
Abstract
As mitigation of brain aging continues to be a key public health priority, a wholistic and comprehensive consideration of the aging body has identified immunosenescence as a potential contributor to age-related brain injury and disease. Importantly, the nervous and immune systems engage in bidirectional communication and can exert profound influence on each other. Emerging evidence supports numerous impacts of innate, inflammatory immune responses and adaptive T cell–mediated immunity in neurological function and diseased or injured brain states, such as stroke. Indeed, a growing body of evidence supports key impacts of brain-resident immune cell activation and peripheral immune infiltration in both the post-stroke acute injury phase and the long-term recovery period. As such, modulation of the immune system is an attractive strategy for novel therapeutic interventions for a devastating age-related brain injury for which there are few readily available neuroprotective treatments or neurorestorative approaches. However, the role of B cells in the context of brain function, and specifically in response to stroke, has not been thoroughly elucidated and remains controversial, leaving our understanding of neuroimmune interactions incomplete. Importantly, emerging evidence suggests that B cells are not pathogenic contributors to stroke injury, and in fact may facilitate functional recovery, supporting their potential value as novel therapeutic targets. By summarizing the current knowledge of the role of B cells in stroke pathology and recovery and interpreting their role in the context of their interactions with other immune cells as well as the immunosenescence cascades that alter their function in aged populations, this review supports an increased understanding of the complex interplay between the nervous and immune systems in the context of brain aging, injury, and disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The world’s population is aging (Vaupel 2010). Specifically in the USA, by the year 2050, nearly 85 million people are projected to be over 65 years of age, constituting more than 20% of the nation’s total population (Ortman et al. 2014). Notably, these projections are markedly increased relative to the current number of 45 million older adults who comprise just 13.7% of the population. Furthermore, these same projections estimate that the number of individuals comprising the oldest-old cohort (ages 85+) will more than triple, growing to include 18 million people in the coming decades. Given that aging represents a multifactorial process that affects the functionality of all bodily systems and is associated with profound alterations in quality of life, these dramatic shifts in population age distribution will have important human health impacts both on an individual and national level.
Of key clinical importance is brain aging, age-related brain injury, and neurodegenerative disease. Indeed, brain aging is associated with a host of functional changes including genomic and epigenetic alterations, impaired proteostasis mechanisms, dysregulated mitochondrial function and energy metabolism, disruptions to ionic homeostasis that impact cell signaling cascades, inflammation, impaired neurogenesis, accumulation of molecular and cellular damage, and cell death (Lopez-Otin et al. 2013; Mattson and Arumugam 2018). Brain aging and its associated consequences represent a key component of a number of neurological injuries and neurodegenerative diseases. For example, stroke, the main risk factor for which is advanced age, is the fifth leading cause of death and a major contributor to long-term disability. The pathological sequelae associated with stroke, or the post-occlusion reperfusion process, is heterogeneous and complex but generally include cellular metabolism dysfunction brought about by energy supply deficiencies, disruption to ion homeostasis, accumulation of cell-damaging species, excitotoxicity, cell death, the activation of the brain-resident microglia, and recruitment of peripheral immune cells to the infarcted tissue to clear dead/damaged tissues (Xing et al. 2012). Yet at present, despite decades of intense investigation focused on probing the underlying neurobiological causes with the hopes of revealing novel neurorestorative treatments and preventative measures, pharmacological or mechanical endovascular thrombolytic interventions remain the only approved therapeutic approach for the treatment of stroke (Bhogal et al. 2018; Gurman et al. 2015). Furthermore, the clinical applications of the few known neuroprotective/neurorestorative approaches, such as therapeutic hypothermia and transcranial magnetic stimulation, are extremely limited (Dionisio et al. 2018; Tahir and Pabaney 2016).
A more comprehensive understanding of the biological contributors to brain aging, as well as additional strategies for the effective treatment of stroke and other age-related neurological diseases, is urgently needed. Emerging data suggest key roles for the immune system, the body’s defensive response against infection, injury, and disease. Importantly, senescence cascades that occur in the immune system may potentially compound changes associated with brain aging and age-related neurological injury or disease (Deleidi et al. 2015). This warrants further consideration. This review will discuss the convergence of the aging nervous and immune systems in the context of stroke with a focus on the role of B cells, whose impacts on brain aging and stroke remain understudied. Both the potentially damaging and protective effects of B cells in the context of stroke that have been reported to date and the impact that aging of the B cell system may have on the risk for, occurrence of, and recovery from, cerebral ischemia will be addressed.
Brain-immune interactions impact neurological function in normal and injured, aged, and diseased states
Brief overview of immune system components
The immune system serves as the body’s response against infection, injury, and disease. This complex network of intercommunicating and interactive cells and secretory factors housed in several organs coordinates to mount a rapid and appropriate response to a threat to homeostasis through complex signaling and cell activation/regulation cascades. A thorough discussion of key players and cascades is beyond the scope of the current review; the reader is directed to several excellent texts that describe the function of the innate immune cell, complement, and adaptive immune responses during adulthood (Chaplin 2010; Nicholson 2016).
Briefly, the immune system is a complex network of organs, cells, and secretory factors that mobilize in response to bodily damage or antigen challenge that would do the organism harm. It is composed of the innate arm, which is an immediate, non-specific response (i.e., inflammation) that activates the adaptive arm, a delayed, antigen-specific response. First, following penetration or bypass of surface barriers such as the skin or mucosal membranes, or the production of damage-associated molecular patterns in response to cellular injury, pathogen/injury recognition components detect injury or pathogenic challenge. This subsequently, rapidly, and robustly activates non-specific innate immune leukocytic cells such as the mononuclear phagocytes (i.e., monocytes, macrophages, and dendritic cells), granulocytes (i.e., mast cells, basophils, eosinophils, neutrophils), and innate lymphoid cells (e.g., natural killer cells) to attempt to quash the burgeoning infection and/or injury. Innate immune cells secrete a variety of factors, including cytokines and chemokines, that relay threat information to the rest of the body, recruit additional innate immune cells to the site, and activate secondary immunological cascades. As a consequence of their mobilization, inflammatory cascades are initiated, producing redness, swelling, heat, and enhanced nociceptive sensitivity. A subset of these cells serves as professional antigen-presenting cells that travel from the site of infection to secondary immune organs to directly activate the adaptive immune response through antigen presentation. Bridging innate and adaptive immune responses, the complement system potentiates the recruitment signals initiated by innate immune players, labels non-self-antigens to facilitate immune-induced attack on these cells, and themselves act to mitigate the spread of the infection by promoting membrane dysfunction-induced cell death.
The adaptive immune system, which takes hours to days to mobilize, comprises cell-mediated immunity, facilitated by T cells, and humoral immunity, driven by B cells. The key functions of adaptive immune responses support an antigen-specific response (i.e., bacterial versus viral) by mobilizing cells that are uniquely responsive to a given antigen, and the generation of immunological memory to the encountered antigen. T cell–mediated responses are either cytotoxic, or serve to facilitate and regulate the extent of immune activation (helper). Upon stimulation of the T cell receptor by antigen fragments contained on major histocompatibility complex (MHC) molecules of antigen-presenting cells, activated cytotoxic or killer T cells are deployed to destroy host cells that are infected, damaged, or dysfunctional through the release of cytotoxins that disrupt cell membrane integrity or induce apoptosis. Helper T cells support the immune response through their secretion of cytokines that impact the function of a variety of immune cell types and via their stimulation of B cells to produce antibodies that ultimately supports further T cell activation.
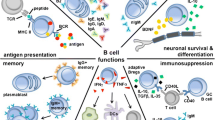
Several excellent reviews and texts have discussed the development, structure, and function of the B cell/humoral immune system in great detail (Allman and Pillai 2008; Hoffman et al. 2016; Mak et al. 2014; Shen and Fillatreau 2015). In brief here, the humoral immune system is designed to address antigen detected in the extracellular fluids. Originating from hematopoietic stem cells (HSCs), for the majority of B cells, development in the bone marrow proceeds through several key stages in which selection checkpoints are in place to ensure proper functionality of the B cell receptor (BCR); self-reactivity must be lacking for cell survival and migration to secondary immune organs as a naïve B lymphocyte. Once activated, B cell functions can be classified into several domains: (1) antibody producers that once created and bound to antigen promote complement signaling as well as antigen neutralization, antigen opsonization (coating with antibodies), and ultimately destruction by other immune cells, (2) antigen presenters, (3) cytokine and trophic factor secretors, (4) immune response regulators, and (5) antigen memory cells that enable a more rapid and specific threat response in future encounters with that specific antigen. A key aspect of B cell function is their necessary involvement in regulating the response of other immune cells. Thus, the humoral immune system mediated by the B cell represents a diverse, potent, complementary, and critical component to the overall immune response.
B cell modulation of brain function, injury, and disease
Although once considered “immune privileged,” a growing body of literature indicates that the central nervous system (CNS) and the peripheral immune systems engage in bidirectional communication and profoundly influence one another during homeostasis and in pathological/diseased states (Pavlov et al. 2018) suggesting that therapeutic targeting of the immune system may be a viable, novel approach to treat brain-related injuries and diseases. Indeed, chronic inflammation mediated by the innate immune response is implicated in a variety of neurodegenerative diseases and new evidence suggests that anti-inflammatory agents may represent a promising therapeutic avenue for their treatment (Lucas et al. 2006). Research interests in the potential for adaptive immune responses to impact neurological function are also increasing. The notion that T cells may be present in healthy brain structures and fluids (i.e., meninges and cerebrospinal fluid) and can also infiltrate into CNS tissue in response to injury or autoimmune disease has fostered major interest in the role of antigen-specific adaptive immunity in normal and abnormal brain function (Filiano et al. 2017; Fletcher et al. 2010; Herkenham and Kigar 2017; Meeker et al. 2012; Rayasam et al. 2018). A growing body of evidence suggests that B cells, too, may play important roles in modulating the function of the brain.
B cells are detectable in very low quantities in meninges, cerebrospinal fluid (CSF), and possibly brain parenchyma under normal conditions, but are trafficked in larger quantities to CNS tissues in response to injury or disease (Anthony et al. 2003; Funaro et al. 2016; Gredler 2012). Indeed, as an example, B cells are emerging as a key mediator of disease progression in multiple sclerosis (MS), a demyelinating autoimmune disorder once considered a disease chiefly of dysfunctional T cells (Fletcher et al. 2010; Funaro et al. 2016), acting via multiple mechanisms to promote pathogenesis (Feng and Ontaneda 2017). The first is through the production of proinflammatory mediators. MS patients exhibit a lymphocyte repertoire characterized by high quantities of lymphotoxin-, GM-CSF-, and TNF-α-expressing proinflammatory B effector cells (Beff) (Bar-Or et al. 2010; Li et al. 2015). This B cell subset is significantly increased during the active phase of MS, during which the patients exhibit overt clinical symptoms (Li et al. 2015). GM-CSF is known to promote myeloid cell activation within the CNS. These myeloid cells can potentiate MS pathology through the production of mediators that promote demyelination, axonal loss, and axonal degeneration (Monaghan and Wan 2020). B cells from MS patients have also been demonstrated to produce both IL-6 and TNF-α, which maintain the proinflammatory milieu within CNS and potentiate damage (Matsushita 2019). Second, B cells have the capacity to act as antigen-presenting cells, which promote the activation and expansion of encephalogenic Th1 and Th17 cells (Häusser-Kinzel and Weber 2019). Additionally, antibodies against myelin oligodendrocyte glycoprotein, proteolipid protein, and myelin basic protein are observed in the lesions of MS patients (Genain et al. 1999). This suggests that B cells may directly contribute to demyelination via antibody-dependent cell-mediated cytotoxicity (Feng and Ontaneda 2017).
Yet, the anti-inflammatory action of certain B cell populations may serve as a protective mechanism in MS. Indeed, more severe experimental autoimmune encephalitis develops in mice whose B cells are defective in IL-10 secretion or exhibit a loss of cells expressing TIM-1, a broad marker for IL-10+ B cells with regulatory activity (Breg) (Cherukuri et al. 2019; Ding et al. 2011; Fillatreau et al. 2002; Xiao et al. 2012). Interestingly, B cell depletion with rituximab, effective at treating MS, reduces T cell hyper-reactivity observed in MS patients and leads to restoration of a balance between Breg and Beff cells (Bar-Or et al. 2010; Li et al. 2015). Thus, emerging findings support the important and potentially distinct effector and regulatory roles for B cells in brain function, behavior, and neurological disease, indicating a need for further exploration of potential roles of diverse B cell subsets in the context of brain function, especially as the brain undergoes senescence.
B cell immunosenescence
As does the nervous system, the immune system undergoes senescence and these age-related changes in functioning may have important impacts in the context of stroke and the aging brain. Indeed, immune cell populations across the lifespan can be dynamic; differ based on biological factors, such as age or sex; and also change in response to a variety of stimuli, such as stress, physiological state, or pregnancy (Graham et al. 2006; Simon et al. 2015). As an individual ages, the immune system skews towards a chronic, low-level proinflammatory state associated with persistent activation of the innate immune system that has been popularly dubbed “inflammaging” (Franceschi and Campisi 2014). Clinically, responsivity to infection during aging is markedly altered and aged individuals have poorer responses to immunization than younger controls (Bulati et al. 2017) due in large part to peripheral aging lymphocyte populations that undergo substantial alterations in both quantity and functionality. Furthermore, aged microglia, the resident immune cells of the central nervous system (CNS), exhibit a more reactive proinflammatory response profile and diminished phenotypic flexibility in response to changing neurobiological needs. Importantly, changes in the immune response during aging can have profound negative consequences for brain function. For instance, acute infection has been shown to induce confusion and delirium in elderly patients (van Gool et al. 2010). Acute infection also increases the risk for incipient stroke (Grau et al. 1995) and active infection at the time of stroke significantly impairs blood-brain barrier (BBB) integrity, functional outcome, and infarct volume (Doll et al. 2015a; Doll et al. 2015b). Finally, in the long term, a higher infection burden is associated with accelerated cognitive decline and neurodegeneration (Katan et al. 2013; Strandberg et al. 2005; Sy et al. 2011). Thus, a thorough understanding of how the immune system undergoes senescence is of key clinical importance not only for the management of immune challenges in the elderly but also given that this process may have important implications in the aged CNS.
The number and function of B cells undergoes profound alterations during aging. For example, reports evaluating lymphocyte levels among populations of distinct age groups have noted alterations in counts of B cell subtypes and a less diverse B cell repertoire with advancing age, possibly due to a marked reduction in B cell lymphopoiesis among aged individuals. The processes affecting B cell senescence have been meticulously described in two excellent reviews (Bulati et al. 2017; Riley 2013). To summarize here (Fig. 1), bone structure and mineral density declines have been reported during aging beginning in mid-life (Berger et al. 2008). While counts of HSCs remain consistent, the ability of these cells to promote the development of lymphoid lineage cell populations is altered (Bulati et al. 2017; Riley 2013). It is hypothesized that the age-related skewing of the bone marrow microenvironment towards an inflammatory state that depletes the lymphoid HSC pool and inhibits lymphopoiesis (i.e., RANTES, tumor necrosis factor (TNF)-α) coupled with a loss of signaling (namely bioactive IL-7) that stimulates B cell precursor development and promotes a diminished proliferative response to these signals underlies this change, at least in part. An additional consequence of aging bone marrow downstream of HSC dysregulation is a decline in numbers of common lymphoid progenitor (CLP) cells, the majority of which eventually develop into B cells. This decline in CLPs is associated with declines in expression of key transcription factors (i.e., EA47/EA2, EBF1, Pax5, and Bob1) related to critical phases of B cell precursor development (i.e., BCR formation, isotype class switch recombination, and somatic hypermutation). Furthermore, aging is associated with reduced expression of pro-survival factors and developing B cells are more susceptible to pro-apoptotic signals. This explains the well-documented reduction of pro- and pre-B cells among aged individuals and has important consequences for both the diversity of the antibody repertoire and for the functional phenotypes of the immature B cell population. Indeed, the few immature B cells produced tend to have compromised BCR function that shifts the B cell repertoire towards more antigen-experienced, age-associated B cell (ABC) phenotypes. Functionally, these cells are largely anergic (lacking a response to an antigen), proinflammatory, or even self-reactive, which may have important consequences for immunity in old age. Interestingly, ABCs secrete TNF-α and respond strongly to Toll-like receptor stimulation, representing a potential protective mechanism against bacterial and viral infection in the aged organism given the age-associated reduction in functional capacity of T cell–mediated immunity. However, reports have also noted a diminished effectiveness of vaccination, and an increased risk for infection among older cohorts. Whether the complex pattern of change in the B cell system represents an adaptive response to aging or a pathological side effect of immunosenesence is not yet known. What is clear is that, coupled with significant changes in innate and adaptive immune responses, the biological impacts of altered B cell functional repertoires that occur during immune aging cannot be ignored.

Immunosenescence cascade of B cells. With aging, bone mineral density declines and the bone marrow microenvironment becomes more proinflammatory. Hematopoietic stem cells (HSCs) found in aged bone marrow promote myeloid rather than lymphoid lineage cell population development, thereby depleting the lymphoid HSC pool and inhibiting lymphopoiesis due to a decline in numbers of common lymphoid progenitor cells (CLPs). Poor cell developmental signaling (namely bioactive IL-7), reduced expression key transcription factors (EA47/EA2, EBF1, Pax5, and Bob1), and diminished proliferative response to these signals among B cell precursor cells fails to stimulate critical phases of development (i.e., B cell receptor (BCR) formation, isotype class switch recombination (CSR), and somatic hypermutation (SHM)) and results in reduced numbers of pro- and pre-B cells. This coupled with a reduced expression of pro-survival factors, a greater susceptibility to pro-apoptotic signals, and the emergence of anergic, self-reactive, age-associated B cells (ABCs) with an antigen-experienced proinflammatory phenotype shifts the diversity of the antibody repertoire and for the functional phenotypes of the immature B cell population. The altered B cell compartment in aging has important consequences both in the context of immunity in the aging body and response to neurological insults, such as stroke, in the aging brain
The clinical burden of stroke in the aging brain
Stroke, defined as an interruption of cerebral blood flow induced by either vessel blockage or hemorrhage that deprives brain tissue of necessary oxygen and energy, is the second-leading cause of death worldwide, accounting for nearly 12% of all deaths globally (Mozaffarian et al. 2016). This number represents a significant health disparity considering that the global burden of stroke is shouldered disproportionately by lower income countries; regional health disparities that are also clearly evident within the US “stroke belt,” a comparatively impoverished region marked by the presence of stroke comorbidities and limited health care access, mirror this global trend (Global Burden of Cardiovascular Diseases Collaboration et al. 2018). While stroke survival has encouragingly improved in the past few decades (Lackland et al. 2014), researchers, clinicians, and patients now face the daunting challenge of addressing a complex constellation of post-stroke health consequences. Indeed, stroke remains a leading cause of long-term disability with costs of post-stroke care estimated to be $140,048/patient (Rosamond et al. 2007). Research innovations to reduce the burden of this devastating brain injury are urgently needed.
To study stroke preclinically, a variety of stroke induction models are employed and have been extensively reviewed elsewhere (Fluri et al. 2015). In brief, to approximate ischemic stroke, middle cerebral artery occlusion (MCAO) is the most common experimental stroke model employed in rodents in which an intraluminal filament occludes the middle cerebral artery, thereby resulting in the loss of ~ 50–75% blood flow to the cortex or striatum (Longa et al. 1989; Macrae 2011). In the permanent (pMCAO) approach, the filament remains lodged in the artery for the duration of experimental timeframe until tissue collection. Conversely, in a transient (tMCAO) model, blood flow to the brain is blocked by the filament for a specific period of time, generally 30 to 120 min, and is then removed thereby permitting re-entry of blood into the artery and consequently reperfusion injury. Problematically, these approaches are limited with regard to mortality rates, lesion size, or area, and that the incisions required for occlusion of the MCA may initiate inflammatory cascades that are not representative of the injury that occurs in human populations. Alternative focal stroke models are available. Chemical occlusion approaches, such as the injection of the vasoconstrictive peptide, endothelin-1 (ET-1) to restrict blood flow to 30–50% of normal (Biernaskie et al. 2001), or the photothrombotic approach in which light focused on a section of skull activates the photoreactive Rose Bengal (Watson et al. 1985), obviate some of these concerns. In contrast to these focal stroke models, global 2, 3, or 4 vessel occlusion models restrict blood flow, oxygenation, and nutrient supply to large sections of the brain (Cechetti et al. 2010; Traystman 2003). Finally, balloon insertion/inflation, collagenase injection, autologous blood injection, and endovascular puncture approaches can be effective approaches to experimentally recapitulate the pathological cascades initiated by intracerebral and subarachnoid hemorrhages (MacLellan et al. 2010; Titova et al. 2009).
Importantly, aging is a principal non-modifiable risk factor for the occurrence of stroke (Boehme et al. 2017) such that the risk for stroke after 55 years of age doubles with each decade of life; the average age of a first-time stroke occurs is typically ~ 70 years of life. These trends are problematic for the USA considering that, as the Baby Boomer generation continues to age and predicted lifespan durations rise, the size of the elderly population at risk for experiencing a stroke will only increase, thereby perpetuating challenges faced by an already taxed health care system as well as exacerbating a substantial economic burden for both the public and private sectors. Yet, the majority of preclinical studies assessing mechanisms of injury and viability of novel therapeutic agents targeting these mechanisms are conducted in young adult animals, whose brains may not successfully recapitulate the response profile of an aged brain following stroke (Buga et al. 2013; Popa-Wagner et al. 2018; Sohrabji et al. 2013). This may be especially true with respect to the immune response to ischemic injury. For example, a variety of immune responses in the brain, including glial reactivity, lymphocyte recruitment, apoptosis, and infarct development, are accelerated following stroke in an aged versus young organism (Badan et al. 2003; Popa-Wagner et al. 2007). Furthermore, work from the McCullough group indicated that brain permeation of peripheral immune cells, reactive oxygen species production from microglia, and risk for hemorrhagic transformation were all increased in aged animals exposed to tMCAO (Ritzel et al. 2018). This leads to higher mortality rates in spite of significantly smaller infarct sizes relative to young adult mice subjected to an identical experimental stroke. Importantly, bone marrow transfer from young hosts attenuated these changes and improved stroke-induced functional deficits, indicating that immunomodulation is a viable approach to addressing age-related post-stroke outcome disparities. Taken together, the converging evidence suggests that stroke in an aged organism represents a distinct injury in a distinct biological system compared with that of a young adult; additional consideration for how aging impacts other biological systems known to influence the function of the brain, such as the immune system, is clearly warranted if we are to gain a comprehensive understanding of the risk factors for, pathology of, and recovery from, stroke.
Neuroimmune responses during acute injury and chronic recovery and their impacts on the pathophysiology of stroke
Stroke represents a complex disease pathology that is profoundly influenced by a variety of factors including the ischemic versus hemorrhagic nature of the stroke, the localization of the infarct, and whether reperfusion was achieved and how quickly it took place following the insult (Bramlett and Dietrich 2004). In brief, in ischemic stroke cases, at the cellular level, severe reductions in cerebral blood flow at the ischemic site deprive neurons of crucially needed oxygen and glucose, resulting in the depletion of ATP reserves and causing an energy crisis in local brain tissues. Ionic disruption, metabolic stress, and cell death in the ischemic core are evident within hours of initial insult. Surrounding the necrotic core is the ischemic penumbra, in which tissues experience secondary impacts of stroke-induced pathological cascades affecting the core; this region represents an area at which post-stroke neuroprotective interventions can be directed.
In addition to promoting perturbations in the vasculature and neuronal death, ischemia initiates an inflammatory cascade that is now thought to promote, but also protect against, stroke-associated sequelae (Jayaraj et al. 2019); these observations have initiated a number of investigations probing the stroke-induced immune responses (Rayasam et al. 2018). Following the ischemic event, necrotic cells and the subsequent generation of cell debris can activate CNS astrocytes and microglia by acting as damage-associated molecular patterns (Kim et al. 2016). Additionally, ischemia and blood reperfusion result in the production of reactive oxygen species (ROS), which can increase cell death and subsequent inflammation (Jayaraj et al. 2019). For example, the concentration of nitric oxide is increased following ischemic stroke. Nitric oxide induces cytotoxicity and increases the expression of adhesion molecules (Shirley et al. 2014). The activation of glial cells and the production of additional proinflammatory mediators promote the trafficking of peripheral leukocytes into the CNS. During the acute phase, the activation of glial cells and the recruitment of peripheral leukocytes into the CNS exacerbate stroke pathology and worsen stroke outcome (Kamel and Iadecola 2012). However, during the chronic phase, the CNS-resident and CNS-infiltrated immune cells promote tissue repair and improve stroke outcome (Kamel and Iadecola 2012). Understanding the changes that these cells undergo throughout the disease course could provide insight into therapies that curtail the pathogenic functions of these cells during the acute phase of stroke but preserve and even enhance the protective functions of these cells during the chronic phase of stroke. These changes can also provide insight into the mechanisms that potentiate systemic immunosuppression following stroke, which has significant and potentially detrimental consequences for post-stroke patients (detailed below).
Glial cells
Microglia are yolk sac–derived CNS-resident phagocytes that make up approximately 16.6% of cells in the human brain and 12% of cells in the mouse brain (Bachiller et al. 2018; Ginhoux et al. 2010). Microglia predominantly maintain homeostasis in the CNS by acting as sentinels that clear debris from apoptotic cells and preserve the functions of neurons via the repair and pruning of synapses (El Khoury et al. 1998; Hickman et al. 2018). During an ischemic insult, microglia become activated and undergo a series of morphological and functional alternations (Jayaraj et al. 2019). Studies have demonstrated that, upon activation, microglia transition from a resting state, in which they have a highly branched morphology, to an ameboid morphology, which facilitates migration to the site of focal ischemia (Young and Morrison 2018). Activated microglia can be detected in the infarct as early as two hours following ischemia (Kawabori and Yenari 2015). During the acute phase of stroke, murine models have demonstrated that microglia potentiate stroke-associated damage and have a proinflammatory phenotype (Geissmann et al. 2010). Microglia can directly mediate neurotoxicity in the ischemic core through the production of ROS and tumor necrosis factor (TNF)-α (Ritzel et al. 2015; Sawada et al. 1989). Activated microglia also produce matrix metalloproteinases (MMP), which significantly contribute to perturbations in the BBB (Zinnhardt et al. 2015). These so called “M1” microglia therefore promote neuronal death and tissue damage during the acute phase of stroke. There is evidence to suggest that during the chronic phase of stroke, these M1 microglia, in response to cytokine stimulation, switch to an immunomodulatory M2 phenotype. The M2 microglia modulate tissue repair by secreting vascular endothelial growth factor and brain-derived neurotrophic factor (Ma et al. 2017). Additionally, M2 microglia promote the clearance of cell debris via phagocytosis (Ma et al. 2017). The capacity of microglia to alternate between the M1 and M2 phenotype appears to be impacted by age. When aged mice are exposed to traumatic brain injury, immunohistochemical analysis reveals that the lesion size is increased in aged mice compared with young mice. In addition, there are significantly more reactive M1 microglia in the cortex, hippocampus, and thalamus (Kumar et al. 2013). This suggests that age can negatively contribute to long-term stroke outcomes by preventing the phenotypic switch from proinflammatory M1 to immunomodulatory M2 microglia.
Alterations in CNS-infiltrated immune cells following ischemic stroke
The production of cytokines, chemokines, and the increased expression of adhesion molecules promotes peripheral immune cell infiltration into the CNS (Shim and Wong 2016). Using the murine tMCAO model, Gelderblom and colleagues demonstrated that neutrophils are the first peripheral immune cells to infiltrate into the CNS (Gelderblom et al. 2009). Neutrophils migrate to the site of tissue damage via a chemokine gradient and exacerbate inflammation through the production of proinflammatory cytokines. Neutrophils further exacerbate stroke severity via the production of MMPs, which decrease the integrity of the BBB and promote immune cell infiltration from the periphery (Ruhnau et al. 2017). Neutrophil numbers usually peak around 72 h following the ischemic event, then begin to decrease as additional immune cells infiltrate into the CNS (Ruhnau et al. 2017). Following ischemia, the concentration of the chemokine CCL2 is increased in the CNS, which leads to the recruitment of CCR2-expressing monocytes (Chu et al. 2014). Once in the CNS, monocytes can differentiate into either proinflammatory or anti-inflammatory macrophages, depending on the cytokine signals that they receive (Chu et al. 2014). Proinflammatory macrophages appear morphologically and functionally similar to reactive microglia, and therefore, it has been difficult to distinguish between these cell types (Jayaraj et al. 2019). To better ascertain the timing of macrophage migration following ischemic stroke, Schilling and colleagues utilized green fluorescent protein transgenic bone marrow chimeras to identify when the macrophages began phagocytosing neuronal debris following tMCAO induction. This study revealed that macrophages do not accumulate in the infarcted area until 3–4 days following the induction of focal ischemia (Schilling et al. 2005). It is thought that during the acute phase of stroke, proinflammatory macrophages act in conjunction with reactive microglia to produce proinflammatory mediators such as TNF-α, interleukin (IL)-1, ROS, and MMPs. Additionally, these cells can also phagocytose healthy neurons in the penumbra in an attempt to clear dead and dying cells (Schilling et al. 2005). Therefore, during the acute phase of stroke, macrophages are thought to exacerbate inflammation within the CNS. Evidence from murine models of ischemic stroke suggest that during the chronic phase of stroke (6–7 days following focal ischemia induction), macrophages acquire an “M2” or alternatively activated phenotype. These cells are thought to resolve inflammation via the production of IL-10, TGFβ, arginase, and growth factors. These M2 macrophages also phagocytose necrotic debris, thereby further resolving inflammation (Kanazawa et al. 2017). Therefore, therapies should aim to modulate the function of macrophages to promote their tissue repair and immune modulatory function, rather than block their migration into the CNS.
The functional alterations that lymphocytes undergo following ischemic stroke are not as well characterized as neutrophils and monocytes. Lymphocyte migration into the CNS occurs following neutrophils and monocyte infiltration, during the chronic phase of stroke (Shim and Wong 2016). T cells are not only thought to promote inflammation and tissue damage but also tissue repair following ischemic stroke (Gu et al. 2015). Cytotoxic CD8+ T cells may induce cell death and exacerbate tissue damage through the release of perforin and granzyme B (Arumugam et al. 2005). Furthermore, Th1 cells may potentiate inflammation and tissue damage via the production of IL-2, interferon (IFN)-γ, and TNF-α (Arumugam et al. 2005). On the contrary, Liesz and colleagues demonstrated that the depletion of T regulatory cells significantly increases ischemia-associated tissue damage. It was further demonstrated that neuroprotection is conferred by T regulatory cells through IL-10 signaling (Liesz et al. 2009).
A number of reports have indicated that T cells are the primary mediators of stroke-induced pathological sequelae and that B cells do not play a crucial role in stroke-induced neuropathology. Early reports noted that lymphocyte (T and B cells) deficiency in rodents is protective in experimental stroke as indicated by reduced infarct size relative to wild-type stroked mice (Hurn et al. 2007; Kleinschnitz et al. 2010; Schuhmann et al. 2017; Yilmaz et al. 2006) and follow-up investigations attributed beneficial effect of lymphocyte deficiency to the absence of T cells. For example, Yilmaz et al. (2006) noted that infarct volumes were significantly attenuated among CD4+- and CD8+-deficient mice relative to wild-type controls, while infarct sizes between B cell–deficient mice and wild-type controls were equivalent. Furthermore, neither genetic (JHD−/−) nor induced B cell deficiency (via anti-CD20 antibody) impacted infarct volumes nor brain-immune response endpoints as compared with wild-type mice subjected to the same experimental stroke (Schuhmann et al. 2017). In this same study, adoptive transfer of T cells into Rag1−/− mice reversed the beneficial effects of lymphocyte deficiency on infarct volume while lesion sizes among Rag1−/− mice given B cell–adoptive transfer did not differ from stroked control Rag1−/− mice that did not receive cell transfer (Schuhmann et al. 2017), replicating previous findings (Kleinschnitz et al. 2010). B cell depletion in these studies was assessed 1–3 days following focal ischemia induction; given that B cell infiltration does not typically occur until several days following focal ischemia induction, B cells may play an important role during the chronic phase of stroke, and this remains to be clarified. Nevertheless, conclusions drawn from these collective findings suggested that B cells did not substantially contribute to stroke neuropathology.
Yet, emerging findings noting B cell presence in brain is markedly enhanced in diseased/injured states where BBB integrity is compromised (Anthony et al. 2003; Doyle et al. 2015; Hickey 2001) suggest important and potentially protective roles for the B cell in the context of stroke (Doyle and Buckwalter 2017; Seifert et al. 2018; Selvaraj et al. 2016). For instance, Ren et al. (2011) noted worsened functional deficits, larger infarct volumes, and higher mortality rates among mice genetically deficient in B cells (μMT−/−) subjected to 60-min tMCAO, suggesting a protective role of the humoral immune system in the acute injury period following ischemia induction. Recent work from the Stowe group suggests that the protective effects of B cells following stroke may be related to their ability to migrate to multiple ipsi- and contralateral injury sites within the brain parenchyma, support neuronal viability, maintain dendritic complexity, and promote neurogenesis via an IL-10-dependent mechanism (Ortega et al. 2020). Additional support for IL-10 as the mechanistic underpinning of B cell–mediated neuroprotection in stroke comes from findings that systemic or central administration of B cells derived from wild-type mice, but not from IL-10-deficient mice, 24 h prior to stroke limited infarct size, inflammatory cell recruitment, functional deficits, and mortality rates (Chen et al. 2012; Ren et al. 2011). Later, IL-10-producing B10 regulatory B cells were found to accumulate in the ipsilateral cortex following stroke in wild-type mice (Chen et al. 2012), while administration of these cells to stroked μMT−/− or wild-type mice reduced infarct volume, attenuated the proinflammatory phenotype, limited the infiltration of other activated immune cells into brain, and simultaneously enhanced the activity of other regulatory immune cell subsets centrally (Bodhankar et al. 2013; Bodhankar et al. 2014). Others have noted that lesion volumes could be altered by the methodical manipulation of IL-10 levels or expression (Grilli et al. 2000), representing just some of the evidence implicating a beneficial role for IL-10 in acute brain injury (Garcia et al. 2017; Ortega et al. 2020). In addition to the neuroprotective effects post-stroke, a role for B cells in underlying the beneficial impacts of pre-stroke protective interventions has also been demonstrated. Indeed, repeated hypoxic preconditioning reduces the severity of injury, potentially via inducing an immunosuppressed phenotype in B cells prior to stroke and then increasing recruitment of B cells to the ischemic cortex while inhibiting diapedesis of T cells, monocytes, activated macrophages/microglia, and neutrophils (Monson et al. 2014). These findings mirror those seen in clinical populations, as circulating B cell numbers were negatively correlated with infract volumes, stroke severity, and long-term poor outcome (Urra et al. 2009; Wang et al. 2017). Lastly, while thrombolytic treatment has no impact on B cell counts post-stroke (Wang et al. 2017), IL-10+ B cell transfer at 4 as well as 24 h post-stroke still conferred protective effects on lesion size, post-stroke immunological cascades, and neurological deficits (Bodhankar et al. 2015), well beyond the accepted therapeutic window of tissue plasminogen activator (Gurman et al. 2015).
Differences in the method of B cell depletion employed may reconcile the differing conclusions regarding the role of B cells in stroke. Interestingly, most studies reporting exacerbated lesion sizes utilized the μMT strain of B cell–deficient mice from Jackson Laboratories (Bodhankar et al. 2013; Chen et al. 2012; Ren et al. 2011), while a study that noted no difference in infarct size utilized the JHD−/−- strain supplied by Taconic (Schuhmann et al. 2017). Importantly, in addition to profoundly reduced B cell numbers, μMT−/− mice may exhibit reduced numbers of T cells and T cell functionality in response to immune challenge may be impaired (Bergmann et al. 2001; Homann et al. 1998). JHD−/− mice appear to exhibit normal T cell numbers (Taconic 2020) although T cells from these animals may exhibit some deficiencies in their responsivity to immune challenge (Macaulay et al. 1998). Moreover, while anti-CD20 treatments generally spare T cells (Uchida et al. 2004), a consequence of B cell depletion is that complex synergistic T cell-B cell interactions known to take place in normal immune function (Crotty 2015) are also attenuated; consideration of this loss of interactive function is important given prior work noting reduced infarct sizes in SCID and Rag1−/− strains of mice. Furthermore, in vivo depletion via anti-CD20 treatments can regionally specifically spare certain subtypes of B cells, namely innate-like T cell–independent B1 cells (Ait-Oufella et al. 2010; Hamaguchi et al. 2005). Finally, data collected in the experimental autoimmune encephalomyelitis (EAE) model of MS noted beneficial effects of anti-CD20-mediated B cell depletion only when the model used to induce EAE resulted in activated, antigen-presenting B cells that polarized proinflammatory T cells but exacerbated EAE when the induction model left B cells in an inactive state (Weber et al. 2010). This insight, combined with the observation that early immunomodulatory actions of IL-10-secreting B cells may appropriately restrain the proinflammatory responses aimed at clearing dead and dying cells in the infarcted area to promote sparing of penumbral neurons whose functions may be recovered over time (Bodhankar et al. 2013; Bodhankar et al. 2014; Chen et al. 2012; Ortega et al. 2020; Ren et al. 2011), suggests that the relationship between stroke and the actions of distinct B cell populations in the acute injury period is more complex that previously appreciated. More investigations addressing the role that shifting ratios of immune cells have in the context of stroke, both in their individual impacts on the brain in various activation states and in their ability to interact with each other to influence nervous system function, are needed.
Although these collective findings indicate that B cells may not robustly contribute to stroke neuropathology in the acute injury period, and may in fact exert neuroprotective actions depending on their phenotype, findings from the Buckwalter laboratory (2015) have indicated potential roles for B cells in the development of delayed post-stroke cognitive impairment. Indeed, mice exposed to distal MCAO display activated B cells secreting antibodies such as IgG, IgM, and IgA in the infarcted brain, impaired hippocampal long-term potentiation, and disrupted cognitive endpoints; genetic or pharmacological depletion of B cells prevented these effects. In human patients, brain tissues revealed an accumulation of B cells and auto-reactive IgG staining in some patients who developed post-stroke cognitive impairment. Furthermore, higher titers of myelin basic protein antibodies predicted worsened mini-mental status exam performance among patients who exhibited cognitive decline in the months following their stroke (Becker et al. 2016). Taken together, evidence collected to date supports that B cells may exert neuroprotective actions in the acute injury period, though these effects may be subtype-specific and short-lived (Fig. 2).

Dual roles for B cells in the acute and chronic periods following stroke. The pathological sequelae associated with stroke-induced restriction in blood flow and the post-occlusion reperfusion process induces neuronal metabolic dysfunction leading to an energy crisis in infarcted brain tissue. Depletion of ATP reserves, disruption to ion homeostasis, accumulation of cell-damaging reactive oxygen species, and excitotoxic states in brain parenchymal cell serve as danger/damage signals to induce an activated, proinflammatory phenotype in resident microglia. This proinflammatory state recruits peripheral immune cells, including B cells, to injured CNS tissues through leaky blood-brain barrier openings. During the acute phase, the activation of glial cells and the recruitment of peripheral leukocytes into the CNS exacerbate stroke pathology and worsen stroke outcome. Yet, B cell recruitment does not seem to contribute to stroke neuropathology and may actually serve protective roles during the acute injury period via interleukin (IL)-10-dependent immunoregulation. During the chronic recovery phase, CNS-resident and peripheral-originating CNS-infiltrated immune cells promote tissue repair and improve stroke outcome. However, B cell production of auto-reactive antibodies against various CNS antigens may contribute to the development of delayed post-stroke cognitive impairment. While immunomodulation represents a viable novel therapeutic approach for the treatment of stroke, additional studies are required to better understand the function of lymphocytes following ischemic stroke
Impact of stroke on immune suppression and susceptibility to infection
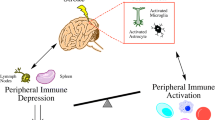
Following the ischemic event, there is systemic immune response that is characterized by an increase in circulating IL-6 and CXCL1. This systemic inflammatory response peaks 4 h following stroke, and promotes the activation of peripheral immune cells, which will migrate to the site of focal ischemia within 72 h (Chapman et al. 2009). As a compensatory mechanism to curtail inflammation within the CNS, this period of systemic inflammation is followed by a period of immune suppression, which greatly increases susceptibility to infection (Shim and Wong 2016). The mechanisms that promote immune suppression are likely to be multifactorial and have still not been fully explored. One potential mechanism through which immune suppression occurs may be through alteration in peripheral immune cell function following ischemic stroke (Shim and Wong 2016). Neutrophils are the first innate immune cells to become activated during infection (de Oliveira et al. 2016). Interestingly, blood samples from stroke patients revealed that the neutrophils from stroke patients have a decreased ability to undergo netosis as well as a decreased ability to generate respiratory bursts compared with healthy controls, suggesting that alternations in neutrophil function may render stroke patients more susceptible to infection (Ruhnau et al. 2014). Additionally, monocytes from ischemic stroke patients express significantly lower levels of human leukocyte antigen (HLA)-DR (Haeusler et al. 2012). This is important because an effective immune response against pathogens is contingent on the capacity of antigen-presenting cells to process and present antigens in the context of the MHC to T lymphocytes (Kotsias et al. 2019). This deficiency in innate immune cell function is accompanied by splenic atrophy, which occurs 4 days following tMCAO induction (Ross et al. 2007). Annexin V and TUNEL staining revealed that splenic atrophy occurs in part due to an increase in apoptosis and an increase in cell migration to the CNS (Ross et al. 2007). Systemically, splenic atrophy results in a significant decrease in the concentration of proinflammatory cytokines; a significant decrease in both splenic and circulating B cells; and, interestingly, a significant increase in the number of circulating CD4+ Foxp3-expressing regulatory T cells (Ross et al. 2007). Additionally, there is evidence to suggest that there is a shift from a systemic Th1 to a Th2 response, which is thought to decrease the concentration of proinflammatory cytokines, thereby protecting the brain from additional damage (Engelbertsen et al. 2013). However, this phenotypic switch further increases immunosuppression. These systemic alterations in immune cell function mediate immune suppression following ischemic stroke yet the precise mechanisms that trigger these functional changes remain to be elucidated.
One potential mechanistic explanation for how post-stroke immunosuppression occurs may be that the brain dampens inflammation via the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (Mracsko et al. 2014). The activation of the HPA axis results from stress caused by the ischemic event, and this signaling axis promotes the production of glucocorticoids (Mracsko et al. 2014; Shim and Wong 2016). The production of glucocorticoids can be further exacerbated by the increased production of proinflammatory cytokines following the activation of glial cells and the infiltration of peripheral immune cells (Burford et al. 2017). Glucocorticoids are thought to inhibit nuclear factor-κΒ expression immune cells and increase the systemic production of IL-10 (Coutinho and Chapman 2011). Glucocorticoids can suppress the production of IFN-γ by CD4+ T cells and promote T lymphocyte apoptosis (Theodorou et al. 2008). They can also reduce MHC II expression by peripheral dendritic cells (Kitajima et al. 1996). This suggests that glucocorticoids play a major role in suppressing the peripheral immune response following ischemic stroke.
When the sympathetic nervous system is activated in response to stress caused by ischemic stroke, catecholamines are produced (Shim and Wong 2016). Catecholamines act on α- r β-adrenoreceptors to promote apoptosis and promote immune suppression (Schulze et al. 2014). Specifically, catecholamines inhibit the production of IL-6 by monocytes/macrophages; promote an increase in IL-33 production by dendritic cells, which promotes a Th2 response; and inhibit the production of IFN-γ by CD4+ T cells (Ramer-Quinn et al. 1997; Straub et al. 1997; Yanagawa et al. 2011). Interestingly, blockade of α- or β-adrenoreceptors via pharmacological inhibitors prevents splenic atrophy and reduces infarct size following induction of tMCAO (Ajmo Jr. et al. 2009). Additionally, a retrospective analysis of stroke patients that were administered β-blockers found that these patients exhibited significantly less disability, had a lower neurological deficit score, and had an overall better prognosis compared with patients that did not receive β-blockers (Dziedzic et al. 2007). This suggests that therapies which target either the HPA axis or the sympathetic nervous system could potentially ameliorate systemic immunosuppression and prevent infection. Such therapies would have to be carefully modified to curtail harmful inflammation in the CNS, while still preventing peripheral immunosuppression.
Remaining issues and concluding remarks
An important remaining issue to be addressed relates to the age of the study populations from which these findings are drawn. As discussed above, the majority of studies investigating the role of the immune cells in the response to stroke were conducted in young adult animals. Importantly, initial findings in aged animals suggest that splenectomy prior to stroke confers protection against infarct development and behavioral deficit manifestation in 18–22-month-old mice (Chauhan et al. 2018), mirroring the protective effects reported with lymphocyte-deficient young adult animals. Furthermore, CD4+ T cell depletion 3 days after stroke attenuated injury-induced increases in IFN-γ levels and behavioral deficits in 18–20-month-aged mice (Harris et al. 2020), indicating that T cells may play an important neuropathological role in stroke at multiple ages. Whether similar observations to those reported in younger counterparts would be realized with cell depleting strategies targeting other immune cells or adoptive immune cell transfers in older organisms subjected to experimental stroke has yet to be thoroughly tested but is nonetheless warranted given the profound alterations in immune function with increasing age and the emerging evidence that supports many immune cells exert critical effects in brain.
Given the known interactions between the nervous and immune systems in response to brain injury, investigation of immunomodulation as a novel therapeutic approach for stroke recovery is warranted. Evidence suggests that the magnitude and duration of the immune response in the acute injury phase and during the long-term chronic phase following stroke may be tied to functional recovery. Indeed, a recent study by Tsai et al. (2019) utilizing elastic net regularized regression modeling to evaluate patterns of long-term post-stroke immunological responses revealed that an elongated and robust acute inflammatory phase driven by elevated STAT3 transcription factor activation was associated with greater long-term cognitive decline. Resolution of this inflammatory cascade appears to be crucial in the realization of post-stroke recovery and cells that comprise the lymphocyte compartment may be ideal therapeutic targets to improve long-term stroke outcome. A number of immunomodulatory approaches targeting a variety of components of the post-stroke immune response, including antioxidant treatments and restriction of lymphocyte trafficking to injury sites, are currently undergoing clinical trials (Zera and Buckwalter 2020); the results of these studies are eagerly awaited. The strong support preclinically for the potent neuromodulatory and neuroprotective actions of IL-10-secreting immune cells (such as Bregs) supports their consideration as another novel therapeutic target. Until then, additional preclinical studies are required to better understand the function of various immune populations following ischemic stroke.
In conclusion, as emerging findings address the impact of how the nervous and immune systems converge to implicate important interactive effects in the context of brain injury and disease, more research addressing not only the role of any individual immune cell type but also the complex interactions between these cells and those of the brain is needed. Evidence collected to date suggests dual protective but also potentially detrimental roles for immune cells in the pathophysiology of stroke, effects that may be timing specific, dependent on the underlying characteristics of the organism in which stroke occurs, and contingent on the cell subtype involved. Proper titration of the magnitude and timing of inflammatory, modulatory, and immunosuppressive cascades may serve as effective novel intervention approaches to promote stroke survival and long-term functional recovery. As more studies addressing this important issue continue to be conducted in the coming years, consideration of immunosenence in the context of brain aging will likely reveal novel components of the post-stroke pathological cascade and could reveal potentially new therapeutic and/or preventative targets for the treatment of this devastating brain injury. Finally, more broadly, this knowledge may also support innovations in the study of other age-related brain diseases that continue to plague our health care system; we enthusiastically await advances in each of these important areas.
References
Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;207:1579–87. https://doi.org/10.1084/jem.20100155.
Ajmo CT Jr, Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, et al. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol. 2009;218:47–55. https://doi.org/10.1016/j.expneurol.2009.03.044.
Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–57. https://doi.org/10.1016/j.coi.2008.03.014.
Anthony IC, Crawford DH, Bell JE. B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain. 2003;126:1058–67.
Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuromolecular Med. 2005;7:229–42. https://doi.org/10.1385/nmm:7:3:229.
Bachiller S, Jiménez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, et al. Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front Cell Neurosci. 2018;12. https://doi.org/10.3389/fncel.2018.00488.
Badan I, Buchhold B, Hamm A, Gratz M, Walker LC, Platt D, et al. Accelerated glial reactivity to stroke in aged rats correlates with reduced functional recovery. J Cereb Blood Flow Metab. 2003;23:845–54. https://doi.org/10.1097/01.WCB.0000071883.63724.A7.
Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67:452–61. https://doi.org/10.1002/ana.21939.
Becker KJ, Tanzi P, Zierath D, Buckwalter MS. Antibodies to myelin basic protein are associated with cognitive decline after stroke. J Neuroimmunol. 2016;295-296:9–11. https://doi.org/10.1016/j.jneuroim.2016.04.001.
Berger C, et al. Change in bone mineral density as a function of age in women and men and association with the use of antiresorptive agents. CMAJ. 2008;178:1660–8. https://doi.org/10.1503/cmaj.071416.
Bergmann CC, Ramakrishna C, Kornacki M, Stohlman SA. Impaired T cell immunity in B cell-deficient mice following viral central nervous system infection. J Immunol. 2001;167:1575–83.
Bhogal P, Andersson T, Maus V, Mpotsaris A, Yeo L. Mechanical thrombectomy-a brief review of a revolutionary New Treatment for Thromboembolic Stroke. Clin Neuroradiol. 2018;28:313–26. https://doi.org/10.1007/s00062-018-0692-2.
Biernaskie J, Corbett D, Peeling J, Wells J, Lei H. A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn Reson Med. 2001;46:827–30.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. IL-10-producing B-cells limit CNS inflammation and infarct volume in experimental stroke. Metab Brain Dis. 2013;28:375–86. https://doi.org/10.1007/s11011-013-9413-3.
Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. Treatment of experimental stroke with IL-10-producing B-cells reduces infarct size and peripheral and CNS inflammation in wild-type B-cell-sufficient mice. Metab Brain Dis. 2014;29:59–73. https://doi.org/10.1007/s11011-013-9474-3.
Bodhankar S, Chen Y, Lapato A, Vandenbark AA, Murphy SJ, Saugstad JA, et al. Regulatory CD8(+)CD122 (+) T-cells predominate in CNS after treatment of experimental stroke in male mice with IL-10-secreting B-cells. Metab Brain Dis. 2015;30:911–24. https://doi.org/10.1007/s11011-014-9639-8.
Boehme AK, Esenwa C, Elkind MS. Stroke risk factors, genetics, and prevention. Circ Res. 2017;120:472–95. https://doi.org/10.1161/CIRCRESAHA.116.308398.
Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–50. https://doi.org/10.1097/01.WCB.0000111614.19196.04.
Buga AM, Di Napoli M, Popa-Wagner A. Preclinical models of stroke in aged animals with or without comorbidities: role of neuroinflammation. Biogerontology. 2013;14:651–62. https://doi.org/10.1007/s10522-013-9465-0.
Bulati M, Caruso C, Colonna-Romano G. From lymphopoiesis to plasma cells differentiation, the age-related modifications of B cell compartment are influenced by “inflamm-ageing”. Ageing Res Rev. 2017;36:125–36. https://doi.org/10.1016/j.arr.2017.04.001.
Burford NG, Webster NA, Cruz-Topete D. Hypothalamic-pituitary-adrenal axis modulation of glucocorticoids in the cardiovascular system. Int J Mol Sci. 2017;18. https://doi.org/10.3390/ijms18102150.
Cechetti F, Worm PV, Pereira LO, Siqueira IR, C AN. The modified 2VO ischemia protocol causes cognitive impairment similar to that induced by the standard method, but with a better survival rate. Braz J Med Biol Res. 2010;43:1178–83. https://doi.org/10.1590/s0100-879x2010007500124.
Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125:S3–23. https://doi.org/10.1016/j.jaci.2009.12.980.
Chapman KZ, Dale VQ, Dénes A, Bennett G, Rothwell NJ, Allan SM, et al. A rapid and transient peripheral inflammatory response precedes brain inflammation after experimental stroke. J Cereb Blood Flow Metab. 2009;29:1764–8. https://doi.org/10.1038/jcbfm.2009.113.
Chauhan A, Al Mamun A, Spiegel G, Harris N, Zhu L, McCullough LD. Splenectomy protects aged mice from injury after experimental stroke. Neurobiol Aging. 2018;61:102–11. https://doi.org/10.1016/j.neurobiolaging.2017.09.022.
Chen Y, Bodhankar S, Murphy SJ, Vandenbark AA, Alkayed NJ, Offner H. Intrastriatal B-cell administration limits infarct size after stroke in B-cell deficient mice. Metab Brain Dis. 2012;27:487–93. https://doi.org/10.1007/s11011-012-9317-7.
Cherukuri A, Ding Q, Sharma A, Mohib K, Rothstein DM. Regulatory and effector B cells: a new path toward biomarkers and therapeutic targets to improve transplant outcomes? Clin Lab Med. 2019;39:15–29. https://doi.org/10.1016/j.cll.2018.10.011.
Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG. Role of CCR2 in inflammatory conditions of the central nervous system. J Cereb Blood Flow Metab. 2014;34:1425–9. https://doi.org/10.1038/jcbfm.2014.120.
Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2–13. https://doi.org/10.1016/j.mce.2010.04.005.
Crotty S. A brief history of T cell help to B cells. Nat Rev Immunol. 2015;15:185–9. https://doi.org/10.1038/nri3803.
de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378–91. https://doi.org/10.1038/nri.2016.49.
Deleidi M, Jäggle M, Rubino G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front Neurosci. 2015;9:172. https://doi.org/10.3389/fnins.2015.00172.
Ding Q, et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J Clin Invest. 2011;121:3645–56. https://doi.org/10.1172/JCI46274.
Dionisio A, Duarte IC, Patricio M, Castelo-Branco M. The use of repetitive transcranial magnetic stimulation for stroke rehabilitation: a systematic review. J Stroke Cerebrovasc Dis. 2018;27:1–31. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.09.008.
Doll DN, Engler-Chiurazzi EB, Lewis SE, Hu H, Kerr AE, Ren X, et al. Lipopolysaccharide exacerbates infarct size and results in worsened post-stroke behavioral outcomes. Behav Brain Funct. 2015a;11:32. https://doi.org/10.1186/s12993-015-0077-5.
Doll DN, Hu H, Sun J, Lewis SE, Simpkins JW, Ren X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke. 2015b;46:1681–9. https://doi.org/10.1161/STROKEAHA.115.009099.
Doyle KP, Buckwalter MS. Does B lymphocyte-mediated autoimmunity contribute to post-stroke dementia? Brain Behav Immun. 2017;64:1–8. https://doi.org/10.1016/j.bbi.2016.08.009.
Doyle KP, et al. B-lymphocyte-mediated delayed cognitive impairment following stroke. J Neurosci. 2015;35:2133–45. https://doi.org/10.1523/JNEUROSCI.4098-14.2015.
Dziedzic T, Slowik A, Pera J, Szczudlik A. Beta-blockers reduce the risk of early death in ischemic stroke. J Neurol Sci. 2007;252:53–6. https://doi.org/10.1016/j.jns.2006.10.007.
El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 1998;19:S81–4. https://doi.org/10.1016/s0197-4580(98)00036-0.
Engelbertsen D, Andersson L, Ljungcrantz I, Wigren M, Hedblad B, Nilsson J, et al. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler Thromb Vasc Biol. 2013;33:637–44. https://doi.org/10.1161/atvbaha.112.300871.
Feng JJ, Ontaneda D. Treating primary-progressive multiple sclerosis: potential of ocrelizumab and review of B-cell therapies. Degener Neurol Neuromuscul Dis. 2017;7:31–45. https://doi.org/10.2147/dnnd.S100096.
Filiano AJ, Gadani SP, Kipnis J. How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat Rev Neurosci. 2017;18:375–84. https://doi.org/10.1038/nrn.2017.39.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–50. https://doi.org/10.1038/ni833.
Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. https://doi.org/10.1111/j.1365-2249.2010.04143.x.
Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445–54. https://doi.org/10.2147/DDDT.S56071.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–9. https://doi.org/10.1093/gerona/glu057.
Funaro M, Messina M, Shabbir M, Wright P, Najjar S, Tabansky I, et al. The role of B cells in multiple sclerosis: more than antibodies. Discov Med. 2016;22:251–5.
Garcia JM, et al. Role of interleukin-10 in acute brain injuries. Front Neurol. 2017;8:244. https://doi.org/10.3389/fneur.2017.00244.
Geissmann F, Gordon S, Hume DA, Mowat AM, Randolph GJ. Unravelling mononuclear phagocyte heterogeneity. Nat Rev Immunol. 2010;10:453–60. https://doi.org/10.1038/nri2784.
Gelderblom M, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–57. https://doi.org/10.1161/strokeaha.108.534503.
Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–5. https://doi.org/10.1038/5532.
Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–5. https://doi.org/10.1126/science.1194637.
Global Burden of Cardiovascular Diseases Collaboration, Roth GA, Johnson CO, Abate KH, Abd-Allah F, Ahmed M et al. The burden of cardiovascular diseases among US states, 1990-2016. JAMA Cardiol. 2018;3:375–89. https://doi.org/10.1001/jamacardio.2018.0385.
Graham JE, Christian LM, Kiecolt-Glaser JK. Stress, age, and immune function: toward a lifespan approach. J Behav Med. 2006;29:389–400. https://doi.org/10.1007/s10865-006-9057-4.
Grau AJ, et al. Recent infection as a risk factor for cerebrovascular ischemia. Stroke. 1995;26:373–9. https://doi.org/10.1161/01.Str.26.3.373.
Gredler VRM. B cells accumulate in the cerebrospinal fluid in inflammatory neurological diseases. J Cytol Histol. 2012;S1:001.
Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, et al. Interleukin-10 modulates neuronal threshold of vulnerability to ischaemic damage. Eur J Neurosci. 2000;12:2265–72.
Gu L, Jian Z, Stary C, Xiong X. T cells and cerebral ischemic stroke. Neurochem Res. 2015;40:1786–91. https://doi.org/10.1007/s11064-015-1676-0.
Gurman P, Miranda OR, Nathan A, Washington C, Rosen Y, Elman NM. Recombinant tissue plasminogen activators (rtPA): a review. Clin Pharmacol Ther. 2015;97:274–85. https://doi.org/10.1002/cpt.33.
Haeusler KG, et al. Immune responses after acute ischemic stroke or myocardial infarction. Int J Cardiol. 2012;155:372–7. https://doi.org/10.1016/j.ijcard.2010.10.053.
Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, et al. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005;174:4389–99.
Harris NM, et al. Depletion of CD4 T cells provides therapeutic benefits in aged mice after ischemic stroke. Exp Neurol. 2020;326:113202. https://doi.org/10.1016/j.expneurol.2020.113202.
Häusser-Kinzel S, Weber MS. The role of b cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front Immunol. 2019;10. https://doi.org/10.3389/fimmu.2019.00201.
Herkenham M, Kigar SL. Contributions of the adaptive immune system to mood regulation: mechanisms and pathways of neuroimmune interactions. Prog Neuropsychopharmacol Biol Psychiatry. 2017;79:49–57. https://doi.org/10.1016/j.pnpbp.2016.09.003.
Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–24.
Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nature Neurosci. 2018;21:1359–69. https://doi.org/10.1038/s41593-018-0242-x.
Hoffman W, Lakkis FG, Chalasani G. B cells, antibodies, and more. Clin J Am Soc Nephrol. 2016;11:137–54. https://doi.org/10.2215/CJN.09430915.
Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol. 1998;72:9208–16.
Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–805. https://doi.org/10.1038/sj.jcbfm.9600482.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16:142. https://doi.org/10.1186/s12974-019-1516-2.
Kamel H, Iadecola C. Brain-immune interactions and ischemic stroke: clinical implications. Arch Neurol. 2012;69:576–81. https://doi.org/10.1001/archneurol.2011.3590.
Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017;18. https://doi.org/10.3390/ijms18102135.
Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, Elkind MS. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology. 2013;80:1209–15. https://doi.org/10.1212/WNL.0b013e3182896e79.
Kawabori M, Yenari MA. The role of the microglia in acute CNS injury. Metab Brain Dis. 2015;30:381–92. https://doi.org/10.1007/s11011-014-9531-6.
Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol. 2016;25:241–51. https://doi.org/10.5607/en.2016.25.5.241.
Kitajima T, Ariizumi K, Bergstresser PR, Takashima A. A novel mechanism of glucocorticoid-induced immune suppression: the inhibiton of T cell-mediated terminal maturation of a murine dendritic cell line. J Clin Investig. 1996;98:142–7. https://doi.org/10.1172/jci118759.
Kleinschnitz C, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115:3835–42. https://doi.org/10.1182/blood-2009-10-249078.
Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation International review of cell and molecular biology. 2019;348:69–121. https://doi.org/10.1016/bs.ircmb.2019.07.005.
Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34:1397–411. https://doi.org/10.1016/j.neurobiolaging.2012.11.013.
Lackland DT, et al. Factors influencing the decline in stroke mortality: a statement from the American Heart Association/American Stroke Association. Stroke. 2014;45:315–53. https://doi.org/10.1161/01.str.0000437068.30550.cf.
Li R, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. 2015;7:310ra166. https://doi.org/10.1126/scitranslmed.aab4176.
Liesz A, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–9. https://doi.org/10.1038/nm.1927.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039.
Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–40. https://doi.org/10.1038/sj.bjp.0706400.
Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247–72. https://doi.org/10.1016/j.pneurobio.2016.01.005.
Macaulay AE, DeKruyff RH, Umetsu DT. Antigen-primed T cells from B cell-deficient JHD mice fail to provide B cell help. J Immunol. 1998;160:1694–700.
MacLellan CL, Silasi G, Auriat AM, Colbourne F. Rodent models of intracerebral hemorrhage. Stroke. 2010;41:S95–8. https://doi.org/10.1161/STROKEAHA.110.594457.
Macrae IM. Preclinical stroke research--advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol. 2011;164:1062–78. https://doi.org/10.1111/j.1476-5381.2011.01398.x.
Mak T, Saunders ME, Jett BD. B cell development, activation and effector functions. In: Mak T, Saunders ME, Jett BD, editors. Primer to the immune response. 2nd ed: Academic Cell; 2014. p. 111–42.
Matsushita T. Regulatory and effector B cells: friends or foes? J Dermatol Sci. 2019;93:2–7. https://doi.org/10.1016/j.jdermsci.2018.11.008.
Mattson MP, Arumugam TV. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 2018;27:1176–99. https://doi.org/10.1016/j.cmet.2018.05.011.
Meeker RB, Williams K, Killebrew DA, Hudson LC. Cell trafficking through the choroid plexus. Cell Adh Migr. 2012;6:390–6. https://doi.org/10.4161/cam.21054.
Monaghan KL, Wan ECK. The role of granulocyte-macrophage colony-stimulating factor in murine models of multiple sclerosis. Cells. 2020;9. https://doi.org/10.3390/cells9030611.
Monson NL, Ortega SB, Ireland SJ, Meeuwissen AJM, Chen D, Plautz EJ, et al. Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflammation. 2014;11:22. https://doi.org/10.1186/1742-2094-11-22.
Mozaffarian D, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–360. https://doi.org/10.1161/CIR.0000000000000350.
Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immunity. 2014;41:200–9. https://doi.org/10.1016/j.bbi.2014.05.015.
Nicholson LB. The immune system. Essays Biochem. 2016;60:275–301. https://doi.org/10.1042/EBC20160017.
Ortega SB, et al. B cells migrate into remote brain areas and support neurogenesis and functional recovery after focal stroke in mice. Proc Natl Acad Sci U S A. 2020;117:4983–93. https://doi.org/10.1073/pnas.1913292117.
Ortman J, Velkoff VA, Hogan H. An aging nation: the older population in the United States US Department of Commerce, Census Bureau. 2014:1-28
Pavlov VA, Chavan SS, Tracey KJ. Molecular and Functional Neuroscience in Immunity. Annu Rev Immunol. 2018;36:783–812. https://doi.org/10.1146/annurev-immunol-042617-053158.
Popa-Wagner A, Badan I, Walker L, Groppa S, Patrana N, Kessler C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007;113:277–93. https://doi.org/10.1007/s00401-006-0164-7.
Popa-Wagner A, Sandu RE, Cristin C, Uzoni A, Welle KA, Hryhorenko JR, et al. Increased degradation rates in the components of the mitochondrial oxidative phosphorylation chain in the cerebellum of old mice. Front Aging Neurosci. 2018;10:32. https://doi.org/10.3389/fnagi.2018.00032.
Ramer-Quinn DS, Baker RA, Sanders VM. Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production. J Immunol (Baltimore, Md: 1950). 1997;159:4857–67.
Rayasam A, Hsu M, Kijak JA, Kissel L, Hernandez G, Sandor M, et al. Immune responses in stroke: how the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology. 2018;154:363–76. https://doi.org/10.1111/imm.12918.
Ren X, Akiyoshi K, Dziennis S, Vandenbark AA, Herson PS, Hurn PD, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011;31:8556–63. https://doi.org/10.1523/JNEUROSCI.1623-11.2011.
Riley RL. Impaired B lymphopoiesis in old age: a role for inflammatory B cells? Immunol Res. 2013;57:361–9. https://doi.org/10.1007/s12026-013-8444-5.
Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation. 2015;12:106. https://doi.org/10.1186/s12974-015-0329-1.
Ritzel RM, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 2018;136:89–110. https://doi.org/10.1007/s00401-018-1859-2.
Rosamond W, et al. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. https://doi.org/10.1161/CIRCULATIONAHA.106.179918.
Ross AM, Hurn P, Perrin N, Wood L, Carlini W, Potempa K. Evidence of the peripheral inflammatory response in patients with transient ischemic attack. J Stroke Cerebrovasc Dis. 2007;16:203–7. https://doi.org/10.1016/j.jstrokecerebrovasdis.2007.05.002.
Ruhnau J, et al. Stroke alters respiratory burst in neutrophils and monocytes. Stroke. 2014;45:794–800. https://doi.org/10.1161/STROKEAHA.113.003342.
Ruhnau J, Schulze J, Dressel A, Vogelgesang A. Thrombosis, neuroinflammation, and poststroke infection: the multifaceted role of neutrophils in stroke. J Immunol Res. 2017;2017:5140679. https://doi.org/10.1155/2017/5140679.
Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res. 1989;491:394–7. https://doi.org/10.1016/0006-8993(89)90078-4.
Schilling M, Besselmann M, Müller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196:290–7. https://doi.org/10.1016/j.expneurol.2005.08.004.
Schuhmann MK, Langhauser F, Kraft P, Kleinschnitz C. B cells do not have a major pathophysiologic role in acute ischemic stroke in mice. J Neuroinflammation. 2017;14(1):112. https://doi.org/10.1186/s12974-017-0890-x.
Schulze J, Vogelgesang A, Dressel A. Catecholamines, steroids and immune alterations in ischemic stroke and other acute diseases. Aging Dis. 2014;5:327–39. https://doi.org/10.14336/ad.2014.0500327.
Seifert HA, Vandenbark AA, Offner H. Regulatory B cells in experimental stroke. Immunology. 2018;154:169–77. https://doi.org/10.1111/imm.12887.
Selvaraj UM, Poinsatte K, Torres V, Ortega SB, Stowe AM. Heterogeneity of B cell functions in stroke-related risk, prevention, injury, and repair. Neurotherapeutics. 2016;13:729–47. https://doi.org/10.1007/s13311-016-0460-4.
Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol. 2015;15:441–51. https://doi.org/10.1038/nri3857.
Shim R, Wong CH. Ischemia, Immunosuppression and Infection--tackling the predicaments of post-stroke complications. Int J Mol Sci. 2016;17. https://doi.org/10.3390/ijms17010064.
Shirley R, Ord EN, Work LM. Oxidative stress and the use of antioxidants in stroke. Antioxidants (Basel). 2014;3:472–501. https://doi.org/10.3390/antiox3030472.
Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. 2015;282:20143085. https://doi.org/10.1098/rspb.2014.3085.
Sohrabji F, Bake S, Lewis DK. Age-related changes in brain support cells: implications for stroke severity. Neurochem Int. 2013;63:291–301. https://doi.org/10.1016/j.neuint.2013.06.013.
Strandberg TE, Pitkala K, Eerola J, Tilvis R, Tienari PJ. Interaction of herpesviridae, APOE gene, and education in cognitive impairment. Neurobiol Aging. 2005;26:1001–4. https://doi.org/10.1016/j.neurobiolaging.2004.09.008.
Straub RH, Herrmann M, Berkmiller G, Frauenholz T, Lang B, Schölmerich J, et al. Neuronal regulation of interleukin 6 secretion in murine spleen: adrenergic and opioidergic control. J Neurochem. 1997;68:1633–9. https://doi.org/10.1046/j.1471-4159.1997.68041633.x.
Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, et al. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–22. https://doi.org/10.1016/j.ajpath.2011.02.012.
Taconic. Jh constitutive knockout. 2020. https://www.taconic.com/transgenic-mouse-model/jh. Accessed 01 May 2020.
Tahir RA, Pabaney AH. Therapeutic hypothermia and ischemic stroke: a literature review. Surg Neurol Int. 2016;7:S381–6. https://doi.org/10.4103/2152-7806.183492.
Theodorou GL, Marousi S, Ellul J, Mougiou A, Theodori E, Mouzaki A, et al. T helper 1 (Th1)/Th2 cytokine expression shift of peripheral blood CD4+ and CD8+ T cells in patients at the post-acute phase of stroke. Clin Exp Immunol. 2008;152:456–63. https://doi.org/10.1111/j.1365-2249.2008.03650.x.
Titova E, Ostrowski RP, Zhang JH, Tang J. Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol Res. 2009;31:568–81. https://doi.org/10.1179/174313209X382412.
Traystman RJ. Animal models of focal and global cerebral ischemia. ILAR J. 2003;44:85–95. https://doi.org/10.1093/ilar.44.2.85.
Tsai AS, et al. A year-long immune profile of the systemic response in acute stroke survivors. Brain. 2019;142:978–91. https://doi.org/10.1093/brain/awz022.
Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199:1659–69. https://doi.org/10.1084/jem.20040119.
Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158:1174–83. https://doi.org/10.1016/j.neuroscience.2008.06.014.
van Gool WA, van de Beek D, Eikelenboom P. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet. 2010;375:773–5. https://doi.org/10.1016/S0140-6736(09)61158-2.
Vaupel JW. Biodemography of human ageing. Nature. 2010;464:536–42. https://doi.org/10.1038/nature08984.
Wang Y, et al. Frequencies of circulating B- and T-lymphocytes as indicators for stroke outcomes. Neuropsychiatr Dis Treat. 2017;13:2509–18. https://doi.org/10.2147/NDT.S148073.
Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. https://doi.org/10.1002/ana.410170513.
Weber MS, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010;68:369–83. https://doi.org/10.1002/ana.22081.
Xiao S, et al. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell Ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci U S A. 2012;109:12105–10. https://doi.org/10.1073/pnas.1120914109.
Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke. 2012;7:378–85. https://doi.org/10.1111/j.1747-4949.2012.00839.x.
Yanagawa Y, Matsumoto M, Togashi H. Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain Behav Immunity. 2011;25:1427–33. https://doi.org/10.1016/j.bbi.2011.04.012.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–12. https://doi.org/10.1161/CIRCULATIONAHA.105.593046.
Young K, Morrison H. Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J Vis Exp. 2018. https://doi.org/10.3791/57648.
Zera KA, Buckwalter MS. The local and peripheral immune responses to stroke: implications for therapeutic development. Neurotherapeutics. 2020. https://doi.org/10.1007/s13311-020-00844-3.
Zinnhardt B, et al. Multimodal imaging reveals temporal and spatial microglia and matrix metalloproteinase activity after experimental stroke. J Cereb Blood Flow Metab. 2015;35:1711–21. https://doi.org/10.1038/jcbfm.2015.149.
Acknowledgments
The authors wish to thank JW Simpkins, DM Rothstein, D Corbin, J Gilbert, S Seigan, and B White for their contributions to the review of the document.
Funding
This work was supported by NIH K01 MH117343 (PI = Engler-Chiurazzi), WVU Office of Research (PI = Engler-Chiurazzi), NIH P20 GM109098 (PI = Simpkins), NIH R01 NS099918 (PI = Huber), NIH U54 GM104942 (PI = Hodder), Praespero Innovation Award Program (PI = Wan), American Heart Association Scientific Development Grant 16SDG31170008 (PI = Ren), NSF 1916894 (PI = Ren).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Engler-Chiurazzi, E.B., Monaghan, K.L., Wan, E.C.K. et al. Role of B cells and the aging brain in stroke recovery and treatment. GeroScience 42, 1199–1216 (2020). https://doi.org/10.1007/s11357-020-00242-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-020-00242-9