
Abstract
The photocatalytic reduction of carbon dioxide (CO2) to produce methanol (CH3OH) is a promising strategy for producing clean energy. The catalyst, the aqueous medium, and the UV light are key parameters for the formation of the most relevant pair (e−/h+) and the specific selectivity towards the desired product (methanol). The use of Ga2O3 and V2O5 in the photocatalytic reduction of CO2 to produce methanol has been little studied. However, the combination of these oxides is important to generate synergies and decrease the band energy, enhancing the photocatalytic activity in CO2 reduction. In this work, V2O5-Ga2O3 combined photocatalysts have been prepared and investigated for the photocatalytic reduction of CO2. These photocatalysts were characterized by spectroscopic and microscopic techniques. The results showed that textural properties such as surface area and morphology do not influence the photocatalytic activity. However, species such as Ga2p3/2 and Ga2p1/2 identified by XPS enhanced the photocatalytic activity, most likely due to the formation of vacancies and the reduction of the bandgap in the combined oxides, as compared to single oxides. The contribution of these factors in pair interactions (e−/h+) with CO2 to generate methanol is demonstrated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The increase of CO2 emissions due to the use of fossil hydrocarbons is one of the main causes of climate change. Due to the increasing and frequent use of these fuels, CO2 emissions are unavoidable. By controlling CO2 emissions, this environmental problem can be avoided by converting the CO2 into other useful or non-toxic compounds (Sun et al. 2019). Although converting CO2 to a reusable hydrocarbon can benefit humans and the environment, it requires a significant amount of energy (Ghosh et al. 2021). The transformation of CO2 has been the subject of research by means of chemical, electrochemical, and photochemical methods. Methane, methanol, formic acid, alkyl carbamates, cyclic carbonates, dimethyl carbonate, and formic acid esters are some of the products obtained (Chang et al. 2016; Dou et al. 2007; Li et al. 2011; Wang et al. 2016; Yaashikaa et al. 2019).
Photochemical reduction of CO2 to methanol using photocatalysts is a feasible alternative for renewable energy and CO2 emissions reduction (Li et al. 2020; Nabil et al. 2021). The photocatalytic process involves three steps: (1) sunlight capture, (2) electron-hole pair photogeneration and surface migration, and (3) surface reactions. TiO2 has potential as a photocatalyst due to its non-toxicity and low cost (Barba-Nieto et al. 2020); however, its wide UV-activated bandgap and accelerated electron-hole pair recombination rate result in low activity and yield. Thus, research has focused on developing optimal semiconducting materials such as TiO2 for the reduction of CO2 to hydrocarbons (Zhao et al. 2017). Impurities introduced into the semiconducting material can either donate electrons near the conduction band or accept electrons near the valence band, producing excited states. The gap between these energy states and the nearest energy band is often called the binding energy of the dopant site (Efhart and Albe 2015).
In addition, the use of organic semiconductors such as metal-organic frameworks (MOFs) and covalent organic frameworks (COFs) for photocatalysis is currently arousing great interest due to their morphological and structural properties, such as tunable porous structure, physicochemical stability, and large specific surface area (Zhang et al. 2022). To carry out charge separation, these organic semiconductors must be excited by light, like inorganic semiconductors. Their band gap (Eg) is also a function of the method used to prepare them and their final composition (Liu et al. 2022). Their use in the photoreduction of CO2 has resulted in significant yields and valuable products such as formic acid, methane, carbon monoxide, and methanol (Zhang et al. 2022). Despite their impact, they still have high costs and problems with their stability and conductivity.
For example, mixing Ga2O3 with V2O5 can lead to structural changes and shifts in the bandgap energies. Gallium oxide (Ga2O3) is used in the preparation of a variety of sensors, optoelectronic devices, luminescent materials, and catalysts (Jamwal and Kiani 2022). Ga2O3 is an insulator with a broad bandgap of about 4.9 eV at room temperature. However, when chemically reduced, it becomes an n-type semiconductor (Pescarmona et al. 2007). Several authors have reported the use of Ga2O3 in the reduction of CO2, obtaining alcohols as the main product (Gallo et al. 2020). The Ga2O3-V2O5 mixture has been shown to enhance conductivity through the production of highly reactive e−/h+ pairs as species (Sharma and Sunkara 2002), since V2O5 can potentially change oxidation states and is stable as V2+, V3+, V4+, and V5+. Ga2O3 is a highly active photocatalyst in oxidation reactions due to its ability to absorb ultraviolet light (Binet et al. 1994). Furthermore, it can form oxygen vacancies and reactive species such as O2•− and OH− radicals (Zhao et al. 2007).
In addition, vanadium with an oxidation state of V5+ can be found in a tetrahedral form as VO4, in a pentahedral form as VO5, and in an octahedral form as VO6, and can produce polyoxyanions (Bilal et al. 2015). Surface species of vanadium V5+ are the most abundant and occur as monomeric VO4 species. In all these forms, vanadium acts as an excellent catalyst (Lamoureux et al. 2016). In this work, V2O5− Ga2O3 were used as photocatalysts and tested for chemically reducing CO2 to produce methanol. The V2O5 photocatalyst was synthesized and characterized with Ga2O3 in different amounts to evaluate whether textural and physicochemical properties influence the catalytic activity and to determine the level of efficiency of higher Ga2O3 content.
Experimental section
Preparation of the catalysts
Two different mixtures of vanadium and gallium oxides were obtained by varying the weight ratio of Ga:V metals at 40 and 60%, considering 7 g of V2O5 as 100%. Ga2O3 was considered in varying amounts, the same for gallium nitrate and constant for vanadium oxide. Ga2O3 was prepared from its precursor gallium nitrate in the V2O5 and Ga2O3 references, while ammonium metavanadate was used as the source for vanadium oxide.
V2O5
A total of 100 ml of a 1M NaOH solution was added to 7 g of ammonium metavanadate (NH4VO3) (99%, Aldrich), and the temperature was raised to 70 °C with continuous stirring until the water evaporated. The precipitate dried at room temperature for 7 h. The sample was then calcined at 400 °C for 18 h. Finally, the calcined samples were crushed to obtain a fine powder.
V2O5-XGa2O3
To prepare the mixed oxides, 3.936 and 5.891 g of Ga(NO3)3 (99%, Aldrich) were mixed separately with 7 g of V2O5. Then, 100 ml of 1M NaOH solution was added, and the temperature was increased to 70 °C with continuous stirring until the water evaporated. The precipitate dried at room temperature for 7 h. The samples were then calcined at 400 °C for 18 h. Finally, the calcined samples were ground to obtain a fine powder.
Ga2O3
A total of 100 ml of 1M NaOH solution was added to 7 g of Ga(NO3)3 (99%, Aldrich), and the temperature was raised to 70 °C with continuous stirring until the water evaporated. The residue dried at room temperature for 7 h. The sample was then calcined at 400 °C for 18 h. Finally, the calcined samples were crushed to obtain a fine powder.
Characterization of catalysts
N2 adsorption-desorption isotherms were obtained at −196.15 °C using Belsorp II equipment. Prior to the nitrogen adsorption measurements, solid catalysts were thermally treated at 200 °C for 12 h under vacuum. The surface area was obtained from the adsorption isotherms, and the average pore diameter and pore volume were determined from the desorption isotherms. X-ray diffraction patterns (XRD) were found in a 2θ region between 10° and 60° using Cu Kα radiation (l = 0.15418 nm) through an Empyrean Multi-Purpose Research X-ray diffractometer. Fourier-transform infrared spectroscopy (FTIR) was recorded in an FTIR spectrophotometer (Perkin Elmer Spectrum Two model) with ATR controlled by Windows XP. Samples were scanned over the spectral region of 4000 to 500 cm1. The photoluminescence (PL) spectrum of the synthesized materials was carried out in a FS-2 Fluorescence Spectrometer Scinco equipment with an excitation wavelength of 254 nm in the range from 300 to 700 nm. X-ray-emitted photoelectron spectroscopy analysis (XPS) was performed in a K-alpha spectrophotometer (Thermo Scientific) equipped with a monochrome Al Kα (1478 eV) anode as the X-ray power source.
Catalytic tests
The catalytic activity of Ga2O3-V2O5 photocatalysts in the CO2 photoreduction was evaluated by the following procedure: 30 mg of each photocatalyst was treated at 300 °C to eliminate surface impurities. Then, the photocatalyst was placed in a glass reactor containing 50 ml of a 0.1M NaOH solution with magnetic stirring. A 300 W mercury pen ray lamp (Analytik Jena US company) with a length of 53.8 mm and a total length of 117.3 mm (4.62”) was later introduced. The lamp emits Mercury spectrum with a primary energy at 254 nm and rated voltages of 800 V and 270 V at start-up and in operation, respectively. Immediately, compressed CO2 (99.99%, CeeKay Supply, Inc.) was bubbled into the sealed glass reactor through a mass flow controller for 20 min until saturation. When these conditions are reached, the bubbling ceases and photochemical reduction is achieved. All reactions were conducted for 120 min at room temperature and atmospheric pressure (in Mexico at approximately 0.80 atm.). The CO2 photoreduction reaction was monitored every 15 min for 120 min by FTIR. As the unique product identify was methanol, the intense absorption band at 1378 cm−1 was used to identify and quantify the reaction product. The methanol concentration was determined using a calibration curve constructed from the IR spectra obtained with different methanol concentrations (Aldrich, 99%) versus absorbance. Through this analysis, an appropriate absorbance concentration ratio was obtained.
The following equation was used to determine the conversion of methanol.
where C0 is the initial concentration of methanol at time zero, and Ct is the concentration at time t.
1H and 13C nuclear magnetic resonance spectroscopy was also used to identify the CO2 reduction-derived liquid products on a Bruker Avance III NMR spectrometer operating at 400 MHz 1H. 1H and 13C nuclear magnetic resonance (NMR) spectra analysis and collection were performed on the Mestrenova software.
Results and discussion
N2 adsorption-desorption isotherms analysis
Table 1 summarizes the textural properties of V2O5, Ga2O3, and V2O5-Ga2O3 samples, such as the BET area, diameter, and pore volume obtained from N2 adsorption-desorption isotherms. The surface area of V2O5 was 8.62 m2/g, which when gallium oxide was added decreased to 1.40 m2/g in the V2O5-40Ga2O3 sample, to 2.90 m2/g in the V2O5-60Ga2O3 sample, similarly to the Ga2O3 value of 1.6 m2/g. Besides, the pore diameter of V2O5 decreased from 46.47 to 27.03 nm due to the presence of Ga2O3 molecules. Also, the pore volume decreased with the addition of Ga2O3 (Groen et al. 2003).
FTIR spectroscopy
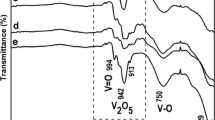
Bands were collected in the FTIR spectra over the 500–1000 cm−1 region for different V2O5-Ga2O3 photocatalysts (Fig. 1a), assigned to the Ga-O bond stretching vibrations of the Ga2O3 lattice. Bands at 529,624, 650, and 818 cm−1 were assigned to Ga2O3, and bands at 909 and 960 cm−1 were accredited to Ga2O3 deformation modes also appreciated. Vanadium content was also visible in Fig. 1, showing peaks between 750 and 1000 cm−1 due to the V-O terminal oxygen of V2O5. A V-O stretching vibrational band was located at 997 cm−1. The peak at 767 cm−1 was attributed to a V-O-V vibrational band, and the peak at 569 cm−1 was assigned to V3O, and the one at 874 cm−1 was attributed to V2O. The signal near 1028 cm−1 was accredited to the presence of V2O5 with an orthorhombic shape, having a symmetrical stretching of the V=O bond.

FTIR spectra of Ga2O3, V2O5-Ga2O3, and V2O5 photocatalysts a in the 4000–500 cm−1 interval and b in the 1500–500 cm−1 interval, respectively
In the spectral results of Ga2O3, V2O5, V2O5-40Ga2O3, and V2O5-60Ga2O3, the characteristic bands of Ga2O3 and V2O5 described above were observed. Ga2O3 bands intensified with the increasing amount of gallium oxide (Deshmane et al. 2010). Figure 1b shows a broadened region between 500 and 1500 cm−1 of Fig. 1a. An intense signal is observed between 960 and 914 cm−1 for Ga2O3 and is intense for pure gallium oxide. In the case of V2O5, the vibrational modes associated with V5+=O (1025 cm−1) and V4+=O (925 cm−1) were observed, which together with the vibrational mode of vanadium oxygen (V=O) can be used to find an approximation of the reduction ratio (e = V4+/(V5+ + V4+)) of V2O5. Around the 1020 cm−1 region, the stress motions corresponding to the V=O bond are reflected (Reddy et al. 2001).
X-ray diffraction
Figure 2 shows the X-ray diffraction patterns of Ga2O3 and V2O5 and their combinations. The XRD pattern can be indexed to V2O5 (JCPDS no. 60-0767). Previous work indicates that peaks at 15.6°, 20. 4°, 21.9°, 26.3°, 31.2°,32.6°, 34.5°, 41.4°, 42.3°, 45.6°, 47.5°, 49.0°, 51.4°, 52.3°, 55.8°, 61.2°, and 62.3° reflect the following planes (020), (001), (011), (110), (040), (101), (130), (002), (012), (141), (060), (102), (200), (061), (201), (240), and (170), respectively, of vanadium oxide (Manríquez-Ramírez et al. 2022). Diffractograms of V2O5 photocatalysts are in good agreement with the standard card for the V2O5 phase and with the results of Manríquez-Ramírez et al. (2022). In gallium oxide results, all peaks could be indexed to gallium oxide (β-Ga2O3) with lattice parameters of (Å) = 12,227, b (Å) = 3,039, c (Å) = 5,808, starting α = 90°, β = 103.82°, and γ = 90° (JCPDS 00-041-1103). Signals at 2θ = 18.949°, 38.420°, 59.184°, and 82.375° correspond to reflections (201), (402), (603), and (804) (Shi and Qiao 2020). The reflections in diffractograms corresponding to the Ga2O3 sample agree with the card number JCPDS 00-041-1103. In V2O5-Ga2O3 photocatalysts, both Ga2O3 and V2O5 phases were observed, showing that Ga2O3 peaks increase with higher G2O3 content and peaks of the V2O5 support are observed intensely (Playford et al. 2014). No additional peaks from other phases formed between V2O5 and Ga2O3 were observed.

X-ray diffraction patterns of different Ga2O3, V2O5-Ga2O3, and V2O5 photocatalysts
Scanning electron microscopy
SEM images of Ga2O3, V2O5, V2O5-40Ga2O3, and V2O5-60Ga2O3 catalysts are shown in Fig. 3. Catalysts exhibited morphologies consisting of particle clusters with irregular geometries. V2O5-60Ga2O3 catalysts have the smallest particles, and V2O5-40Ga2O3 catalysts increased in size. Larger particles were visualized on Ga2O3 catalysts, compared to V2O5 catalysts.

SEM images of different catalysts: a Ga2O3, b V2O5, c V2O5-40Ga2O3, d V2O5-60Ga2O3
X-ray photoelectron spectroscopy
Figure 4 reports the full XPS spectra of Ga2O3 and V2O5-Ga2O3 catalysts. The peaks related to different levels of the C, O, Ga, and V atoms are observed at 531.8 eV (O1s), 20.29 eV (Ga3d), 424 eV (Ga LLM Auger peak), 1117 eV (Ga 2p3/2), 1144 eV (Ga2p1/2), 284.8 eV (C1s), and 517 eV (V2p3/2 and O Auger peaks). The peak of each chemical element is well-defined.

Full XPS spectra of Ga2O3 and two different V2O5-Ga2O3 photocatalysts
Figure 5 shows the XPS spectra of Ga3d and Ga3p of all Ga2O3-containing catalysts. Figure 5a presents the Ga3d signal at 20.29 eV for all catalysts. Figure 5b presents Ga 2p3/2 and Ga2p1/2 signals at 1117 eV and 1144 eV, where the intensity of these peaks increases as the gallium oxide content increases. Figure 5a shows the deconvoluted XPS spectra of Ga3d; a peak centered at 20.38 eV corresponds to Ga3+ of Ga-O bonds (Carli and Bianchi 1994). The Ga2O3 spectrum achieved a binding energy value at 20.38 eV corresponding to Ga3d, and signals at 19.38 and 18.84 eV are associated with small amounts of metallic gallium.

XPS spectra of Ga2O3 and V2O5-Ga2O3 photocatalysts: a Ga3p and b Ga2p
In metal oxides, the lowest energy band is associated with the metal-oxygen bond (in our work, the Ga-O-Ga bond). In contrast, the highest energy band is assigned to hydroxide (OH−), absorbed oxygen (O2)2− type compounds, although it may also be due to absorbed water, thus assigning signals at 21.38 and 23.48 eV to these species, respectively. The XPS spectra of 2p3/2 and 2p1/2 binding energies of Ga at 1118.0 and 1144.9 eV, corresponding to Ga3+, are shown in Fig. 5b.
Figure 6 shows the XPS spectra for V2p of all vanadium oxide samples. Signals found at 517 and 524.5 eV correspond to V2p3/2 and V2p1/2, respectively. The intensity of these signals is positively correlated and increases with the amount of V2O5. Other peaks at 516 and 517 eV are V4+ and V5+, respectively. The ratio of V4+ and V5+ is related to oxygen defects and to the increase in V2O5. Due to its close proximity, oxygen (O1s) is also observed at 529.6 eV. This is consistent with reported values (Ryou et al. 2020; Silversmit et al. 2006).

XPS spectra of V2p3/2 and V2p1/2 of V2O5 and different V2O5-Ga2O3 photocatalysts
In the samples of the spectrum, peaks possibly corresponding to V2 and O1s appear intense. Figure 6 shows signals from 530 to 532 eV for carboxylic groups CH3-C=O, peaks at 533 and 533.9 eV are accredited to water, and a peak at 532 eV is attributed to surface oxygen at 530.8 eV, a prominent feature of the Ga-O bond and with the highest oxidation state of Ga (Ga3+).
Band gap energies and photoluminescence determination
These oxides are of outstanding importance due to their high conductivity and transparency in the UV-visible region of the electromagnetic spectrum and the energy range of the visible spectrum (1.6–3.1 eV), as well as their chemical stability. Figure 7 shows the valence band spectra of the samples measured by XPS (Benmoussa et al. 2003). The band gap energy value for pure V2O5 was about 3.20 eV. It decreased with the contribution of G2O3 until it reached the visible region. Previous research suggests that the visible emission is a result of surface defects and oxygen vacancies in the crystalline structure of V2O5-Ga2O3 materials (Cezar et al. 2014).

a Band energy and b photoluminescence spectra of Ga2O3, V2O5, and two different V2O5-Ga2O3 photocatalysts
Our V2O5-G2O3 catalysts show continuous absorption in the visible range, which is more intense when the G2O3 content is high. The Eg data obtained from Fig. 7 are reported in Table 2. The determined energy values show that, with the addition of gallium oxide, the energy band shifts to the visible region. Since the electronic motion depends on the gap between the valence band and the conduction band, when semiconductors such as gallium oxide or vanadium oxide are irradiated, ultraviolet light is needed for the electrons in the valence band to be transferred to the conduction band. When combined, the bandgap energy is lowered, allowing the electron-hole pair recombination and/or oxidation-reduction reactions to occur. Doped oxides are excellent candidates for photocatalysis applications.
Photoluminescence (PL) analysis provided information on charge transfer, energy gap, and surface defects. The PL emission spectra of Ga2O3, V2O5, V2O5-40Ga2O3, and V2O5-60Ga2O3 samples show peaks at 467 and 475 nm (see Fig. 7b). For Ga2O3, the most substantial emission peak is intense at 475 nm due to a faster rate of hole-electron recombination. The intensity of this band decreases at the following order V2O5<V2O5-40Ga2O3<V2O5-60Ga2O3 as a result of the slower hole-electron pair recombination rate. It is produced by the presence of electronic states or combinations caused by the addition of Ga2O3 in V2O5. A slight emission peak at 610 nm corresponds to the excitation of ionized oxygen vacancies: lower electron-hole recombination and higher catalytic activity.
Catalytic reaction
In this work, catalytic activity tests were carried out by the photocatalytic reduction of CO2. Considering that CO2 is a high-impact atmospheric pollutant, its reduction and transformation into products of interest such as methanol is an attractive solution. The activity of V2O5 and Ga2O3 catalysts and their combinations was tested by photocatalysis using NaOH and ultraviolet light under room temperature and atmospheric pressure conditions. The V2O5-60Ga2O3 catalyst showed a conversion of about 75% at the end of the 110 min reaction, which decreased to 60% with the reduced amount of Ga2O3 (Fig. 8). Figure 8 shows that the lowest conversions are obtained for V2O5 and Ga2O3 catalysts, with conversions of 50% and 42%, respectively. The mixture of oxides produced a higher activity due to the synergy between oxides. In addition, the decrease in the band gap energy led to an increase in the catalytic activity (Tsuneoka et al. 2010; Teramura et al. 2008; Teramura et al. 2014). The conversion of each photocatalyst was determined by FTIR as a function of time, but there was no measurement of selectivity. Although conversion provides information on the catalytic activity, light absorption and charge separation play a significant role in the activity and efficiency of the photocatalyst, resulting in improved yields and lifetime of the photocatalyst. In the same way, suitable catalytic sites for redox chemical processes that lead to the desired products and with low activation barriers are required.

Time-dependent conversion from the CO2 reduction to obtain methanol using V2O5-Ga2O3, V2O54, and Ga2O3 photocatalysts
Electrons (e−) migrate from the conduction band, producing holes (h+) in the valence band of a semiconductor. In a carrier material of the resulting charges (e− and h+), reaction can occur with the electron donor or acceptor species adsorbed on the catalyst surface. In the reaction mechanism of the photocatalytic reduction of CO2, oxidizing species located on the surface of the excited photocatalyst are of great importance. Photocatalytic excitation generates vacancies, which are oxidant species driving the oxidation process, and free hydroxyl radicals are adsorbed on the catalyst surface (Yamamoto et al. 2015; Yamamoto et al. 2014). Photoluminescence is another factor that measures the electronic mobility of materials. As observed in this CO2 reduction reaction, the addition of Ga2O3 slows down the electron-hole recombination rate, which favors the interaction with the medium and increases the photocatalytic activity.
In addition, the decrease in the band energy is essential for CO2 photoreduction. This may be due to vacancies formed in the presence of Ga2p3/2 and Ga2p1/2 species, which were observed by XPS to have high binding energies, and to neighboring oxygen vacancies, which showed a high attraction effect (Ga-O). When exposed to UV light, holes are created on the surface of the material which can be easily transferred to vacancies containing adjacent oxygen. This is confirmed by XPS, since Ga2p3/2 and Ga2p1/2 species form vacancies with their neighboring oxygen, which increases the catalytic activity. The catalytic activity is improved by increasing the number of these species. A significant effect on the activity is the size of the structures; they decrease with the addition of Ga2O3 and perhaps a slight increase in surface area with more Ga2O3 in the catalyst.
When reducing CO2 reduction to methanol, the following mechanism is proposed to produce alcohol. The formation of OH-radicals aids in the decomposition of the CO2 molecule when ultraviolet light hits the key using a NaOH solution as a reaction medium. OH-radicals in an aqueous solution can act as a strong “sweeper” of holes and OH-radicals, reducing the recombination of electron-hole pairs and increasing the photocatalytic activity, for which the following mechanism is proposed until the formation of methanol species.
In the first stage, the reaction starts in the NaOH reactive medium and catalysts with UV light to generate HO∗-radicals that propitiate active e−/H+ species, which react with CO2 dissolved in aqueous medium to form intermediate species of CO2−. These species in turn react with the (H*) vacancies to generate the CO species, which react with (H*), and these react with H* in consecutive stages until methanol is formed. The formation of HO*-radicals, which are important for the reaction to occur, is the main step in the photocatalytic reduction of CO2.
In addition, CO is an important intermediate or byproduct, which depends on the binding mode of CO on the surface of photocatalysts. Effective photogenerated electrons reach surface-active sites and catalytic reduction reactions occur, while photogenerated holes are consumed by H2O or additional sacrificial reagents to generate oxidation products that are desorbed from the photocatalyst surface. The most selective product of this mechanism is methanol. Both Ga2O3 and V2O5 are materials with oxidative photochemical properties that result in synergy and an increase in the activity and selectivity of the products.
Figure 9 schematizes in a general way the aim of this work and the mechanisms of converting CO2 to methanol using different photocatalysts based on V2O5 and Ga2O3. First, the combination of V2O5 and Ga2O3 resulted in V2O5-Ga2O3 photocatalysts with lower Eg than single oxides. This is represented by the connection (in blue color) of the two circles representing V2O5 (brown circle) and Ga2O3 (green circle). As a result, less energy is required for these catalysts to be photo-excited and for the electron-hole pairs to be generated. Once separated in both the valence and conduction bands by light exposure, the positively charged holes will interact with water molecules H2O to form H+ and O2 species. Conversely, CO2 molecules interact with H+ ions to form methanol, the only identified reaction product.

Schematic representation of combining V2O5 and Ga2O3 oxides to reduce the band gap, la photo-formation of the electron-hole pairs, and the general reaction of photoreduction of CO2 to methanol formation
FTIR and 13C NMR techniques were used to identify the reaction products (Figs. 10, 11, and 12); each methanol could be identified as the only reaction product.

Spectra obtained on time function during the CO2 reduction reaction using a Ga2O3, b V2O5, c V2O5-40Ga2O3, and d V2O5-60Ga2O3. Spectrum was taken each 15 min after t = 0

FTIR of the reaction products from the CO2 reduction using a without a catalyst and irradiated with light, where t is the time in min., and b with the (i) Ga2O3, (ii) V2O5, (iii) V2O5-40Ga2O3, and (iv) V2O5-60Ga2O3 photocatalysts

a 13C NMR and b 1H NMR that identify methanol as the reaction product obtained from the photocatalytic reduction of CO2 using V2O5, Ga2O3 V2O5-40Ga2O3, and V2O5-60Ga2O3 catalysts
Figure 10 shows the FTIR monitoring of the CO2 reduction reaction over time, showing that at t = 0, the solution is at saturation with CO2. Peaks corresponding to the CO2 in the solution at the beginning of the reaction are visible. From 0 to 30 min, in all FTIR spectra, CO2 signals disappeared and new bands were revealed due to the formation of alcohol. This monitoring was performed in situ using a FTIR equipment combined with ATR. The sensitivity of the equipment allowed detecting with great precision of the product formation corresponding to primary alcohol origination. Vibrations assigned to the methanol molecule can be seen in the time-dependent FTIR spectra of all the catalysts. The bands at 3318 cm−1 and 2815 cm−1 are associated with stretching vibrations of O-H and C-H, respectively. The bands at 1150 cm−1 and 1055 cm−1 are due to C-O-C stretching vibration. The band at 1230 cm−1 is attributed to C-O stretching vibrations, the band at 1226 cm−1 is assigned to CH3, and the band at 1378 cm−1 is accredited to the O-H bond of the methanol.
In the blank experiment, in the absence of a catalyst and irradiated with light (Fig. 11a), CO2 is reduced and carbonate mineralization takes place. Figure 11b shows the FTIR of the CO2 reduction using different catalysts after 110 min of reaction. As the most important signals and groups, the broad band of the CO group is located in the region between 3700 and 3000 cm−1. CH groups of methanol are present between 3000 and 2700 cm−l. Another important signal is found at 1378 cm−1 due to the methanol group. Also, catalysts V2O5-60Ga2O3 and V2O5-40Ga2O3 show more defined and intense signals.
Finally, the 13C and 1H NMR spectra identified the reaction products (Fig. 12). The single peak at δ = 51.0 ppm from the 13C spectra corresponded to the carbon atom of the methanol (Fig. 12a). The 1H NMR showed two important signals at 3.4 ppm and 4.7 ppm, assigned to hydrogen atoms of −CH3 and OH groups of the methanol molecule, respectively (Fig. 12b). These findings agree with those obtained by FTIR analysis. The presence of methanol was confirmed by the NMR spectrum obtained only for the products with all catalysts.
Conclusions
The co-precipitation method allowed obtaining catalysts based on vanadium and gallium oxide, confirmed by spectroscopic and microscopic studies. The synthesis and characterization of the different catalysts led to the observation that both textural and morphological properties are parameters that influence the catalytic activity, together with their chemical composition and the synergy that occurred when combined. Lastly, the use of photocatalysis as a clean reaction allowed the activation of these photocatalysts by UV light and the generation of electronic motions (e− and h+), reactive species capable of converting CO2 molecules into less harmful products.
The catalytic activity in reducing CO2 is selectively transformed to methanol, useful for converting it to products of interest such as methanol. Photocatalysis using NaOH and ultraviolet light at mild temperature and pressure was used to measure the activity of V2O5 and Ga2O3 catalysts and their mixtures. The conversion was about 75% for the V2O5-60Ga2O3 catalyst and decreased with lower amounts of Ga2O3. The lowest conversions of 50 and 42% were observed for V2O5 and Ga2O3 catalysts, respectively.
The mixture of oxides produced a higher activity due to the synergy among them. In addition, the decrease of the energy band occurred due to the photoactivation of V2O5-Ga2O3, and the material was illuminated by a photon of energy equal to or higher than the difference between the valence and the conduction band (band gap energy) and the different Gallium species obtained.
Data availability
All data obtained and analyzed during the present study are reported in this article.
References
Barba-Nieto I, Caudillo-Flores U, Fernández-García M, Kubacka A (2020) Sunlight-operated TiO2-based photocatalysts. Molecules 25:1–27. https://doi.org/10.3390/molecules25174008
Benmoussa M, Outzourhit A, Jourdani R, Bennouna A (2003) Structural, optical and electrochromic properties of sol–gel V2O5 thin films. Act Passive Electron Compon 26:245–256 10.1080=08827510310001162
Bilal AB, Khan GR, Asokan K (2015) Role of substrate effects on the morphological, structural, electrical and thermoelectrical properties of V2O5 thin films. RSC Adv 5:52602–52611. https://doi.org/10.1039/C5RA07542D
Binet L, Gourier D, Minot C (1994) Relation between electron band structure and magnetic bistability of conduction electrons in β-Ga2O3. J Solid State Chem 113:420–433. https://doi.org/10.1006/jssc.1994.1390
Carli R, Bianchi CL (1994) XPS analysis of gallium oxides. Appl Sur. Sc 74:99–102. https://doi.org/10.1016/0169-4332(94)90104-X
Cezar AB, Graff IL, Varalda J, Schreiner WH, Mosca DH (2014) Oxygen-vacancy-induced room temperature magnetization in lamellar V2O5 thin films. J Appl Phys 116:163904. https://doi.org/10.1063/1.4899249
Chang XX, Wang T, Gong JL (2016) CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ Sci 9:2177–2196. https://doi.org/10.1039/C6EE00383D
Deshmane CA, Jasinski JB, Carreon MA (2010) Microwave-assisted synthesis of nanocrystalline mesoporous gallium oxide. Microporous Mesoporous Mater 130:97–102. https://doi.org/10.1016/j.micromeso.2009.10.018
Dou XY, Wang JQ, Du Y, Wang E, He LN (2007) Guanidinium salt functionalized PEG: an effective and recyclable homogeneous catalyst for the synthesis of cyclic carbonates from CO2 and epoxides under solvent-free conditions. Synlett 19:3058–3062. https://doi.org/10.1055/s-2007-992362
Erhart P, Albe K (2015) Dopants and dopant–vacancy complexes in tetragonal lead titanate: a systematic first principles study. Comput Mater Sci 103:224–230. https://doi.org/10.1016/j.commatsci.2015.02.029
Gallo A, Snider JL, Sokaras D, Nordlund D, Kroll T, Ogasawara H, Kovarik L, Duyar MS, Jaramillo TF (2020) Ni5Ga3 catalysts for CO2 reduction to methanol: exploring the role of Ga surface oxidation/reduction on catalytic activity. Appl Catal B: Environ 267:1–11. https://doi.org/10.1016/j.apcatb.2019.118369
Ghosh S, Modak A, Samanta A, Kole K, Jana S (2021) Recent progress in materials development for CO2 conversion: issues and challenges. Mater Adv. 2:3161–3187. https://doi.org/10.1039/D1MA00107H
Groen JC, Peffer LAA, Perez-Ramıirez J (2003) Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis. Microporous Mesoporous Mater 60:1–17. https://doi.org/10.1016/S1387-1811(03)00339
Jamwal NS, Kiani A (2022) Gallium oxide nanostructures: a review of synthesis, properties and applications. Nanomaterials 12:1–31. https://doi.org/10.3390/nano12122061
Lamoureux B, Singh VR, Jovic V, Kuyyalil J, Su T-Y, Smith KE (2016) Structural and electronic properties of thermally evaporated V2O5 epitaxial thin films. Thin Solid Films 615:409–414. https://doi.org/10.1016/j.tsf.2016.07.062
Li D, Kassymov M, Cai X, Zang S, Jiang HL (2020) Photocatalytic CO2 reduction over metal-organic framework-based materials. Coord Chem Rev 412:1–16. https://doi.org/10.1016/j.ccr.2020.213262
Li J, Qi X, Wang L, He Y, Deng Y (2011) New attempt for CO2 utilization: one-pot catalytic syntheses of methyl, ethyl and n-butyl carbamates. Catal Commun 12:1224–1227. https://doi.org/10.1016/j.catcom.2011.04.008
Liu X, Verma G, Chen Z, Hu B, Huang Q, Yang H, Ma S, Wang X (2022) Metal-organic framework nanocrystal-derived hollow porous materials: Synthetic strategies and emerging applications. The Innovation 3(5):100281. https://doi.org/10.1016/j.xinn.2022.100281
Manríquez-Ramírez ME, Trejo-Valdez M, Castro LV, Estrada-Flores M, Ortiz-Islas E (2022) Application of CeO2-V2O5 catalysts in the oxidative desulfurization of 4,6-dimethyl dibenzothiophene as a model reaction to remove sulfur from fuels. Mater Res Bull 153:111864. https://doi.org/10.1016/j.materresbull.2022.111864
Nabil S, Hammad AS, El-Bery HM, Shalaby EA, El-Shazly AH (2021) The CO2 photoconversion over reduced graphene oxide based on Ag/TiO2 photocatalyst in an advanced meso-scale continuous-flow photochemical reactor. Environ Sci Pollut Res 28:36157–36173. https://doi.org/10.1007/s11356-021-13090-7
Pescarmona PP, Janssen KPF, Jacobs PA (2007) Novel transition-metal-free heterogeneous epoxidation catalysts discovered by means of high-throughput experimentation. Chem Eur J 13:6562–6572. https://doi.org/10.1002/chem.200700074
Playford HY, Hannon AC, Tucker MG, Dawson DM, Ashbrook SE, Kastiban RJ, Sloan J, Walton RI (2014) Characterization of structural disorder in γ-Ga2O3. J Phys Chem C 118:16188–16198. https://doi.org/10.1021/jp5033806
Reddy BM, Ganesh I, Reddy EP, Fernández A, Smirniotis PG (2001) Surface characterization of Ga2O3−TiO2 and V2O5/Ga2O3−TiO2 catalysts. J Phys Chem B 105:6227–6235. https://doi.org/10.1021/jp010763o
Ryou H, Hee Yoo T, Yoon Y, Lee IG, Shin M, Cho J, Cho BJ, Hwang WS (2020) Hydrothermal synthesis and photocatalytic property of Sn-doped Β-Ga2O3 nanostructure. ECS J Solid State Sci Technol 9:045009. https://doi.org/10.1149/2162-8777/ab8b4b
Sharma S, Sunkara MK (2002) Direct synthesis of gallium oxide tubes, nanowires, and nanopaintbrushes. J Am Chem Soc 124:12288–12293. https://doi.org/10.1021/ja027086b
Shi F, Qiao H (2020) Influence of hydrothermal reaction time on crystalqualities and photoluminescence properties of b-Ga2O3 nanorods. J Mater Sci: Mater Electron 31:20223–20231. https://doi.org/10.1007/s10854-020-04542-w
Silversmit G, Depla D, Poelman H, Marin GB, De Gryse R (2006) An XPS study on the surface reduction of V2O5(0 0 1) induced by Ar+ ion bombardment. Surf Sci 600:3512–3517. https://doi.org/10.1016/j.susc.2006.07.006
Sun Y, Lin Z, Peng SH, Sage V, Sun Z (2019) A critical perspective on CO2 conversions into chemicals and fuels. J Nanosci Nanotechnol 19:3097–3109. https://doi.org/10.1166/jnn.2019.16588
Teramura K, Tsuneoka H, Shishido T, Tanaka T (2008) Effect of H2 gas as a reductant on photoreduction of CO2 over a Ga2O3 photocatalyst. Chem Phys Lett 467:191–194. https://doi.org/10.1016/j.cplett.2008.10.079
Teramura K, Wang Z, Hosokawa S, Sakata Y, Tanaka T (2014) A Doping technique that suppresses undesirable H2 evolution derived from overall water splitting in the highly selective photocatalytic conversion of CO2 in and by water. Chem Eur J 20:9906–9909. https://doi.org/10.1002/chem.201402242
Tsuneoka H, Teramura K, Shishido T, Tanaka T (2010) Adsorbed species of CO2 and H2 on Ga2O3 for the photocatalytic reduction of CO2. J Phys Chem C 114:8892–8898. https://doi.org/10.1021/jp910835k
Wang JQ, Zhang YG (2016) Boronic acids as hydrogen bond donor catalysts for efficient conversion of CO2 into organic carbonate in water. ACS Catal 6:4871–4876. https://doi.org/10.1021/acscatal.6b01422
Yaashikaa PR, Senthil Kumara P, Varjanic SJ, Saravanan A (2019) A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J CO2 Util 33:131–147. https://doi.org/10.1016/j.jcou.2019.05.017
Yamamoto M, Yoshida T, Yamamoto N, Nomoto T, Yamamoto Y, Yagi S, Yoshida H (2015) Photocatalytic reduction of CO2 with water promoted by Ag clusters in Ag/Ga2O3 photocatalyst. J Mater Chem A 3:16810–16816. https://doi.org/10.1039/C5TA04815J
Yamamoto M, Yoshida T, Yamamoto N, Yoshida H, Yagi S (2014) In-Situ FT-IR study on the mechanism of CO2 reduction with water over metal (Ag or Au) loaded Ga2O3 photocatalysts. J Surf Sci Nanotechnol 12:299–303. https://doi.org/10.1380/ejssnt.2014.299
Zhang Y, Liu H, Gao F, Tan X, Cai Y, Hu B, Huang Q, Fang M, Wang X (2022) Application of MOFs and COFs for photocatalysis in CO2 reduction, H2 generation, and environmental treatment. Energy Chem 4:100078. https://doi.org/10.1016/j.enchem.2022.100078
Zhao SY, Wang SP, Zhao YJ, Ma XB (2017) An in situ infrared study of dimethyl carbonate synthesis from CO2 and methanol over well-shaped CeO2. Chin Chem Lett 28:65–69. https://doi.org/10.1016/j.cclet.2016.06.003
Zhao Y, Frost RL, Martens WN (2007) Synthesis and characterization of gallium oxide nanostructures via a soft-chemistry route. J Phys Chem C 111:16290–16299. https://doi.org/10.1021/jp075575y
Acknowledgements
The authors would like to thank the National Polytechnic Institute for funding through internal project SIP-20220593.
Author information
Authors and Affiliations
Contributions
Conceptualization: Ma Elena Manríquez. Methodology: Rodrigo Espinosa and Adrian Farid Bustos. Formal analysis and investigation: Rodrigo Espinosa, Ma Elena Manríquez, Martín Trejo-Valdez, Francisco Tzompantzi, Adrian Farid Bustos, and Emma Ortiz-Islas. Writing—original draft preparation: Emma Ortiz-Islas and Ma Elena Manríquez. Writing—review and editing: Emma Ortiz-Islas. Funding acquisition: Ma Elena Manríquez. Resources: Ma Elena Manríquez. Supervision: Emma Ortiz-Islas and Ma Elena Manríquez.
Corresponding author
Ethics declarations
Ethical approval
Not applicable
Consent to participate
All authors agreed to participate in the preparation of this article.
Consent for publication
All authors agreed to the publication of this article.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: George Z. Kyzas
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Espinosa, R., Manríquez, M.E., Trejo-Valdez, M. et al. Synthesis and characterization of V2O5-Ga2O3 photocatalysts and their application on the photocatalytic reduction of CO2. Environ Sci Pollut Res 30, 54119–54129 (2023). https://doi.org/10.1007/s11356-023-26155-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-023-26155-6