
Abstract
In recent years, Prorocentrum donghaiense, as a dominant species, has ranked first in terms of cumulative number and area of algal blooms in the East China Sea. In this study, the D1–D2 region of the large ribosomal subunit of P. donghaiense was used as the target gene, and specific primers DH–FP/DH–RP were designed according to the species-specific region of the target gene. An easy, sensitive and visual detection method refered to as polymerase chain reaction-nucleic acid chromatography strip (PCR-NACS) was established for P. donghaiense. The optimized parameters of the PCR amplification system are as follows: primer concentration, 0.15 μM; annealing temperature, 62 °C; and Mg2+ concentration, 1.5 mM. The specificity test showed that PCR-NACS was exlusively specific for the detection of the target algae. The sensitivity test show that the lowest detection limit (LDL) of PCR-NACS was 2.7 × 10−2 ng·μL−1 for genomic DNA and 3.58 × 102 copies·μL−1 for plasmid DNA, respectively. The tests using both genomic DNA and plasmid DNA as templates showed that the sensitivity of PCR-NACS was 10 times higher than that of ordinary PCR. The stability test showed that the interfering algal species did not affect the detection of the target algae by PCR-NACS. In addition, the test with simulated natural samples containing target algae showed that the LDL of PCR-NACS could reach 1.27 × 101 cells·mL−1. In summary, the PCR-NACS established in this study may provide a new method for easy identification of P. donghaiense in natural water samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Harmful algal blooms (HABs) are anomalous phenomena in which marine microalgae proliferate or accumulate on a large scale in seawater under specific trophic, hydrological, and climatic conditions, thus negatively affecting the aquatic ecosystem (Anderson et al. 2012; Sha et al. 2021). The number and extent of outbreaks of HABs are gradually increasing globally in the context of the eutrophication of ocean waters and the continously increased global temperature (Patricia et al. 2005; Lin et al. 2020; Anderson et al. 2021). HABs have a serious impact on human economic activities and life safety. For example, HABs are often accompanied by a decrease in dissolved oxygen in seawater and an imbalance in the proportion of nutrients, which usually leads to mass mortality of aquaculture organisms (Sun et al. 2020; Trottet et al. 2021). The decay and decomposition of algae will produce sulfide, which may destroy marine environment and cause economic loss to coastal tourism (Bechard, 2020). In addition, many harmful microalgae can produce toxins that pose a serious threat to human food security with the enrichment effect of the food web (Landsberg 2002; Kantiani et al. 2010; Grattan et al. 2016).
Among the recorded HABs, the blooms caused by marine dinoflagellates are the most common ones (Lu and Jeanette 2001; Patricia et al. 2005; Turner et al. 2021). Dinoflagellates are highly adaptable and can preferentially compete for sunlight or nutrients through vertical migration, and can also form cyst in response to adverse environment. These environmental adaptation strategies allow them quickly become the dominant vegetative cell population when competing with other coexisting organisms (Zhou et al. 2001). P. donghaiense, also known as P. shikokuense (Shin et al. 2019a), which belong to the genus Prorocentrum in the Dinophyceae, was first identified from the East China Sea by Lu and Jeanette (2001). As the dominant species responsible for the algal blooms in spring in the East China Sea (Lin et al. 2014), large-scale P. donghaiense-forming blooms have occured almost every year since 2000, for example, in the Zhoushan waters in 2002 and the East China Sea in 2004 (Zhou et al. 2003; Chen et al. 2006). The cell density in areas with dense blooms can reach 107 cells·L–1, with an area of 1500 km2 and a duration of up to 1 month. In some cases, the depth of blooms can reach 10 m. Such a large area and long-term HABs are also rare worldwide, severely destroying the marine ecological environment and affecting marine fishery production (Lu et al. 2005; Lin et al. 2014). Subsequently, the HABs formed by P. donghaiense also appeared in South Korea, Japan, Turkey, and the Black Sea (Lu et al 2003; Chen et al. 2006; Shin et al. 2019b). Although P. donghaiense does not produce toxins, its enormous biomass can cause serious damage or even death to farmed animals and cause direct economic losses to marine fisheries as high as millions of dollars each year (Tang et al. 2006; Zhang et al. 2016). Therefore, to reduce the threats caused by P. donghaiense to economic activities, seafood safety, and marine environment, the establishment of early monitoring and detection techniques for this alga is particularly important.
Up to date, the detection of harmful algae mainly relied on the microscopic examination of the morphology and surface structure of target cells. Unfortunately, correct identification of P. donghaiense is not easy due to its small size and high morphological similarity with other Prorocentrum. In particular, both professional taxonomic knowledge and experience are required to distinguish it from congenetic algal species (Cai et al. 2006; Zhang et al. 2016). The high frequency of P. donghaiense-forming HABs has led to increasing demands for the monitoring and detection of this harmful alga. However, faced with dealing with a large number of natual samples and ultra-high testing intensity, the time-consuming and cumbersome microscopic examination cannot meet the real-time monitoring aimed at determing the population structure and cell concentration of harmful algae. Therefore, with the development of molecular biology, more and more molecular detection techniques have been applied to the detection of harmful algae, including fluorescence in situ hybridization (FISH) (Chen et al. 2011; Meek and Dolah 2016), sandwich hybridization assay (SHA) (Doll et al. 2014), quantitative PCR (qPCR) (Eckford-Soper and Daugbjerg 2015; Hatfield et al. 2019), multiplex PCR (mPCR) (Nagai 2011; Al-Tebrineh et al. 2012; Sun et al. 2019), and isothermal amplification technology (IAT) (Qin et al. 2019; Liu et al. 2021).
Nagai et al. (2016) recently established a novel detection mthod referred to as Kaneka DNA chromatography chip (KDCC), which is essentially a DNA-DNA hybridization technology. Following the same principle with the KDCC, this study aims to develop a nucleic acid chromatography test strip (NACS) based on PCR for P. donghaiense. NACS can be divided into two main stages, namely, nucleic acid amplification and nucleic acid chromatography (Fig. 1). In the first stage, a pair of specific primers (5′➔3′) (forward primer and reverse primer) composed of tag sequences, spacer, and specific sequences are employed to perform PCR amplication. Among them, the specific sequences that are complementary to the target fragment are responsible for initiating nucleic acid amplification, and the spacer can stop the extension of DNA polymerase. The tag sequences of forward primer and reverse primer can hybridize with colloidal gold probe and detection probe, respectively. Therefore, specific primers can produce DNA fragments with single-stranded tag sequences at both ends. In the second stage, the PCR products are chromatographed on the test strip by capillary action, i.e., the PCR products that are first added to sample pad will successively move across conjugate pad, detection membrane, and absorbent pad on the test strip. When the PCR products flow to conjugate pad, their Tgn tag hybridize complementarily with the colloidal gold probe cTgn to form a PCR product-colloidal gold complex. Next, the resultant complex and colloidal gold probe that is not bound to the PCR products continue to move to the detection membrane. Here, another tag (Tsp) of the PCR products is complementary to the detection probe cTsp at the detection point, thereby forming a “detection probe-PCR product-colloidal gold” sandwich complex that can form a visible purple-red spot. In addition, the free colloidal gold probe cTgn that is not bound to the PCR products continues to flow to control point and complementarily binds to the control probe Tgn, forming a visible purple-red spot. In general, analysis of PCR-amplified products by nucleic acid chromatography can be completed within 10 min. If both detection and control spots appear on the detection membrane, the test result can be judged to be positive, namely, target PCR products are included in the analyzed sample. In contrast, if only the control spot appears on the detection membrane, the test result can be judged to be negative, namely, the analyzed sample does not contain target PCR product. In addition, if control spot does not appear on the detection membrane, the NACS detection is invalid.

Schematic diagram of the principle for PCR-NACS. a Nucleic acid amplification: PCR amplification is performed using specific primers with tag sequences to obtain DNA double strands with tag sequences at both ends. b Nucleic acid test strip chromatography: the PCR products are added to the sample pad of the NACS, and the sample will move along the NACS by capillary action. PCR products will first combine with the colloidal gold probe cTgn, and then the PCR products with the colloidal gold particles will hybridize with cTsp at the detection point to produce a purple-red spot, displaying a positive result. When no target PCR product is contained in the sample, no visible spot will be produced on the test spot, displaying a negative result. When no visible spot is produced on the control spot, the NACS analysis is invalid
NACS has the advantages of high sensitivity, rapidity, low cost, and simplicity. In particular, NACS is generally less dependent on large-scale instruments, so it has been widely used in many fields since its establishment, including the diagnosis of cannabis genes (Yamamuro et al. 2018), the identification of harmful microalgae (Chen et al. 2020), and the detection of pathogenic microorganisms such as herpes virus (Sano et al. 2018) and mycobacteria (Chikamatsu et al. 2019; Yoshida et al. 2021). In this study, we further explore the application of NACS to detect harmful algae by using P. donghaiense as target. For this purpose, the large ribosomal subunit (LSU rDNA) D1–D2 region of P. donghaiense was first cloned. Second, mainly using LSU rDNA D1-D2 as target gene, two specific primers DH–FP/DH–RP were designed to establish the PCR system, followed by the subsequent optimization. Finally, the PCR-NACS detection system was established and optimized, and its specificity, sensitivity, stability, and practicability were evaluated.
Materials and methods
Algal culture
P. donghaiense and 22 other control microalgae used for specificity verification (Table 1) are kept in the Applied Marine Biotechnology Laboratory, School of Marine Science and Technology, Harbin Institute of Technology (Weihai). All of the microalgal species were isolated by micropipette, kindly donated as gifts or purchased from commercial company. The algal species were cultured in a light incubator (GXZ-380B, Ningbo Jiangnan Instrument Factory) with f/2 or f/2-Si medium (Guillard 1975) with a salinity of 36 psu and pH of 8.0 ± 0.1 that was prepared with naturally aged seawater after precipitation. The culture conditions were as follows: culture temperature, 20 ± 2 °C; light intensity, 66 mmol photons m−2·s−1; light and dark cycle, 12 h:12 h.
Cloning and sequencing of target algae DNA
Target cells were collected from 45 mL of algal culture during the exponential growth period by centrifugation (12,000 g for 10 min at 4 °C). The algal cells were subjected to genomic DNA extraction using the Ezup Column-type Plant Genomic DNA Extraction Kit (Sangon Biotech, Shanghai, China) according to the manusfacturer’s instructions. A preliminary assessment of quality and concentration of the obtained genomic DNA performed by analysis by 1.5% AGE. The exact concentration and purity of the extracted DNA were finally determined by a NAS-99 micro-nucleic acid spectrophotometer (Thermo Fisher Scientific, MA, USA). The genomic DNA sample was stored at − 20 °C before use.
Using the extracted DNA as a template, the D1 − D2 region of LSU rDNA of P. donghaiense was PCR-amplified using the primers (D1/D2) and conditions described in Scholin et al. (1994). The PCR products were analyzed by 1.5% AGE and purified by using Sanprep Type DNA Gel Extraction Kit (Sangon Biotech, Shanghai, China). The purified PCR products were ligated with pMD™ 18-T vector (TaKaRa, Dalian, China) to form recombinant plasmid, which was further applied to transform the competent Escherichia coli cells. The positive clones were identified by colony PCR and were inoculated into Luria–Bertani liquid medium. After a shaking culture at 37 °C for 12 h, the resultant cell culture was directly delivered to Sangon Biotech (Shanghai) Co., Ltd for sequencing.
Design and synthesis of primers and probes
The obtained DNA sequence with the part of primer removed were analyzed by the online tool BLASTn implemented in the website of national center for biotechnology information (https://www.ncbi.nlm.nih.gov/). The obtained sequence has a high similarity (100%) with the LSU rDNA sequences of P. donghaiense with the accession number of AY833513.1 deposited in GenBank indicating target sequence was sucessfully obtained. At the same time, all of the homologous gene sequences from other algal species which belong to the genus Prorocentrum deposited in GenBank were downloaded. Both the target and homologous gene sequences were aligned by ClustalW Multiple alignment implemented in BioEdit software to find out the species-specific regions of the target algae by visual inspection. Based on the identified specific region of target species, the software Primer Premier 5.0 was used to design the specific sequences of forward primer (DH-F) and reverse primer (DH-R), respectively. Specific primers for PCR-NACS consist of 3 parts, namely, tag sequences (forward primer tag sequence Tgn and reverse primer tag sequence Tsp), spacer 9 (iSp9), and specific sequences (DH-F and DH-R). Among them, specific sequence and Tag sequences were connected by spacer 9. Both Tgn and Tsp are designed with Primer Premier 5.0 using the actin gene of Penaeus vannamei (Accession Number: LOC113819259) as a basic sequence.
Three probes were involved in NACS: (1) capture probe cTgn, which is reversely complementary to Tgn, and labeled at the 5′-end with a sulfhydryl group that can combine with colloidal gold particles to form so-called colloidal gold probe; (2) detection probe cTsp, which is inversely complementary to Tsp and can bind with PCR products; (3) control probe Tgn, which is inversely complementary to cTgn and can bind with the colloidal gold probe that is not bound to PCR products. In addition, a polyA (16) tail labeled with biotin was added to the 5′-end of cTsp and Tgn, respectively. By mixing the probe containing biotin with anti-biotin antibody, both cTsp and Tgn can be firmly fixed on the detection membrane via biotin-anti-biotin complex. The purpose of adding the polyA (16) tail is to prevent the probes from being embedded in the biotin-anti-biotin antibody complex, thereby affecting the binding of probe with PCR products or colloidal gold probe. The detailed information for all of the probes and primer are shown in Table 2, which were synthesized by Sangon Biotech (Shanghai) Co., Ltd.
Preparation and optimization of colloidal gold probe
The colloidal gold particles (approximately 20 nm in diameter) were prepared by citric acid reduction method (Liu and Lu 2006). In brief, 100 mL of ultrapure water was first added into a 500-mL clean beaker, followed by dropwise addition of 1 mL of chloroauric acid solution (1%) under the light proof condition. Next, the solution was heated to boil, and 2.8 mL of trisodium citrate solution (1%) was added quickly in a whole. The solution was continously stirred for 15 min, with the color gradually changing from colorless to gray and then to deep red. Finally, the obtained colloidal gold solution was naturally cooled to room temperature and stored in a refrigerator at 4 °C avoiding exposure to light. For the further preparation of colloidal gold probe, 1 mL of the colloidal gold solution was first centrifuged at 12000 g for 20 min at room temperature. Next, after removing the supernatant, 6 μL of capture probe cTgn (100 μM) and 94 μL of TE buffer (pH = 8.0) were added to the precipitate and mixed evenly by pipetting up and down. After standing for 12 h at 4 °C, 100 μL PBS solution (0.2 M NaCl, 20 mM Na2HPO4/NaH2PO4 buffer, pH 7.0) was added. The mixture was allowed to stand for 24 h at 4 °C, followd by centrifugation at room temperature at 12,000 g for 10 min. After discarding the supernatant, 200 μL of ultrapure water was added to remove the capture probes that were not bound to the colloidal gold particles by centrifugation under the same conditions. Finally, the resultant precipitate, i.e., colloidal gold probe, was resuspended with 1 mL of embedding buffer (20 mM Na3PO4, 5% BSA, 0.25% Triton X-100, and 8% sucrose) and stored at 4 °C under the light proof condition.
Given that colloidal gold particles can be stabilized by nucleic acid, the amount of capture probe that was used to prepare colloidal gold probe was optimized under the premise of the effect protection of colloidal gold particles by capture probe and saving cost. For this purpose, the capture probe with the final concentrations of 0.6, 0.5, 0.4, 0.3, 0.2, 0.1, and 0 μM was added to 1 mL of colloidal gold solution to prepare colloidal gold probe, respectively. By directly observing the color of colloidal gold probe solution and further comparing the performance of NACS with the prepared colloidal gold probe, the optimal amount of capture probes was determined.
Preparation of NACS
The proposed NACS consists of 5 modules, i.e., sample pad, conjugate pad, detection membrane, absorbent pad, and backing (Fig. 2). The dimensions [length (cm) × width (cm)] of each module are as follows: sample pad (glass fiber SB06), 1.5 × 0.4; conjugate pad (glass fiber RB65), 0.7 × 0.4; detection membrane (nitrocellulose membrane CN140), 2.9 × 0.4; absorbent pad (absorbent paper CH27), 1.5 × 0.4; and backing (rubber plate SM31–40), 6 × 0.4. All of the original materials that were used to prepare NACS were manufactured by Shanghai Liangxin Technology Co., Ltd. Some of them were required to be pretreated before being used to assemble the NACS. For the preparation of sample pad, the glass fiber SB06 was immersed in the sample pad buffer (0.05 M Tris–HCl, 0.15 M NaCl, 0.25% Triton X-100, pH 8.0) for 20 min, dried at 37 °C for 60 min and then stored at room temperature. For the preparation of conjugate pad, the colloidal gold probe solution was dispersed by ultrasonic treatment with an ultrasonic cell disruptor (JY-92 II, Ningbo Xinzhi Biotechnology Co., Ltd.) by setting at 40% duty cycle and 52 W for 10 s under the light proof condition. The colloidal gold probe pretreated with ultrasonic dispersion was evenly sprayed on the glass fiber RB65 (50 μL for single NACS). The prepared conjugate pad was dried at 37 °C for 2 h, and then stored at room temperature. For the preparation of detection membrane, nitrocellulose membrane was successively immersed in ultrapure water for 12 min and in 20 × SSC (NaCl, 17.5322 g; Na3C6H5O7-2H2O, 8.823 g; ddH2O, 100 mL; pH 7.0) for 9 min, and dry in the air. Next, 0.75 μL of mixture consisting of detection probe or control probe and anti-biotin antibody (10 µM) (Cell Signaling) in a volume ratio of 10:1 was spotted on the detection and control point, respectively. The detection membrane was then UV cross-linked (SCIENTZ 03, Ningbo Xinzhi Biotechnology Co., Ltd.) for 2 min at a wavelength of 254 nm and an intensity of 90 J·cm−2 with the detection and control probes immobilized. The pretreated sample pad, conjugate pad, detection membrane, coupled with absorbent pad and backing, were assembled and cut, resulting in single NACS with a length of 6 cm, a width of 0.4 cm, and an overlap part of 2 mm between sample pad, colloidal gold pad, detection membrane, and absorbent pad.

Schematic diagram of the assembling of NACS
PCR conditions and optimization
The PCR system with a total volume of 12.5 μL consists of 1 × PCR buffer (1.25 μL), 25 mM Mg2+ (0.75 μL), 2.5 mM dNTP Mixture (1 μL), 10 μM primers (DH–FP and DH–RP, each 0.25 μL), 5.0 U·µL−1 Taq DNA Polymerase (0.15 µL), genomic DNA (1 µL), and ultrapure water (7.85 µL). PCR amplification was performed in a thermal cycler (Bioer Technology, Hangzhou, China) with the following procedure: 94 °C for 5 min; 94 °C for 1 min, 58 °C for 50 s, 72 °C for 50 s, 29 cycles; 72 °C for 7 min. After PCR amplification, an aliquot of 5 µL of PCR products were analyzed with 1.5% AGE.
The PCR conditions, including annealing temperature, primer concentration, and Mg2+ concentration were optimized successively to improve the amplification efficiency. For this purpose, comparative PCR was performed with different primer concentrations (0.10, 0.15, 0.20, 0.25, 0.30, and 0.35 μM), annealing temperatures (52, 54, 56, 58, 60, 62, and 64 °C), and Mg2+ concentrations ( 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 mM), respectively.
Specificity test for PCR-NACS
A total of 22 control marine microalgae that are commonly distributed along the Chinese coast were randomly selected to verify the specificity of PCR-NACS for P. donghaiense. In brief, genomic DNAs were first extracted from all of the control microalgae by using the Ezup column-based plant genomic DNA extraction kit (Sangon Biotech, Shanghai, China). Next, all of the obtained genomic DNAs were used as templates to perform universal PCR with the primers D1/D2, respectively, aimed at excluding the possible interference of DNA quality on the subsequent experiment. Finally, using the same DNA templates that are verified to be universal PCR positive, specific PCR with the primers DH–FP/DH–RP were performed using the optimized PCR conditions in the “PCR conditions and optimization” section. The PCR products were analyzed by 1.5% AGE and NACS, respectively.
Sensitivity test of PCR-NACS
The sensitivity of PCR-NACS was tested with the gradient-diluted genomic DNA from P. donghaiense and the recombinant plasmid DNA containing the inserted LSU D1-D2 rDNA of P. donghaiense as test samples, respectively. The recombinant plasmids were extracted from the culture of the screened positive clones in the “Cloning and sequencing of target algae DNA” section using the SanPrep Spin Column and Collection Tube (Plasmid) Kit (Sangon Biotech, Shanghai, China). First, genomic DNA and recombinant plasmid DNA were diluted with nuclease-free water to obtain a series of solutions in a tenfold concentration gradient, respectively. Next, all of the prepared genomic DNA and recombinant plasmid DNA solutions were used as templates to perform PCR, respectively, with nuclease-free water as a blank control. Finally, the PCR products were analyzed by 1.5% AGE and NACS to comparatively evaluate the sensitivity of PCR-NACS.
Stability test of PCR-NACS
Chlorella vulgaris, as a relatively common algal species in the natural marine water, was selected as the interfering algal species to test the stability of the developed PCR-NACS. In brief, the genomic DNA of C. vulgaris was isolated using the same method as described in “Cloning and sequencing of target algae DNA” and its concentration was measured using a NAS-99 micro-nucleic acid spectrophotometer. Next, the genomic DNA from both P. donghaiense and C. vulgaris were adjusted to the same concentration of 20 ng·µL−1, and then mixed in a ratio of 100:1, 10:1, 1:1, 1:10, and 1: 100, respectively. Using the genomic DNA mixtures as test samples, comparative PCR-AGE and PCR-NACS analysis were finally performed to evaluate the stability of PCR-NACS.
Practicality test of PCR-NACS
The practicality of PCR-NACS was evaluated by the test with the simulated natural samples containing the target algal cells. For this purpose, natural samples were first collected from Jinhai Bay waters of Weihai City, Shandong Province. Microscopic examination was conducted to confirm that P. donghaiense cells were not contained in the natrual water samples. Next, the algal cultures of P. donghaiense in the exponential growth period was fixed with Luger’s solution (final concentration 1.5%) and then counted with a plankton counting chamber to determine the cell density. By mixing the algal culture with known cell density (1.27 × 106 cells·mL−1) with natural sample, the initial simulated natural samples with the cell concentration of 1.27 × 105 cells·mL−1 were prepared. The initial simulated natural samples were further diluted in a tenfold gradient with natural seawater to obtain a series of simulated natural samples with the cell density range of 1.27 × 105 cells·mL−1 − 1.27 × 100 cells·mL−1. Finally, crude DNA extracts were prepared from the above simulated natural samples using the One-Tube Plant DNA Extraction Kit (Sangon Biotech, Shanghai, China), and used as templates for PCR and PCR-NACS assays. The cell density of the sample that shows a positive result and contains the lowest cell density can be regarded as the lowest detection limit (LDL) for cell of PCR-NACS.
Results
Optimization of capture probe conentration
The optimization of capture probe conentration is shown in Fig. S1. The color of the colloidal gold solution remains unchanged with the probe concentration range of 0.6 − 0.3 μM, displaying a good dispersion of the colloidal gold particles. In contrast, when the probe concentration ranged from 0.2 to 0 μM, the color of the colloidal gold solution gradually darkened and the colloidal gold particle precipitate appeared. These results indicate that not all colloidal gold particles were bound with capture probe when the capture probe concentration was less than 0.3 μM. The excess colloidal gold particles aggregate to form precipitate in the PBS solution. In consideration of the possible loss during the preparation of colloidal gold probe and the chromatographic conditions of the corresponding NACS, the optimal concentration of capture probe was set to 0.4 μM.
Optimization of PCR
The primer concentration, annealing temperature, and Mg2+ concentration were comparatively tested to improve the PCR amplification efficiency. The optimization results of primer concentration are shown in Fig. S2a. Obviously, the yield of the PCR products was low when the primer concentration was 0.10 μM. There was no significant difference in the yield of PCR products when the primer concentration ranged from 0.15 to 0.30 μM. By contrast, the yield of PCR product decreased significantly when the primer concentration was 0.35 μM. Based on these results, in combination with the aim of saving money and reducing the yield of primer dimer, the primer concentration was set to 0.15 μM. The optimization results of annealing temperature are shown in Fig. S2b. When the annealing temperature was 52 °C, no target DNA band could be detectd by AGE, indicating that PCR amplification cannot effectively occur. In contrast, when the annealing temperature was increased from 54 to 62 °C, the amplified DNA band gradually became more and more brighter, indicating that the yield of PCR products increases gradually. In addition, when the annealing temperature was increased to 64 °C, the yield of PCR product decreased. Therefore, the optimal annealing temperature for PCR was set to 62 °C. The optimization results of Mg2+ concentration are shown in Fig. S2c. PCR amplification did not occur when Mg2+ concentration is 0.5 mM. In contrast, the yield of PCR product gradually increased with the Mg2+ concentration range of 1.0 − 1.5 mM. However, the yield of PCR products decreased with the Mg2+ concentration range of 2.0 − 3.0 mM, indicating that high concentration of Mg2+ could inhibit PCR amplification. Therefore, the optimal Mg2+ concentration was set to 1.5 mM. In conclusion, the optimal PCR amplification conditions that were used in the subsequent PCR amplification were as follows: primer concentration, 0.15 μM; annealing temperature, 62 °C; and Mg2+ concentration, 1.5 mM.
Optimization of detection probe concentration
Preliminary PCR was performed using the optimized amplification conditions, and the resultant amplification products were analyzed by 1.5% AGE and NACS. Trace primer dimer could be detected with the PCR product from blank control by AGE analysis (Fig. S3a). Correspondingly, a false positive result was detected with the PCR product from blank control by NACS assay (Fig. S3b). According to principle of NACS, it can be inferred that the primer dimer formed by the primers with tag sequences in the PCR can also bind with the colloidal gold probe cTgn and the detection probe cTsp, which therefore produce the “false positive” result. The generation of primer dimer may be resulted from excessive concentration of primers. However, given that the optimal primer concentration has been determined in the optimization of PCR conditions, we attempted to avoid false positive result by adjusting the concentration of detection probe on the detection point. For this purpose, a series of NACSs were prepared by setting the concentration of the detection probe to 10.0, 7.5, 5.0, and 2.5 μM. Then, these NACSs were applied to analyze the PCR product of blank control (Fig. S3c). False positive results could still be detected with the probe concentration range of 10.0 − 5.0 μM. In contrast, false positive result disappeared when the probe concentration was set to 2.5 μM. Furthermore, the detection of PCR product of blank control by using the NACS with the optimized probe concentration was performed in triplicate, all of which did not produce false positive result (Fig. S4). Based on the above results, the concentration of detection probe and control probe that was used for the preparation of NACS in the subsequent tests was 2.5 and 10 μM, respectively.
Specificity of PCR-NACS
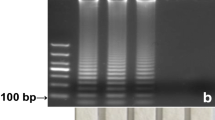
Using the genomic DNAs from the 23 algal species listed in Table 1 as template, PCR with the universal primers D1/D2 targeting the LSU rDNA were first performed. The results (Fig. 3a) showed that successful amplification could occur with all of the genomic DNAs, which therefore can be thought to be competent for the subsequent PCR-NACS. Next, specific PCR with the specific primers DH–FP/DH–RP that is designed for P. donghaiense using the mentioned genomic DNA as template was performed, with the amplified products analyzed by AGE and NACS. As shown in Fig. 3b and c, respectively, only the genomic DNA from P. donghaiense could produce amplification product with an expected size of 249 bp. Correspondingly, only the detection of the genomic DNA from P. donghaiense by PCR-NACS could generate a control point and a detection point, displaying a positive result. All of the above results indicated that the designed primers in this study have good amplification specificity and the established PCR-NACS assay is specific for P. donghaiense.

Specificity test of PCR and PCR-NACS for P. donghaiense. L1–23 are respectively the PCR and PCR–NACS test of algal species shown in Table 1 in a top-to-bottom order; L24: Blank control. a Validity test of DNA templates with the primers D1 and D2; M: DL1000 DNA marker. b Electrophoresis analysis of PCR products with specific primers DH–FP and DH–RP; M: DL500 DNA marker. c The results of PCR-NACS
Sensitivity of PCR-NACS
The tenfold serially diluted genomic DNA from P. donghaiense (2.7 × 101–2.7 × 10−4 ng·μL−1) were used as test samples for PCR-AGE and PCR-NACS analysis. Positive PCR amplification could be detected by AGE with the genomic DNA with the concentration range of 2.7 × 101 − 2.7 × 10−1 ng·μL−1. In contrast, PCR-amplified product could not be detected by AGE when the concentration of genomic DNA was below 2.7 × 10−1 ng·μL−1 (Fig. 4a). Therefore, the LDL of PCR-AGE for genomic DNA can be determined as 2.7 × 10−1 ng·μL−1. At the same time, it can be seen that the LDL of PCR-NACS for genomic DNA was determined as 2.7 × 10−2 ng μL−1 (Fig. 4b). Similarly, a series of plasmid DNA solutions that were serially tenfold diluted to form the final concentration range of 3.58 × 106 − 3.58 × 101 copies μL−1 were analyzed by PCR-AGE and PCR-NACS, respectively. As shown in Fig. 5, PCR-AGE and PCR-NACS displayed a LDL of 3.58 × 103 copies·μL−1 (Fig. 5a) and 3.58 × 102 copies·μL−1 (Fig. 5b) for plasmid DNA, respectively. In conclusion, the sensitivity test using both genomic DNA and plasmid DNA as template showed that the PCR-NACS assay established in this study was 10 times sensitive than PCR-AGE.

Sensitivity test of PCR (a) and PCR-NACS (b) with the genomic DNA from P. donghaiense. M: DL1000 DNA marker; L1–L6: the concentration of genomic DNA is 2.7 × 101 ng·μL−1, 2.7 × 100 ng·μL−1, 2.7 × 10−1 ng·μL−1, 2.7 × 10−2 ng·μL−1, 2.7 × 10−3 ng·μL−1, and 2.7 × 10−4 ng·μL−1, respectively; L7: blank control

Sensitivity test of PCR (a) and PCR-NACS (b) with the recombinant plasmid containing the inserted LSU D1-D2 of P. donghaiense. M: DL1000 DNA marker; L1–6: the concentration of plasmid is 3.58 × 106 copies·μL–1, 3.58 × 105 copies·μL–1, 3.58 × 104 copies·μL–1, 3.58 × 103 copies·μL–1, 3.58 × 102 copies·μL–1, 3.58 × 101 copies·μL–1, respectively; L7: blank control
Stability of PCR-NACS
To verify the stability of the established PCR-NACS, the genomic DNAs from the target and interfering algal species were mixed in different ratios and used as test samples for PCR-AGE and PCR-NACS analysis, respectively. All of these samples could produce target DNA band, without detectable non-specific PCR product (Fig. 6a), despite that PCR amplification efficiency varied with the mixture with different genomic DNA ratios. Accordingly, PCR-NACS analysis of each mixed sample showed positive results (Fig. 6b). The above results indicated that the performance of the established PCR-NACS was stable and was not affected by the interfering algal species.

Stability test of PCR (a) and PCR-NACS (b). M: DL1000 DNA marker; L1–5: the concentration ratio of the genomic DNA from target algae to that of interfering algaes (Chlorella vulgaris) is 100:1, 10:1, 1:1, 1:10, and 1:100, respectively; L6: blank control
PCR-NACS detection of simulated natural samples
The results of PCR-NACS test of the simulated natural samples are shown in Fig. 7. Sucessful PCR amplification could occur with the simulated samples with the cell density range of 1.27 × 105 − 1.27 × 102 cells·mL−1. Thus, the cellular LDL for PCR was 1.27 × 102 cells·mL−1 (Fig. 7a). In comparison, PCR-NACS displayed a cellular LDL of 1.27 × 101 cells·mL−1 (Fig. 7b). In conclusion, PCR-NACS again displayed a 10 times hiher sensitivity than PCR-AGE.

Comparative analysis of simulated samples by PCR (a) and PCR-NACS (b). M: DL1000 DNA marker; L1–6: the simulated samples containing P. donghaiense with a cell density of 1.27 × 105, 1.27 × 104 cells·mL−1, 1.27 × 103 cells·mL−1, 1.27 × 102 cells·mL−1, 1.27 × 101 cells·mL−1, and 1.27 × 100 cells·mL−1, respectively; L7: blank control
Discussion
The excessive accumulation of nitrate in the Yangtze River Estuary has been considered as one of the main cause for P. donghaiense-forming algal blooms in the East China Sea (Zhou et al. 2008). Compared with diatoms, P. donghaiense as a dinoflagellate has a much stronger ability to utilize dissolved organic phosphorus under the phosphorus-limited conditions. P. donghaiense thus has an advantage over diatoms to compete for survival, and thus becomes one of the dominant HABs-forming species in the East China Sea (Ou et al. 2008; Yu et al. 2018). The HABs caused by P. donghaiense have seriously affected the cultivation of economic marine animals, community structure of marine organisms, and bio-diversity of other phytoplanktons (Lin et al. 2014; Wang et al. 2020). It is therefore necessary to establish more rapid and convenient detection methods for early warning of P. donghaiense, and then effective and preventive measures can be taken before the outbreak of HABs.
At present, rDNA genes have been widely used to investigate the molecular phylogeny of microalgae. The rDNA genes mainly include the internal transcribed spacer (ITS), large subunit ribosomal DNA (LSU rDNA), and small subunit ribosomal DNA (SSU rDNA). Among them, LSU rDNA is more highly variable than SSU rDNA and more conservative than ITS. Nevertheless, LSU rDNA has been considered as a good molecular marker for classification and identification from species to genera since it is composed of alternating conserved regions and hypervariable regions. For example, Shin et al. (2019a) demonstrated that P. shikokuense and P donghaiense belong to the same species by the molecular identification method using LSU rDNA as genetic marker. In this study, the PCR-NACS assay for P. donghaiense was established by using LSU rDNA as the target, and its specificity was verified by cross-reactivity test. The results again indicated that the LSU rDNA sequence can be used as a good target for the establishment of molecular detection method for P. donghaiense.
The PCR-NACS assay established in this study has a relatively high sensitivity. According to the detection principle for PCR-NACS, a false positive product may be produced by NACS if even trace primer dimers were included in the PCR products. Thererefore, the PCR conditions including the primer concentration, annealing temperature, Mg2+ concentration, and the concentration of the detection probe on the test strip were optimized to eliminate the influence of primer dimer on the determination of detection results. Fortunately, the false positive effect of primer dimer on the NACS was successfully eliminated while ensuring the efficiency and sensitivity of the assay, which is crucial for the practical application of the established detectin method.
Sensitivity is one of the most important parameters to evaluate the practicability of a detection method. Aimed at providing an early warning for HABs, the established detection method is required to be able to successfully detect harmful algae even at low cell density. The LDLs of the established PCR-NACS assay were 2.7 × 10−2 ng·μL−1 for target algal genomic DNA and 3.58 × 102 copies·μL–1 for plasmid DNA, both of which were 10 times more sensitive than ordinary PCR. In addition, the cell sensitivity of the PCR-NACS assay was also evaluated. PCR-NACS displayed a LDL of 1.27 × 101 cells·mL−1, which is comparable to that of sandwich hybridization integrated with nuclease protection assay (NPA-SH) (11.0 cells·mL−1) (Zhen et al. 2009) and mPCR (60.0 cells·mL−1) (Sun et al. 2019), 10 times lower than that of qPCR (100.0 cells·mL−1) (Yuan et al. 2012), and higher than that of loop-mediated isothermal amplification-lateral flow dipstick (LAMP-LFD) (1.0 cells·mL−1) (Qin et al. 2019) and reverse transcription-LAMP (RT-LAMP) (0.6 cells·mL−1) (Chen et al. 2013). Nevertheless, given that cell density of P. donghaiense-forming HABs is generally higher than 103 cells·mL−1 (Lin et al. 2015), the established PCR-NACS assay can meet the early warning requirement of P. donghaiense.
The ocean is a complex ecosystem consisting of numerous organisms, among which the phytoplankton, zooplankton, and microorganisms may interfere with the specific detection of target species. Therefore, whether it is anti-interference or not is also crucial for the established detection method. For this reason, C. vulgaris, a relatively common microalgae in the ocean, was selected as a non-target species for interference test, i.e., comparative PCR-NACS was performed on the genomic DNA mixtures consisting of the interfering algae and the target algae in different ratios that were used to simulate the enviromental samples containing the non-target algae and target algae in different proportions. The results of the interference tests showed that PCR-NACS were unaffected even when the DNA concentration of target algae is very low and the DNA concentration of interfering algae is very high, which is equivalent to that the cell density of non-target algae cells in water sample is high, while the cell density of target algae is low. In addition, the test with the simulated sample also showed that PCR-NACS was not affected by abiotic substances in seawater. In short, the PCR-NACS developed in this study was proved to have good stability and strong anti-interference ability.
To date, the established molecular methods for the detection of P. donghaiense mainly include qPCR (Zhao et al. 2009; Yuan et al. 2012), NPA-SH (Zhen et al. 2009), FISH (Chen et al. 2011), RT-LAMP (Chen et al. 2013), LAMP-LFD (Zou et al. 2018), and mPCR (Sun et al. 2019). One disadvantage for mPCR is that non-specific amplification producing primer dimer may occur since multiple primer pairs were included in the amplification reaction. Another diadvantage is that mPCR may be less sensitive, which thus limits the application of this method. qPCR not only requires specialized equipment, but also is easily affected by aerosols during the detection process. FISH, which is an actual improved morphology-dependent method relying on a fluorescencent microscope, is time-consuming and cumbersome. In particular, the required sample processing may lead to cell deformation or aggregation, which may affect the hybridization of probe. For NPA-SH, the labeled probes used in the hybridization were rather expensive, the whole detection process is complicated and time-consuming. In addition, NPA-SH is not suit for rapid detection in the field because it requires an ELISA Reader. Although LAMP has the advantage of high sensitivity, the design of LAMP primer targeting six regions of DNA template is complex and demanding. In addition, the cost for LAMP is high because the Bst DNA polymerase that is used in the LAMP is rather expensive.
The PCR-NACS established in this study sucessfully realize the chromatographic analysis of PCR products without any additional treatment by ingeniou primer design, which provides a new idea for simple and rapid detection of P. donghaiense. The developed PCR-NACS assay has several advantages. First, few reagents are required for the preparation of test strip and NACS analysis. For example, in theory, 1 mL of chloroauric acid solution is enough for the preparation of 2000 test strips, and only 7.5 μL of PCR products is required for NACS analysis. Second, PCR-NACS results can be judged with the naked eye, independent of special instruments, such as gel imagers, fluorescence microscopes, ELISA Reader, etc., and the test results can be stored for a long time. Finally, the test strips can be prepared purely by hand, without the need for programmable cutters, film spraying machines, and other special instruments that have been used in the preparation of traditional test strips. The detection procedure is simple, and the test cost is low since no expensive reagent and special instrument is required for the preparation of test strips. In addition, it should also be noted that because PCR instrument is required for the nucleic acid amplification of the developed PCR-NACS assay, it actually cannot be used for on-site detection. However, IATs (e.g., RPA) can be used to replace PCR to improve nucleic acid preparation to truly realize the on-site detection application of nucleic acid chromatography test strips in the future.
Data availability
All data generated or analyzed during this study are included in the MS.
References
Al-Tebrineh J, Pearson LA, Yasar SA, Neilan BA (2012) A multiplex qPCR targeting hepato- and neurotoxigenic cyanobacteria of global significance. Harmful Algae 15:19–25
Anderson DM, Cembella AD, Hallegraeff GM (2012) Progress in understanding harmful algal blooms: paradigm shifts and new technologies for research, monitoring, and management. Annu Rev Mar Sci 4:143–176
Anderson DM, Fensin E, Gobler CJ, Hoeglund AE, Hubbard KA, Kulis DM, Landsberg JH, Lefebvre KA, Provoost P, Richlen ML, Smith JL, Solow AR, Trainer VL (2021) Marine harmful algal blooms (HABs) in the United States: history, current status and future trends. Harmful Algae 102:101975
Bechard A (2020) The economic impacts of harmful algal blooms on tourism: an examination of Southwest Florida using a spline regression approach. Nat Hazards 104:1–17
Cai Q, Li R, Zhen Y, Mi T, Yu Z (2006) Detection of two Prorocentrum species using sandwich hybridization integrated with nuclease protection assay. Harmful Algae 5:300–309
Chen HL, Lü SH, Zhang CS, Zhu DD (2006) A survey on the red tide of Prorocentrum donghaiense in East China Sea, 2004. Ecol Sci 25:226–230
Chen GF, Zhang CY, Zhang BY, Wang GC, Lu DD, Xu Z, Yan PS (2011) Development of a PNA probe for fluorescence in situ hybridization detection of Prorocentrum donghaiense. PLoS ONE 6:e25527
Chen GF, Ma CS, Zhang CY, Zhou J, Wang YY, Wang GC, Zhang BY, Xu Z, Lu DD (2013) A rapid and sensitive method for field detection of Prorocentrum donghaiense using reverse transcription-coupled loop-mediated isothermal amplification. Harmful Algae 29:31–39
Chen QX, Zhang CY, Liu FG, Ma HY, Wang YY, Chen GF (2020) Easy detection of Karlodinium veneficum using PCR-based dot chromatography strip. Harmful Algae 99:101908
Chikamatsu K, Aono A, Kawai A, Hata H, Iwamoto T, Igarashi Y, Takaki A, Yamada H, Mitarai S (2019) Evaluation of Q gene mycobacteria: a novel and easy nucleic acid chromatography method for mycobacterial species identification. J Microbiol Meth 163:105657
Doll C, Main CR, Bianco C, Coyne KJ, Greenfield DI (2014) Comparison of sandwich hybridization assay and quantitative PCR for the quantification of live and preserved cultures of Heterosigma akashiwo (Raphidophyceae). Limnol Oceanogr Meth 12:232–245
Eckford-Soper LK, Daugbjerg N (2015) Development of a multiplex real-time qPCR assay for simultaneous enumeration of up to four marine toxic bloom-forming microalgal species. Harmful Algae 48:37–43
Grattan LM, Holobaugh S, Morris JG (2016) Harmful algal blooms and public health. Harmful Algae 57:2–8
Guillard RRL (1975) Culture of phytoplankton for feeding marine invertebrates. In: Smith WL, Chanley MH (eds) Culture of marine invertebrate animals. Springer, Boston, pp 29–60
Hatfield RG, Bean T, Turner AD, Lees DN, Lowther J, Lewis A, Baker-Austin C (2019) Development of a TaqMan qPCR assay for detection of Alexandrium spp. and application to harmful algal bloom monitoring. Toxicon:X 2:100011–100011
Kantiani L, Llorca M, Sanchís J, Farré M, Barceló D (2010) Emerging food contaminants: a review. Anal Bioanal Chem Re 398:2413–2427
Landsberg JH (2002) The effects of harmful algal blooms on aquatic organisms. Rev Fish Sci 10:113–390
Lin JN, Yan T, Zhang QC, Wang YF, Liu Q, Zhou MJ (2014) In situ detrimental impacts of Prorocentrum donghaiense blooms on zooplankton in the east china sea. Mar Pollut Bull 88:302–310
Lin JN, Song JJ, Yan T, Zhang QC, Zhou MJ (2015) Large-scale dinoflagellate bloom species Prorocentrum donghaiense and Karenia mikimotoi reduce the survival and reproduction of copepod calanus sinicus. J Mar Biol Assoc Uk 95:1071–1079
Lin GH, Xu XF, Wang P, Liang SK, Li YB, Su Y, Li KQ, Wang XL (2020) Methodology for forecast and control of coastal harmful algal blooms by embedding a compound eutrophication index into the ecological risk index. Sci Total Environ 735:139404
Liu FG, Zhang, CY, Zheng HW, Yang YD, Zang, DY, Wang Y, Chen GF (2021) Rapid and sensitive detection of Karlodinium veneficum by a novel double-nick rolling circle amplification. Environ Sci Pollut R 28:42570–42582
Liu JW, Lu Y (2006) Preparation of aptamer-linked gold nanoparticle purple aggregates for colorimetric sensing of analytes. Nat Protoc 1:246–252
Lu DD, Jeanette G (2001) Five red tide species in genus prorocentrum including the description of prorocentrum donghaiense lu sp. nov. from the east china sea. Chin J Oceanol Limn 19:337–344
Lu DD, Qi YZ, Jeanette G, Zhou JZ, Gao YH (2003) Revise for Prorocentrum donghaiense and taxology comparison with Prorocentraceae. Chin J Appl Ecol 14:1060–1064
Lu DD, Goebel J, Qi YZ, Zou JZ, Han XT, Gao YH, Li YG (2005) Morphological and genetic study of Prorocentrum donghaiense Lu from the East China Sea, and comparison with some related Prorocentrum species. Harmful Algae 4:493–505
Meek ME, Dolah FV (2016) Phytoplankton IF-FISH: Species-specific labeling of cellular proteins by immunofluorescence (IF) with simultaneous species identification by fluorescence immunohybridization (FISH). J Microbiol Meth 124:21–27
Nagai S (2011) Development of a multiplex PCR assay for simultaneous detection of six Alexandrium species (dinophyceae) 1. J Phycol 47:703–708
Nagai S, Miyamoto S, Ino K, Tajimi S, Nishi H, Tomono J (2016) Easy detection of multiple Alexandrium species using DNA chromatography chip. Harmful Algae 51:97–106
Ou LJ, Wang D, Huang BQ, Hong HS, Qi YZ, Lu SH (2008) Comparative study of phosphorus strategies of three typical harmful algae in Chinese coastal waters. J Plankton Res 30:1007–1017
Patricia MG, Donald MA, Patrick G, Edna G, Kevin GS (2005) The global, complex phenomena of harmful algal blooms. Oceanography 18:136–147
Qin Y, Chen GF, Zhang CY, Wang YY, Zhou J (2019) Development of loop-mediated isothermal amplification combined with a chromatographic lateral-flow dipstick for rapid detection of Chattonella marina. Harmful Algae 89:101666
Sano S, Miyamoto S, Kawamoto S (2018) Rapid multiplex nucleic acid amplification test developed using paper chromatography chip and azobenzene-modified oligonucleotides. J Biosci Bioeng 126:397–403
Scholin CA, Herzog M, Sogin M, Anderson DM (1994) Identification of group- and strain-specific genetic markers for globally distributed Alexandrium (dinophyceae). ii. sequence analysis of a fragment of the LUS rDNA gene1. J Phycol 30:999–1011
Sha J, Xiong HY, Li CJ, Lu ZY, Zhang JC, Zhong H, Zhang W, Yan B (2021) Harmful algal blooms and their eco-environmental indication. Chemosphere 274:129912
Shin HH, Li Z, Mertens KN, Min HS, Matsuoka K (2019) Prorocentrum shikokuense Hada and P. donghaiense Lu are junior synonyms of P. obtusidens Schiller, but not of P. dentatum Stein (Prorocentrales, Dinophyceae). Harmful Algae 89:101686
Shin HH, Li Z, Seo MH, Soh HY, Lim WA, Park JW (2019b) Harmful dinoflagellate Prorocentrum donghaiense lu is widely distributed along the East China Sea and Korean coastal area. Ocean Sci J 54:685–691
Sun YJ, Chen GF, Zhang CY, Guo CL, Wang YY, Sun R (2019) Development of a multiplex polymerase chain reaction assay for the parallel detection of harmful algal bloom-forming species distributed along the Chinese coast. Harmful Algae 84:36–45
Sun FL, Wang CZ, Wang YS, Tu K, Zheng ZP, Lin XF (2020) Diatom red tide significantly drive the changes of microbiome in mariculture ecosystem. Aquaculture 520:734742
Tang DL, Di BP, Wei GF, Ni IH, Oh IS, Wang SF (2006) Spatial, seasonal and species variations of harmful algal blooms in the South Yellow Sea and East China Sea. Hydrobiologia 568:245–253
Trottet A, George C, Drillet G, Lauro FM (2021) Aquaculture in coastal urbanized areas: a comparative review of the challenges posed by harmful algal blooms. Crit Rev Env Sci Tecnol 52:2888–2929
Turner AD, Lewis AM, Bradley K, Maskrey BH (2021) Marine invertebrate interactions with harmful algal blooms–implications for one health. J Invertebr Pathol 186:107555
Wang H, Hu ZX, Chai ZY, Deng YY, Zhan ZF, Tang YZ (2020) Blooms of prorocentrum donghaiense reduced the species diversity of dinoflagellate community. Acta Oceanol Sin 39:110–119
Yamamuro T, Miyamoto S, Kitamura M, Muro T, Iwata YT, Segawa H, Kuwayama K, Tsujikawa K, Kanamori T, Inoue H (2018) Development of simple and accurate detection systems for Cannabis sativa using DNA chromatography. Forensic Sci Int 291:68–75
Yoshida M, Sano S, Chien J, Fukano H, Suzuki M, Asakura T, Morimoto K, Murase Y, Miyamoto S, Kurashima A, Hasegawa N, Hsueh P, Mitarai S, Ato M, Hoshino Y (2021) A novel DNA chromatography method to discriminate Mycobacterium abscessus subspecies and macrolide susceptibility. EBioMedicine 64:103187
Yu RC, Lü SH, Liang YB (2018) Harmful algal blooms in the coastal waters of china. In: Glibert PM et al (eds) Global ecology and oceanography of harmful algal blooms, 1st edn. Springer International Publishing, Cham, pp 309–316
Yuan J, Mi TZ, Zhen Y, Yu ZG (2012) Development of a real-time PCR method (Taqman) for rapid identification and quantification of Prorocentrum donghaiense. J Ocean U China 11:366–374
Zhang CY, Chen GF, Zhou J, Wang YY, Lu DD (2016) Development of a quantitative PCR for detection and quantification of Prorocentrum donghaiense. J Appl Phycol 28:1683–1693
Zhao LY, Mi TZ, Zhen Y, Li MY, He SY, Sun J, Yu ZG (2009) Cloning of proliferating cell nuclear antigen gene from the dinoflagellate Prorocentrum donghaiense and monitoring its expression profiles by real-time RT-PCR. Hydrobiologia 627:19–30
Zhen Y, Mi TZ, Yu ZG (2009) Detection of several harmful algal species by sandwich hybridization integrated with a nuclease protection assay. Harmful Algae 8:651–657
Zhou MJ, Zhu MY, Zhang J (2001) Status of harmful algal blooms and related research activities in China. Chin Bull Life Sci 13:54–59
Zhou MJ, Yan T, Zou JZ (2003) Preliminary analysis of characteristics of red tide areas in Changjiang River estuary and its adjacent sea. Chin J Appl Ecolo 14:1031–1038
Zhou MJ, Shen ZL, Yu RC (2008) Responses of a coastal phytoplankton community to increased nutrient input from the Changjiang (Yangtze) River. Cont Shelf Res 28:1483–1489
Zou YX, Huang HL, Qiao LL, Dang CY, Pang JH, Lü PW, Wang JQ, Zhu P (2018) Rapid detection of prorocentrum donghaiense based on loop-mediated isothermal amplification combined with lateral flow dipstick. Oceanol Limnol Sin 49:586–593
Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation, China (ZR2020MD081); the National Scientific Foundation of China (No. 31600309, 41476086); HIT Scientific Research Innovation Fund/the Fundamental Research Funds for the Central Universities (No. HIT.NSRIF.201702 and HIT.NSRIF.201709); and HIT Environment and Ecology Innovation Special Funds (No. HSCJ201622).
Funding
This work was supported by Shandong Provincial Natural Science Foundation, China (ZR2020MD081); the National Scientific Foundation of China (No. 31600309, 41476086); HIT Scientific Research Innovation Fund/the Fundamental Research Funds for the Central Universities (No. HIT.NSRIF.201702 and HIT.NSRIF.201709); and HIT Environment and Ecology Innovation Special Funds (No. HSCJ201622).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: Chunyun Zhang and Guofu Chen; Performed the experiments: Jingju Ma; Analyzed the data: Yuanyuan Wang. Contributed reagents/ materials/analysis tools: Fuguo Liu. Wrote the paper: Jinju Ma and Yin Liu.
Corresponding author
Ethics declarations
Ethics approval
All the authors would like to seriously state that (a) the material used in this MS has not been published in whole or in part elsewhere; (b) the paper is not currently being considered for 372 publication elsewhere; (c) all authors have been personally and actively involved in substantive work leading to the report and will hold themselves jointly and individually responsible for its content; and (d) all relevant ethical safeguards have been met in this study.
Consent to participate
There is no participant in the MS.
Consent for publication
All the authors consent to publish all the dada included in the MS.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Vitor Vasconcelos
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, J., Zhang, C., Liu, F. et al. Easy detection of Prorocentrum donghaiense by polymerase chain reaction-nucleic acid chromatography strip. Environ Sci Pollut Res 30, 10346–10359 (2023). https://doi.org/10.1007/s11356-022-22856-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-022-22856-6