
Abstract
In this study, electrochemically activated peroxymonosulfate (EC/PMS) with a sacrificial iron electrode was used for the removal of chloramphenicol (CAP) from water. Compared to electrolysis alone, peroxymonosulfate (PMS) alone, and Fe2+/PMS, EC/PMS significantly enhanced the CAP degradation. Various parameters, such as the applied current, electrolyte concentration, and PMS dose, were investigated to optimize the process. In addition, acidic conditions facilitated the CAP degradation. The presence of Cl− slightly enhanced the CAP degradation, while both HCO3− and NO3− exhibited an inhibitory effect on the CAP degradation. The floccules were also analyzed after the reaction by XPS and XRD. Quenching experiments indicated that both sulfate radicals (SO4●−) and hydroxyl radicals (•OH) were responsible for the CAP degradation. In addition, the degradation products were identified by LC/TOF/MS, and the degradation pathways were proposed accordingly. These results indicated that EC/PMS is a promising treatment process for the remediation of water polluted by CAP.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Various kinds of antibiotics have been produced and applied worldwide for the treatment of human and animal diseases caused by microbial infectious diseases. As a result, a large amount of antibiotics has been discharged into the environment via effluent from hospitals and wastewater treatment plants (WWTPs), pharmaceutical industry wastewater release, improper discharge of treated landfill leachate, etc. (Kümmerer 2009). Residual antibiotics in the water environment pose potential risks to aquatic organisms as well as to human health, and they can also cause the formation of antibiotic-resistant bacteria (ARB) and antibiotic resistance genes (ARG) (Danner et al. 2019; Kovalakova et al. 2020). Due to excessive consumption and incorrect treatment methods, antibiotic pollution in the water environment has raised widespread concern and become a common environmental problem (Yan et al. 2015). As one of the commonly used antibiotics, chloramphenicol (CAP) is a broad-spectrum antibiotic for both humans and animals owing to its high activity against many gram-positive and gram-negative bacteria (Wu et al. 2017). It has been reported that a considerable proportion of CAP is directly excreted in the aquatic environment via urine or feces after its intake by humans or animals (Zhou et al. 2017). Moreover, many antibiotics, including CAP, are stable in water and are resistant to traditional wastewater treatment owing to their physical and chemical properties (Cao et al. 2020; Zhou et al. 2009). Accordingly, nano- to micrograms CAP has been frequently detected in aquatic environments. In Germany, CAP concentration was up to 0.56 μg/L and 60 ng/L, as detected in the effluent of one WWTP and in surface water, respectively (Hirsch et al. 1999). As detected in the Pearl River of China, the concentration of CAP was as high as 266 ng/L and 187 ng/L in the high and low water seasons, respectively (Xu et al. 2007). Therefore, effective techniques are urgently needed for efficient CAP removal.
Many techniques have been extensively adopted for eliminating CAP from water, such as adsorption (Fan et al. 2010), ferrate (Zhang et al. 2021), and solar photoelectro-Fenton (Garcia-Segura et al. 2014). Among these techniques, advanced oxidation processes (AOPs) are promising methods for destruction of many organic contaminants in water, which are based on the generation of highly oxidizing hydroxyl radicals (•OH). Recently, sulfate radical (SO4●−)–based advanced oxidation processes (SR-AOPs) have attracted much interest recently due to their excellent performance in degrading various refractory organic contaminants from water (Oh et al. 2016). In SR-AOPs, organic contaminants can be decomposed to CO2 and H2O as a result of SO4●−, which has a strong oxidation ability (E0 = 2.5~3.1 V), high pH adaptability (2~10), and a long lifetime (30~40 μs) (Qi et al. 2017). Generally, SO4●− can be generated mainly via heat, UV irradiation, and transition metal activation of persulfate (PS) and peroxymonosulfate (PMS). PMS is considered to be a green oxidant that is easily activated for a variety of industrial and consumer applications involving disinfection (Tan et al. 2018). The oxidation ability of PMS at room temperature is low (E0 = 1.82 V) (Wang and Chu 2012), but it can still be activated by various methods to produce SO4●−. Over the past few years, ferrous-activated PMS (Fe2+/PMS) has been adopted as an effective tool for SO4●− production for the removal of organic contaminants. Owing to its low cost, nontoxicity, and high activity, Fe2+ is always selected as an ideal activator of PMS or PS (Huang et al. 2021; Zou et al. 2013). Furthermore, owing to its asymmetrical structure (HO-O-SO3−) and lower bond dissociation energy, PMS is considered to be more readily activated by Fe2+ than PS (Oh et al. 2016).
However, like the Fenton reaction, the Fe2+/PMS process contains the disadvantage of a narrow acidic pH range because Fe2+ tends to transform into iron hydroxides at neutral or alkaline pH; this conversion may hinder the transformation of Fe3+ to Fe2+ (Song et al. 2020). Furthermore, the weak reversibility of the transformation of Fe3+ to Fe2+ reduces the activation efficiency of PMS and the utilization of Fe2+; therefore, a higher dose of ferrous salt is needed to continuously activate PMS, which may also quench SO4●− and produce a large amount of iron sludge (Ali et al. 2020). An effective approach to address such drawbacks is introducing an electric field into the Fe2+/PMS system (Brillas et al. 2009). Electrochemically activated PMS (EC/PMS) that uses Fe as the sacrificial anode is an ideal alternative for PMS activation because it can overcome the drawback of the slow Fe3+/Fe2+ cycle that occurs in the Fe2+/PMS process (Govindan et al. 2014). The EC/PMS process is a combination of electrochemical and Fe2+/PMS processes. In the EC/PMS system, soluble Fe2+ is continuously released to the solution from the surface of the iron electrode through anodic oxidation. Meanwhile, the reduction of Fe3+ occurs at the cathode (Wang and Chu 2011). Compared with the Fe2+/PMS process, EC/PMS can not only control the release of Fe2+ by adjusting the applied current but also accelerate the Fe3+/Fe2+ cycle, thus overcoming the low activation efficiency of PMS and the iron sludge problem caused by addition of excessive Fe2+ in the Fe2+/PMS process. In addition, electrolysis can also directly activate PMS to generate SO4●− (Wang and Chu 2011).
Herein, an electrochemically activated PMS (EC/PMS) process that uses iron as the sacrificial anode was proposed for the abatement of CAP. Several influencing factors, including the applied current, electrolyte concentration, PMS dose, initial solution pH, and common anions, were investigated to determine their effect on the CAP degradation performance. Moreover, the floccules formed during the reaction were characterized by X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). The radical species responsible for CAP degradation were investigated by quenching experiments. In addition, the major degradation products of CAP were identified by LC/TOF/MS, and the degradation pathways of CAP during the EC/PMS process were proposed.
Materials and methods
Materials
Chloramphenicol (CAP, ≥98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Peroxymonosulfate (KHSO5·1/2KHSO4·1/2K2SO4, ≥47%) was purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Anhydrous sodium sulfate (Na2SO4, ≥99%) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Chromatographic reagent grade acetonitrile used as the mobile phase was purchased from J. T. Baker (USA). All other chemicals used in the study were of at least analytical grade. Milli-Q water (18.2•MΩ cm) was used to prepare all the experimental solutions.
Experimental procedure
The experiments were carried out in a 250-mL undivided single-compartment glass cell by using a plate iron anode (2 cm × 4 cm) and a plate cathode (stainless steel) of the same dimensions (Fig. 1). Constant current electrolysis conditions were maintained by a DC-regulated power supply provided by Dongguan Tongmen Electronic Technology Co., Ltd. (China). The two electrode plates were fixed parallel to each other at a distance of 2.5 cm. The reaction temperature was kept at 20 ± 1 °C by immersing the reaction cell in a water bath. In a typical run, a mixed solution of CAP and electrolyte (Na2SO4) was added to the reactor and then stirred by a magnetic stirrer. After adding a certain dose of PMS into the reaction solution and turning on the DC power, the reaction was initiated. The initial solution pH was adjusted by H2SO4 or NaOH. At predetermined time intervals, the samples were withdrawn from the electrolytic cell and filtered through 0.45-μm membranes and then quenched with prefilled methanol immediately before analysis.

Scheme of the experimental equipment
Analysis
The concentration of CAP was determined by Shimadzu HPLC consisting of an LC-20AB pump and an SPD-20A UV detector set at 275 nm. A mixture of acetonitrile/water (60:40, v/v) was used as the mobile phase, and the flow rate was set at 0.8 mL/min. The concentration of PMS was determined by UV spectrophotometry according to the method described by Wacławek et al. (2015). The floccules were characterized by XRD (Rigaku Ultima IV) and XPS (Thermo Scientific K-Alpha) after the reaction. The CAP degradation products were analyzed using ultra-performance liquid chromatography and quadrupole time-of-flight mass spectrometry (Agilent 1290 UPLC/6550 Q-TOF) with a Waters BEH C18 column (2.1 × 100 mm, 1.7 μm). An elution gradient with two mobile phases, MQ water (A) and acetonitrile (B), was programmed as follows (min, %A): (0, 90), (1, 90), (8, 10), (12, 10), (12.1, 90), and (13, 90). The flow rate was 0.3 mL/min. The mass spectrometer was operated in negative ionization mode using an electrospray ionization (ESI) source. The spray voltage, sheath gas temperature, and sheath gas flow were set at −3200 V, 350°C, and 12 L/min, respectively. Full scan mode was set to m/z 50–500.
Results and discussion
Comparative study of different systems
We first investigated CAP degradation in five different systems: (1) EC alone, (2) PMS alone, (3) EC/PMS process, (4) EC/PMS process using stainless steel as the anode and iron as the cathode, and (5) Fe2+/PMS. As shown in Fig. 2, no obvious removal was observed when CAP was treated by PMS oxidation alone because PMS is stable at ambient temperature and its redox potential is very limited (E0 = 1.82 V) (Wang and Chu 2012). EC alone achieved 29.7% CAP removal, implying that the coagulation mechanism plays a role in CAP removal. As expected, approximately 91.0% CAP removal was achieved within 20 min in the EC/PMS system using iron as the anode and stainless steel as the cathode. During the EC/PMS process, Fe2+ was first electrochemically produced from the sacrificial iron anode, which could serve as the activator of PMS to generate sulfate radicals (SO4●−) (Eqs. (1) and (2)), which accelerate CAP degradation. Fe2+ was regenerated on the cathode plate according to Eq. (3), maintaining a relatively proper level of Fe2+ in the system. In addition, after swapping the two electrode plates (i.e., using stainless steel as the anode and iron as the cathode), we also noticed that 14.9% removal of CAP was achieved within 20 min, which indicated that electrolysis can also directly activate PMS to form SO4●− and/or •OH (Eqs. (4) and (5)) (Wang and Chu 2011). We should also notice that direct oxidation of CAP on anode is also expected to occur.

CAP degradation under different systems. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, [Fe2+]0 = 621 μΜ, current = 25 mA, [Na2SO4]0 = 25 mM, pH0 = 7
A comparison between the EC/PMS system and the ferrous ion–activated PMS (Fe2+/PMS) system was also conducted. The concentration of added Fe2+ in the EC/PMS system was calculated through Faraday’s law (Eq. (6)) with a reaction time of 20 min.
where C is the molecular concentration of Fe2+ generated in the system (mol/L), Z is the number of electrons (2 equivalents/mol), F is Faraday’s constant (96,485.3 Coulombs/mol), I is the given current (A), t is the reaction time (s), and V is the volume of electrolyte (L).
As seen, the degradation behavior of CAP in the Fe2+/PMS system exhibited a fast and then a slow stage along with the reaction time. The fast stage was attributed to the relatively higher initial Fe2+ concentration, and the slower stage was caused by the depletion of Fe2+ and inadequate regeneration of Fe3+ to Fe2+, which inhibited the generation of SO4●− (Tan et al. 2018). We also noticed that the final removal efficiency of CAP by the EC/PMS system was higher than that of the Fe2+/PMS system. This phenomenon could be ascribed to the different levels of Fe2+ use in the two systems. In the Fe2+/PMS system, excess Fe2+ competed with CAP for the generated SO4●− as well as rapidly consumed PMS in the initial stage and thus hindered the CAP degradation in the following stage (Eq. (7)) (Rastogi et al. 2009). However, in the EC/PMS system, Fe2+ was gradually released from the iron electrode, and Fe2+ could also be regenerated via cathodic reduction of Fe3+, which maintained CAP degradation over time.
Effect of applied current
The effect of the applied current on CAP degradation by the EC/PMS system is shown in Fig. 3. As the applied current increased from 10 to 25 mA, the removal rate of CAP increased from 74.1 to 91.0% within 20 min. As the applied current further increased to 35 mA, the removal rate of CAP slightly increased from 91.0 to 93.5%. Increasing the applied current density led to more rapid Fe2+ production, which improved the decomposition of PMS to generate more SO4●−, thus enhancing CAP degradation. In addition, a higher applied current also led to a faster generation of SO4●− via an electron transfer reaction. Additionally, more Fe2+ was also generated at a higher applied current; thus, the excess Fe2+ may have acted as a SO4●− scavenger and compete with CAP for SO4●− (Eq. (7)) (Rastogi et al. 2009).

Effect of applied current on CAP degradation. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, [Na2SO4]0 = 25 mM, pH0 = 7
Effect of electrolyte concentration
The effect of electrolyte concentration (Na2SO4) on CAP degradation by the EC/PMS system is shown in Fig. 4. As the Na2SO4 concentration increased from 5 to 25 mM, the removal rate of CAP also increased from 81.5 to 91.0%. The electrical conductivity of the solution was directly influenced by the electrolyte concentration, and increasing the Na2SO4 concentration increased the conductivity of the reaction solution, resulting in a lower energy loss during the drop in ohmic resistance of the solution (Periyasamy and Muthuchamy 2018).

Effect of electrolyte concentration on CAP degradation. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, current = 25 mA, pH0 = 7
Effect of PMS dose
The effect of PMS dose on CAP degradation is shown in Fig. 5a. The removal rate increased from 56.3 to 91.0% as the PMS dose increased from 0.1 to 0.5 mM, and then the removal rate of CAP decreased to 77.1% as the PMS concentration further increased to 1 mM. The increment of CAP removal rate when PMS concentration increased from 0.1 to 0.5 mM is ascribed to the fact that PMS served as the source of SO4●− in the EC/PMS system; thus, more reactive species were produced to degrade CAP at higher PMS concentrations. However, if the PMS concentration was too high, side reactions occurred (Eqs. (8) and (9)), leading to the self-quenching of SO4●− and/or •OH by excessive PMS (Shukla et al. 2010).

a Effect of PMS dose on CAP degradation; b change in [PMS]t/[PMS]0 during the reaction. Experimental conditions: [CAP]0 = 10 μM, current = 25 mA, [Na2SO4]0 = 25 mM, pH0 = 7
It is of interest to observe that the initial degradation rate of CAP (computed as Δc/Δt over the first 2 min, μΜ/min) was decreased from 1.64 to 0.33 μΜ/min with the increment of PMS from 0.1 to 1 mM. This observation can also be explained by the unfavorable consumption of SO4●− and •OH according to Eqs. (8) and (9), leading to the production of the less reactive SO5●− and thus slowing the CAP degradation. However, the overall removal of CAP at the end of the process was still observed at a relatively higher PMS concentration because the PMS, which served as the SO4●− source, was completely consumed at a relatively lower PMS concentration during the 20-min reaction (Fig. 5b).
Effect of initial solution pH
The effect of the initial solution pH on CAP degradation by the EC/PMS system is shown in Fig. 6. The results showed that CAP degradation in the EC/PMS system was highly pH-dependent. As the initial pH increased from 3 to 11, the removal rate of CAP significantly decreased from 94.0 to 22.9%. The results showed that acidic conditions favored CAP degradation, and the optimum pH was 3 in this study, which is consistent with that of Fenton-like oxidation processes (Sheng et al. 1999). Because the speciation of Fe ions is highly dependent on the solution pH, as the pH increased from 3 to 11, the main forms of iron in the solution gradually changed from Fe2+ or Fe3+ to Fe(OH)+, FeOH2+, Fe2(OH)24+, and then finally transformed into Fe(OH)3 (Eqs. (10)–(12)). This transformation resulted in the dramatic decrease in the percentage of free Fe2+, leading to the reduction of SO4●− and slowing down CAP degradation (Lin et al. 2013). In addition, the reduction of iron ions in the cathode was also inhibited under alkaline conditions, which further delayed the degradation of CAP (Lakshmanan et al. 2009).

Effect of initial pH on CAP degradation. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, current = 25 mA, [Na2SO4]0 = 25 mM
Effect of common anions
Anions are ubiquitous in natural water and may affect SO4●−-based oxidation process. Thus, it is necessary to explore the influence of these anions on the CAP degradation performance in the EC/PMS system. The effect of common anions, i.e., chloride (Cl−), bicarbonate (HCO3−), and nitrate (NO3−), on CAP degradation by the EC/PMS system is shown in Fig. 7. Adding Cl− only slightly enhanced the degradation of CAP, and the removal rate of CAP increased from 90.9 to 98.8% as the concentration of Cl− increased from 0 to 10 mM. The addition of HCO3− or NO3− exerted a negative effect on CAP degradation performance, and the removal rates decreased from 90.9 to 26.6% and 66.3%, respectively, when the concentration of HCO3− or NO3− increased from 0 to 10 mM. The inhibition by HCO3− and NO3− could be ascribed to their scavenging effect on SO4●− and/or •OH as the following reactions (Eqs. (13)–(16)) (Ghauch and Tuqan 2012; Keen et al. 2012). However, in the presence of Cl-, Cl- reacted with SO4●− to form Cl● (Eq. (17)), which then reacted with Cl− to generate Cl2●− (Eq. (18)). The reactivities of Cl● and Cl2●− with CAP may be similar to the reactivity between SO4●− and CAP (Lian et al. 2017). Furthermore, the transformation of SO4●− to reactive chlorine species can reduce the possibility of SO4●− scavenging by other SO4●− ions and/or PMS; this reduction in scavenging can compensate for the lower redox potential of Cl● and Cl2●−.

Effect of different concentrations of a Cl−, b HCO3−, and c NO3− on CAP degradation. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, current = 25 mA, [Na2SO4]0 = 25 mM, pH0 = 7
Degradation mechanism in the EC/PMS system
Characterization of floccules formed during the reaction
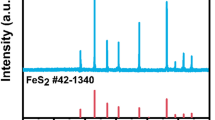
The composition of the floccules was analyzed after the reaction by XPS and XRD, and the results are shown in Fig. 8. According to the XPS full spectrum (Fig. 8a), the presence of Fe 2p, O 1s, and C 1s was observed in the floccules. The charge states of the elements in the formed floccules were determined by scanning the Fe 2p and O 1s orbitals. As shown in Fig. 8b, there are two spin-orbit separation peaks of the Fe 2p3/2 and Fe 2p1/2 core levels at binding energies of 710.4 eV and 723.9 eV, respectively (Xia et al. 2017). The peak positions of the Fe 2p3/2 level of Fe(III), Fe(II), and Fe(0) are at 712.6 eV, 710.9 eV, and 709.9 eV, respectively. The results indicate that Fe(III), Fe(II), and Fe(0) were present in the floccules after the reaction, and their main forms may be Fe2O3, Fe3O4, and FeO(OH) (Xu and Wang 2017). When flocculation occurs during electrolysis, the cathode reduces some of the Fe2+ to form Fe0. The fine O 1s spectrum was also measured, as shown in Fig. 8c. Two intense peaks were observed at 529.6 eV and 530.9 eV. According to previous reports, O2− is found near 529.7~530.1 eV, which confirms the presence of Fe2O3 and Fe3O4 in the floccules (Piumetti et al. 2015). The peak at 530.9 eV indicates the presence of hydroxides, such as FeO(OH), Fe(OH)2, and Fe(OH)3 (Al-Shamsi and Thomson 2013). According to both the XRD patterns (Fig. 8d) and XPS results of the floccules, the main components of the floccules are iron oxides (Fe2O3 and Fe3O4) and iron hydroxides (FeO(OH), Fe(OH)2, and Fe(OH)3). Therefore, in addition to coagulation and/or complexation caused by the formed floccules, PMS activation may occur, generating SO4●− and resulting in the promotion of CAP degradation. (He et al. 2016; Matzek and Carter 2016).

a XPS full spectrum, b Fe 2p spectrum, c O 1s spectrum, and d XRD pattern of the floccules formed during the reaction
Reactive radical species identification
To verify the roles of the different reactive radical species in the EC/PMS system, quenching experiments were conducted in the presence of tert butanol (TBA) and ethanol (EtOH). EtOH (with α-hydrogen) is a well-known radical scavenger for both SO4●− and •OH because it can react quickly with both SO4●− and •OH with rate constants of (1.6–7.7) × 107 and (1.2–2.8) × 109 M−1 s−1, respectively, whereas TBA (without α-hydrogen) mainly quenches •OH because it can react more quickly with •OH ((3.8–7.6) × 108 M−1 s−1) than with SO4●− ((4.0–9.1) × 105 M−1 s−1) (Hu et al. 2020). Figure 9 shows the CAP removal efficiency in the presence of EtOH or TBA. The addition of EtOH significantly reduced the removal rate of CAP, and the removal efficiency of CAP decreased from 91.0 to 21.0% and 16.9% in the presence of 2.5 and 5 mM EtOH, respectively, after a reaction time of 20 min, suggesting that both SO4●− and •OH played an important role in CAP degradation. Moreover, the degradation of CAP was less inhibited by the presence of TBA, and the removal efficiency of TBA decreased from 91.0 to 65.8% and 58.6% in the presence of 2.5 and 5 mM TBA, indicating that compared to SO4●−, •OH played a less important role in CAP degradation in the EC/PMS at neutral pH.

Effect of radical scavengers on CAP degradation. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, current = 25 mA, [Na2SO4]0 = 25 mM, pH0 = 7
Degradation products and transformation pathways
According to the above results, the possible reaction mechanism for the EC/PMS system was proposed, as shown in Fig. 10. In the EC/PMS system, Fe2+ was slowly produced from the sacrificial iron anode through electrochemical corrosion and simultaneously released into the reaction solution (Eq. (1)), which could activate PMS in solution to generate SO4●− and/or •OH according to Eq. (2). The absorbed Fe3+ on the cathode can be reduced to Fe2+ via an electron transfer reaction according to Eq. (3), facilitating the Fe3+/Fe2+ cycle, which reduces the wastage of the anode and maintains Fe2+ and radicals at appropriate concentrations (Li et al. 2018). Additionally, the adsorbed PMS on the cathode could also produce SO4●− and/or •OH (Eqs. (4) and (5)). The formed floccules, such as Fe(OH)3, may also serve as coagulants, which can remove CAP through an enmeshment mechanism.

Reaction mechanism and CAP degradation pathways during the EC/PMS process. Experimental conditions: [CAP]0 = 10 μM, [PMS]0 = 0.5 mM, current = 25 mA, [Na2SO4]0 = 25 mM, pH0 = 7
To further clarify the degradation mechanism, LC/TOF/MS was performed to identify the degradation products of CAP in the EC/PMS system. According to the results, in addition to CAP, eight major degradation products were found, and all these intermediates are summarized in Table 1. Based on the identified degradation products and previous studies, the degradation pathways of CAP were tentatively proposed in Fig. 10.
According to previous studies (Dong et al. 2017; Nie et al. 2018), DP-1 was identified as 2,2-dichloro-N-(1-(4-nitrobenzoyl)vinyl) acetamide, which is a dehydration product of CAP. During SO4●−-induced oxidation, hydroxylation of the aromatic ring may occur by SO4●− attack through electron transfer or by •OH attack through hydroxyl addition; thus, DP-2 was identified as the hydroxylated product of CAP (Dulova et al. 2017). With further oxidation, the hydroxyl group in the side chain of partial CAP transformed into carbonyl groups to yield DP-3. Reactive radicals preferentially attacked the –C–N– bond in the side chain of partial CAP, leading to the formation of DP-4 and 2,2-dichloro-acetamide, followed by further oxidation of 2,2-dichloro-acetamide to yield DP-5 (Nie et al. 2018). Under the attack of •OH, CAP was broken down into dichloro-acetic acid and DP-6. The amino group of DP-6 was further oxidized to the nitro group of DP-7 (Garcia-Segura et al. 2014). The formation of DP-8 indicated that both dehydration and the transformation of hydroxyl groups to aldehyde groups might occur at CAP. Then, these intermediate products were further degraded to smaller molecule compounds and finally mineralized to H2O and CO2.
Conclusion
This study showed that an electrochemically activated peroxymonosulfate (EC/PMS) system can effectively remove CAP from water, and the removal efficiency is better than that of EC alone, PMS alone, and the Fe2+/PMS system. Increasing the applied current and electrolyte concentration enhanced CAP degradation, and the optimal PMS dose for the removal of CAP was 0.5 mM. The removal of CAP is highly pH-dependent, and acidic conditions facilitate CAP degradation. The presence of Cl− slightly promoted CAP degradation, whereas HCO3− and NO3− exhibited inhibitory effects on CAP degradation. The main components of the floccules after the reaction included iron oxides (i.e., Fe2O3 and Fe3O4) and iron hydroxides (i.e., FeO(OH) and Fe(OH)3), as reflected by the XPS and XRD analysis results. The results of quenching experiments indicated that both SO4●− and •OH were responsible for the degradation of CAP. CAP degradation products were identified by LC/TOF/MS, and the primary degradation pathway was proposed.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Ali J, Wenli L, Shahzad A, Ifthikar J, Aregay GG, Shahib II, Elkhlifi Z, Chen ZL, Chen ZQ (2020) Regulating the redox centers of Fe through the enrichment of Mo moiety for persulfate activation: a new strategy to achieve maximum persulfate utilization efficiency. Water Res 181:115862
Al-Shamsi MA, Thomson NR (2013) Treatment of organic compounds by activated persulfate using nanoscale zerovalent iron. Ind Eng Chem Res 52:13564–13571
Brillas E, Sirés I, Oturan MA (2009) Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem Rev 109:6570–6631
Cao Y, Qiu W, Zhao YM, Li J, Jiang J, Yang Y, Pang SY, Liu GQ (2020) The degradation of chloramphenicol by O3/PMS and the impact of O3-based AOPs pre-oxidation on dichloroacetamide generation in post-chlorination. Chem Eng J 401:126146
Danner M-C, Robertson A, Behrends V, Reiss J (2019) Antibiotic pollution in surface fresh waters: occurrence and effects. Sci Total Environ 664:793–804
Dong HY, Qiang ZM, Hu J, Qu JH (2017) Degradation of chloramphenicol by UV/chlorine treatment: kinetics, mechanism and enhanced formation of halonitromethanes. Water Res 121:178–185
Dulova N, Kattel E, Trapido M (2017) Degradation of naproxen by ferrous ion-activated hydrogen peroxide, persulfate and combined hydrogen peroxide/persulfate processes: The effect of citric acid addition. Chem Eng J 318:254–263
Fan Y, Wang B, Yuan S, Wu X, Chen J, Wang L (2010) Adsorptive removal of chloramphenicol from wastewater by NaOH modified bamboo charcoal. Bioresour Technol 101:7661–7664
Garcia-Segura S, Cavalcanti EB, Brillas E (2014) Mineralization of the antibiotic chloramphenicol by solar photoelectro-Fenton: from stirred tank reactor to solar pre-pilot plant. Appl Catal B Environ 144:588–598
Ghauch A, Tuqan AM (2012) Oxidation of bisoprolol in heated persulfate/H2O systems: Kinetics and products. Chem Eng J 183:162–171
Govindan K, Raja M, Noel M, James EJ (2014) Degradation of pentachlorophenol by hydroxyl radicals and sulfate radicals using electrochemical activation of peroxomonosulfate, peroxodisulfate and hydrogen peroxide. J Hazard Mater 272:42–51
He J, Yang XF, Men B, Wang DS (2016) Interfacial mechanisms of heterogeneous Fenton reactions catalyzed by iron-based materials: a review. J Environ Sci 39:97–109
Hirsch R, Ternes T, Haberer K, Kratz KL (1999) Occurrence of antibiotics in the aquatic environment. Sci Total Environ 225:109–118
Hu CY, Hou YZ, Lin YL, Deng YG, Hua SJ, Du YF, Chen CW, Wu CH (2020) Investigation of iohexol degradation kinetics by using heat-activated persulfate. Chem Eng J 379:122403
Huang WX, Fu BM, Fang SY, Wang F, Shao QQ, Du W, Fang F, Feng Q, Cao JS, Luo JY (2021) Insights into the accelerated venlafaxine degradation by cysteine-assisted Fe2+/persulfate: key influencing factors, mechanisms and transformation pathways with DFT study. Sci Total Environ 793:148555
Keen OS, Love NG, Linden KG (2012) The role of effluent nitrate in trace organic chemical oxidation during UV disinfection. Water Res 46:5224–5234
Kovalakova P, Cizmas L, McDonald TJ, Marsalek B, Feng MB, Sharma VK (2020) Occurrence and toxicity of antibiotics in the aquatic environment: a review. Chemosphere 251:126351
Kümmerer K (2009) Antibiotics in the aquatic environment - a review - Part I. Chemosphere 75:417–434
Lakshmanan D, Clifford DA, Samanta G (2009) Ferrous and ferric ion generation during iron electrocoagulation. Environ Sci Technol 43:3853–3859
Lian LS, Yao B, Hou SD, Fang JY, Yan SW, Song WH (2017) Kinetic study of hydroxyl and sulfate radical-mediated oxidation of pharmaceuticals in wastewater effluents. Environ Sci Technol 51:2954–2962
Lin H, Wu J, Zhang H (2013) Degradation of bisphenol A in aqueous solution by a novel electro/Fe3+/peroxydisulfate process. Sep Purif Technol 117:18–23
Li J, Ren Y, Lai LD, Lai B (2018) Electrolysis assisted persulfate with annular iron sheet as anode for the enhanced degradation of 2,4-dinitrophenol in aqueous solution. J Hazard Mater 344:778–787
Matzek LW, Carter KE (2016) Activated persulfate for organic chemical degradation: a review. Chemosphere 151:178–188
Nie MH, Yan CX, Xiong XY, Wen XM, Yang X, Lv ZL, Dong WB (2018) Degradation of chloramphenicol using a combination system of simulated solar light, Fe2+ and persulfate. Chem Eng J 348:455–463
Oh WD, Dong ZL, Lim TT (2016) Generation of sulfate radical through heterogeneous catalysis for organic contaminants removal: current development, challenges and prospects. Appl Catal B Environ 194:169–201
Periyasamy S, Muthuchamy M (2018) Electrochemical oxidation of paracetamol in water by graphite anode: effect of pH, electrolyte concentration and current density. J Environ Chem Eng 6:7358–7367
Piumetti M, Fino D, Russo N (2015) Mesoporous manganese oxides prepared by solution combustion synthesis as catalysts for the total oxidation of VOCs. Appl Catal B Environ 163:277–287
Qi CD, Liu XT, Li Y, Lin CY, Ma J, Li XW, Zhang HJ (2017) Enhanced degradation of organic contaminants in water by peroxydisulfate coupled with bisulfite. J Hazard Mater 328:98–107
Rastogi A, Al-Abed SR, Dionysiou DD (2009) Sulfate radical-based ferrous-peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems. Appl Catal B Environ 85:171–179
Sheng HL, Chi ML, Leu HG (1999) Operating characteristics and kinetic studies of surfactant wastewater treatment by Fenton oxidation. Water Res 33:1735–1741
Shukla P, Fatimah I, Wang SB, Ang HM, Tadé MO (2010) Photocatalytic generation of sulphate and hydroxyl radicals using zinc oxide under low-power UV to oxidise phenolic contaminants in wastewater. Catal Today 157:410–414
Song HR, Yan LX, Wang YW, Jiang J, Ma J, Li CP, Wang G, Gu J, Liu P (2020) Electrochemically activated PMS and PDS: radical oxidation versus nonradical oxidation. Chem Eng J 391:123560
Tan CQ, Dong YJ, Shi L, Chen Q, Yang SW, Liu XY, Ling JF, He XF, Fu DF (2018) Degradation of Orange II in ferrous activated peroxymonosulfate system: efficiency, situ EPR spin trapping and degradation pathway study. J Taiwan Inst Chem E 83:74–81
Wacławek S, Grübel K, Černík M (2015) Simple spectrophotometric determination of monopersulfate. Spectrochim Acta Part A 149:928–933
Wang YR, Chu W (2011) Degradation of 2,4,5-trichlorophenoxyacetic acid by a novel electro-Fe(II)/oxone process using iron sheet as the sacrificial anode. Water Res 45:3883–3889
Wang YR, Chu W (2012) Photo-assisted degradation of 2,4,5-trichlorophenoxyacetic acid by Fe(II)-catalyzed activation of oxone process: the role of UV irradiation, reaction mechanism and mineralization. Appl Catal B Environ 123-124:151–161
Wu D, Sun FQ, Zhou Y (2017) Degradation of chloramphenicol with novel metal foam electrodes in bioelectrochemical systems. Electrochim Acta 240:136–145
Xia XF, Ling L, Zhang WX (2017) Solution and surface chemistry of the Se(IV)-Fe(0) reactions: effect of initial solution pH. Chemosphere 168:1597–1603
Xu L, Wang J (2017) Magnetic nanoscaled Fe3O4/CeO2 composite as an efficient Fenton-like heterogeneous catalyst for degradation of 4-chlorophenol. Environ Sci Technol 46:10145–10153
Xu WH, Zhang G, Zou SC, Li XD, Liu YC (2007) Determination of selected antibiotics in the Victoria Harbour and the Pearl River, South China using high-performance liquid chromatography-electrospray ionization tandem mass spectrometry. Environ Pollut 145:672–679
Yan CX, Yang Y, Zhou JL, Nie MH, Liu M, Hochella MF Jr (2015) Selected emerging organic contaminants in the Yangtze Estuary, China: a comprehensive treatment of their association with aquatic colloids. J Hazard Mater 283:14–23
Zhang Z, Li X, Zhang C, Lu S, Xi Y, Huang Y, Xue Z, Yang T (2021) Combining ferrate(VI) with thiosulfate to oxidize chloramphenicol: influencing factors and degradation mechanism. J Environ Chem Eng 9:104625
Zhou JH, Chen KB, Hong QK, Zeng FC, Wang HY (2017) Degradation of chloramphenicol by potassium ferrate(VI) oxidation: kinetics and products. Environ Sci Pollut Res 24:10166–10171
Zhou JL, Zhang ZL, Banks E, Grover D, Jiang JQ (2009) Pharmaceutical residues in wastewater treatment works effluents and their impact on receiving river water. J Hazard Mater 166:655–661
Zou J, Ma J, Chen LW, Li XC, Guan YH, Xie PC, Pan C (2013) Rapid acceleration of ferrous iron/peroxymonosulfate oxidation of organic pollutants by promoting Fe(III)/Fe(II) cycle with hydroxylamine. Environ Sci Technol 47:11685–11691
Funding
This work was funded by the National Natural Science Foundation of China (51708348); the Scientific and Innovative Action Plan of Shanghai, China (19DZ1208204); and State Key Laboratory of Pollution Control and Resource Reuse Foundation (PCRRF18005).
Author information
Authors and Affiliations
Contributions
Yu-Qiong Gao: conceptualization, methodology, funding acquisition, writing—review and editing
Jin-Qiang Zhou: investigation, visualization, writing—original draft
Han Ning: investigation, validation
Yan-Yan Rao: formal analysis, validation
Nai-Yun Gao: supervision
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Ricardo A. Torres-Palma
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gao, YQ., Zhou, JQ., Ning, H. et al. Electrochemically activated peroxymonosulfate for the abatement of chloramphenicol in water: performance and mechanism. Environ Sci Pollut Res 29, 17866–17877 (2022). https://doi.org/10.1007/s11356-021-17089-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-17089-y