprot.2013.135.
Lee J, Park DY, Park DY, Park I, Chang W, Nakaoka Y, et al. Angiopoietin-1 suppresses choroidal neovascularization and vascular leakage. Invest Ophthalmol Vis Sci. 2014;55(4):2191–9. https://doi.org/10.1167/iovs.14-13897.
Little K, Llorian-Salvador M, Tang M, Du X, O’Shaughnessy O, McIlwaine G, et al. A two-stage laser-induced mouse model of subretinal fibrosis secondary to choroidal neovascularization. Transl Vis Sci Technol. 2020;9(4):3. https://doi.org/10.1167/tvst.9.4.3.
Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165–76. https://doi.org/10.1158/2159-8290.CD-16-0241.
Yao Z, Murali B, Ren Q, Luo X, Faget DV, Cole T, et al. Therapy-induced senescence drives bone loss. Cancer Res. 2020;80(5):1171–82. https://doi.org/10.1158/0008-5472.CAN-19-2348.
White RR, Milholland B, de Bruin A, Curran S, Laberge RM, van Steeg H, et al. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat Commun. 2015;6:6790. https://doi.org/10.1038/ncomms7790.
Zielke N, Edgar BA. FUCCI sensors: powerful new tools for analysis of cell proliferation. Wiley Interdiscip Rev Dev Biol. 2015;4(5):469–87. https://doi.org/10.1002/wdev.189.
Maejima Y, Adachi S, Ito H, Hirao K, Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008;7(2):125–36. https://doi.org/10.1111/j.1474-9726.2007.00358.x.
Blasiak J. Senescence in the pathogenesis of age-related macular degeneration. Cell Mol Life Sci. 2020;77(5):789–805. https://doi.org/10.1007/s00018-019-03420-x.
Baek JH, Lim D, Park KH, Chae JB, Jang H, Lee J, et al. Quantitative proteomic analysis of aqueous humor from patients with drusen and reticular pseudodrusen in age-related macular degeneration. BMC Ophthalmol. 2018;18(1):289. https://doi.org/10.1186/s12886-018-0941-9.
Lozano-Torres B, Estepa-Fernández A, Rovira M, Orzáez M, Serrano M, Martínez-Máñez R, et al. The chemistry of senescence. Nat Rev Chem. 2019;3(7):426–41. https://doi.org/10.1038/s41570-019-0108-0.
Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20(4):576–88. https://doi.org/10.1038/cdd.2012.155.
Ivert L, Keldbye H, Gouras P. Age-related changes in the basement membrane of the retinal pigment epithelium of Rpe65 -/- and wild-type mice. Graefes Arch Clin Exp Ophthalmol. 2005;243(3):250–6. https://doi.org/10.1007/s00417-004-0967-y.
Bailey TA, Alexander RA, Dubovy SR, Luthert PJ, Chong NH. Measurement of TIMP-3 expression and Bruch’s membrane thickness in human macula. Exp Eye Res. 2001;73(6):851–8. https://doi.org/10.1006/exer.2001.1089.
Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. 2012;33(4):487–509. https://doi.org/10.1016/j.mam.2012.06.003.
Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, et al. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018;8(1):2410. https://doi.org/10.1038/s41598-018-20000-4.
Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med. 2016;8(362):362ra144. https://doi.org/10.1126/scitranslmed.aaf9440.
Riessland M, Kolisnyk B, Kim TW, Cheng J, Ni J, Pearson JA, et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell. 2019;25(4):514-30 e8. https://doi.org/10.1016/j.stem.2019.08.013.
Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208–16. https://doi.org/10.1172/JCI95145.
Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15(6):643–60. https://doi.org/10.1007/s10522-014-9532-1.
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–90. https://doi.org/10.1038/ncb2784.
Liu S, Uppal H, Demaria M, Desprez PY, Campisi J, Kapahi P. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci Rep. 2015;5:17895. https://doi.org/10.1038/srep17895.
Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–48. https://doi.org/10.1038/emboj.2011.69.
Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112(46):E6301–10. https://doi.org/10.1073/pnas.1515386112.
Patrone C, Eriksson O, Lindholm D. Diabetes drugs and neurological disorders: new views and therapeutic possibilities. Lancet Diabetes Endocrinol. 2014;2(3):256–62. https://doi.org/10.1016/S2213-8587(13)70125-6.
Valero T. Mitochondrial biogenesis: pharmacological approaches. Curr Pharm Des. 2014;20(35):5507–9. https://doi.org/10.2174/138161282035140911142118.
Mahmoudi S, Xu L, Brunet A. Turning back time with emerging rejuvenation strategies. Nat Cell Biol. 2019;21(1):32–43. https://doi.org/10.1038/s41556-018-0206-0.
Salero E, Blenkinsop TA, Corneo B, Harris A, Rabin D, Stern JH, et al. Adult human RPE can be activated into a multipotent stem cell that produces mesenchymal derivatives. Cell Stem Cell. 2012;10(1):88–95. https://doi.org/10.1016/j.stem.2011.11.018.
Oganesian A, Bueno E, Yan Q, Spee C, Black J, Rao NA, et al. Scanning and transmission electron microscopic findings during RPE wound healing in vivo. Int Ophthalmol. 1997;21(3):165–75. https://doi.org/10.1023/a:1026402031902.
Roider J, Michaud NA, Flotte TJ, Birngruber R. Response of the retinal pigment epithelium to selective photocoagulation. Arch Ophthalmol. 1992;110(12):1786–92. https://doi.org/10.1001/archopht.1992.01080240126045.
Alten F, Eter N. Current knowledge on reticular pseudodrusen in age-related macular degeneration. Br J Ophthalmol. 2015;99(6):717–22. https://doi.org/10.1136/bjophthalmol-2014-305339.
Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20(6):705–32. https://doi.org/10.1016/s1350-9462(01)00010-6.
Kozlowski MR. RPE cell senescence: a key contributor to age-related macular degeneration. Med Hypotheses. 2012;78(4):505–10. https://doi.org/10.1016/j.mehy.2012.01.018.
Dolz-Marco R, Phasukkijwatana N, Sarraf D, Freund KB. Regression of type 2 neovascularization into a type 1 pattern after intravitreal anti-vascular endothelial growth factor therapy for neovascular age-related macular degeneration. Retina. 2017;37(2):222–33. https://doi.org/10.1097/IAE.0000000000001279.
Voigt AP, Whitmore SS, Mulfaul K, Chirco KR, Giacalone JC, Flamme-Wiese MJ, et al. Bulk and single-cell gene expression analyses reveal aging human choriocapillaris has pro-inflammatory phenotype. Microvasc Res. 2020;131:104031. https://doi.org/10.1016/j.mvr.2020.104031.
Voigt AP, Mulfaul K, Mullin NK, Flamme-Wiese MJ, Giacalone JC, Stone EM, et al. Single-cell transcriptomics of the human retinal pigment epithelium and choroid in health and macular degeneration. Proc Natl Acad Sci U S A. 2019;116(48):24100–7. https://doi.org/10.1073/pnas.1914143116.
Lipecz A, Miller L, Kovacs I, Czako C, Csipo T, Baffi J, et al. Microvascular contributions to age-related macular degeneration (AMD): from mechanisms of choriocapillaris aging to novel interventions. Geroscience. 2019;41(6):813–45. https://doi.org/10.1007/s11357-019-00138-3.
Acknowledgements
The authors thank Juhong Kim and Min Kyu Jang for providing technical assistance in obtaining the ERG data and in vivo mouse retinal imaging, respectively.
Funding
This study was supported by the National Research Foundation of Korea and funded by the Ministry of Science and ICT (NRF-2020M3A9D8038188, NRF-2020R1A2C2101941, and NRF-2020M3A9D8038190).
Author information
Authors and Affiliations
Contributions
C.K. and H.C. conceived, designed, and supervised the study. J.C. and H.J. equally contributed to the design, experiments, and data analysis for most of the experiments. C.S., C-W.P., H-Y.L., and N.K. contributed to the in vitro experiments. H.C., S.J., and J-H.R. assisted with and analyzed the results of the TEM experiments. C.K. and H.C. wrote the manuscript with input from all coauthors. H.L. contributed to the method for quantifying the SA-β-gal area. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All experimental procedures and animal care were conducted according to guidelines approved by the Konkuk University IACUC (KU IACUC; approval No. KU18080).
Consent to participate
Not applicable
Consent for publication
Not applicable
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
About this article

Cite this article
Chae, JB., Jang, H., Son, C. et al. Targeting senescent retinal pigment epithelial cells facilitates retinal regeneration in mouse models of age-related macular degeneration. GeroScience 43, 2809–2833 (2021). https://doi.org/10.1007/s11357-021-00457-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00457-4