
Abstract
Aerosol behavior over the Himalayas plays an important role in the regional climate of South Asia. Previous studies at high-altitude observatories have provided evidence of the impact of long-range transport of pollutants from the Indo-Gangetic Plain (IGP). However, little information exists for the valley areas in the high Himalayas where significant local anthropogenic emissions can act as additional sources of short-living climate forcers and pollutants. The valley areas host most economic activities based on agriculture, forestry, and pilgrimage during every summer season. We report here first measurements at a valley site at ~2600 m a.s.l. on the trek to the Gangotri glacier (Gaumukh), in the Western Himalayas, where local infrastructures for atmospheric measurements are absent. The study comprised short-term measurement of aerosols, chemical characterization, and estimation of aerosol radiative forcing (ARF) during the winter and summer periods (2015–2016). The particulate matter mass concentrations were observed to be higher than the permissible limit during the summer campaigns. We obtained clear evidence of the impact of local anthropogenic sources: particulate nitrate is associated with coarse aerosol particles, the black carbon (BC) mass fraction appears undiluted with respect to measurements performed in the lower Himalayas, and in winter, both BC and sulfate concentrations in the valley site are well above the background levels reported from literature studies for mountain peaks. Finally, high concentrations of trace metals such as copper point to anthropogenic activities, including combustion and agriculture. While most studies in the Himalayas have addressed pollution in the high Himalayas in terms of transport from IGP, our study provides clear evidence that local sources cannot be overlooked over the high-altitude Himalayas. The estimated direct clear-sky ARF was estimated to be in the range of −0.1 to +1.6 W m−2, with significant heating in the atmosphere over the high-altitude Himalayan study site. These results indicate the need to establish systematic aerosol monitoring activities in the high Himalayan valleys.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Air quality across the globe is deteriorating due to increasing anthropogenic emissions. Among the air pollutants, atmospheric aerosols play an important role due to their heterogeneous reactions in the atmosphere (George and Abbatt 2010; George et al. 2015). Particulate matter (PM) is one of the key components of atmospheric aerosols. It has complex chemical, physical, and optical properties that can affect our environment both directly and indirectly (Stocker et al. 2013; Seinfeld and Pandis 2016; Zhang et al. 2017). The aerosol direct effect on the atmosphere is its ability to scatter and absorb solar radiation, while its indirect effect is its ability to change cloud microphysics (Jacobson 2000; Penner et al. 2001; Ramanathan and Carmichael 2008; Lohmann and Feichter 2010). Based on its interaction with solar radiation, PM can have a warming or a cooling effect on the climate. For instance, black carbon (BC), a particulate pollutant from combustion, contributes to the warming effect, while particulate sulfates help in the cooling effect (Myhre et al. 2013). Therefore, aerosol radiative forcing (ARF) estimation is a useful tool to assess the adverse impact of aerosols on climate (Ramaswamy et al. 2001).
Different types of PM emission sources can obviously influence the heterogeneity of atmospheric PM. Another factor is the geographic location of the source(s), which can significantly affect the aerosol chemical and physical evolution, including removal from the atmosphere. Therefore, quantification of regional aerosol sources and ARF are of primary interest for scientific and policy perspectives (Yu et al. 2012). Ground-based atmospheric PM has been well studied in metropolitan cities, coastal areas, and arid regions of south Asian countries (Ram and Sarin 2011; Izhar et al. 2016; Prabhu et al. 2019; Soni et al. 2020; Rajeev et al. 2021; Panda et al. 2021). However, studies focused on the mountain terrain of high-altitude Himalayas are limited, partly due to the inaccessibility to the complex mountainous areas (Decesari et al. 2010; Hegde and Kawamura 2012; Arun et al. 2019).
The study of atmospheric aerosols over the Himalayas is of prime importance mainly because (1) the Himalayan region is a major freshwater source in the South Asian region (Soni et al. 2019), (2) the Himalayas help in the orographic lifting of aerosols up to the free troposphere (Bianchi et al. 2021), and (3) increasing catastrophic events such as avalanches, cloud burst, floods, and forest fires could directly and indirectly be affected by air pollution. The Himalayas are the approximately 2500-km-long crescent-shaped mountain ranges situated on the northern edge of South Asia. There is ample evidence available that the transported aerosols from South Asia significantly influence the air quality over the high-altitude Himalayas (Bonasoni et al. 2012; Decesari et al. 2010; Hegde et al. 2007; Kumar et al. 2014; Nasir et al. 2019; Prabhu et al. 2020). Besides the transported aerosols from South Asia to the Himalayas, the limited focus has been attained on the emissions from the local anthropogenic activities over the high-altitude Himalayan valleys.
The Himalayan valleys hold several spiritual temples and tracking routes; thus, millions of people travel over the Himalayas, especially during the summer months. In addition, forest fire activities are also prominent during the summer months. Therefore, the emissions from the local anthropogenic combustion activities and forest fire could also be an important source of atmospheric aerosols. In our previous study, Soni et al. (2019) attempted to identify all possible sources of BC at the neighboring valley location to the Gangotri glacier, which is one of the largest glaciers in India. Study has reported the localized emissions from the surrounding valley were one of the crucial sources of BC, besides the transported one. However, there is a considerable lack of data in the literature concerning aerosol properties in the Gangotri glacier regions of the Himalayas. Added to this are the significant cultural activities embedded in local topography, which can vary the aerosol chemistry and aerosol forcing on a regional scale.
The main objectives of this study are (1) to study ground-based size fractions of PM and BC at two different high-altitude valley sites situated in the lesser and greater Himalayas, (2) to chemically characterize the PM as a function of size and identify possible emission sources, and (3) to estimate direct ARF based on the mass closure of chemical composition of PM.
Methodology
Details of the sampling site
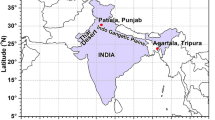
The aerosol measurements were carried out at two different valley sites in the Western Himalayas: (1) The greater Himalayan study site 1, situated in the Dharali village having an altitude of 2600 m a.s.l. (S1-GH), and (2) the lesser Himalayan study site 2, situated in the urbanized town Uttarkashi having an altitude of 1400 m a.s.l. (S2-LH). It is worth mentioning that these study sites are situated in the Bhagirathi eco-sensitive zone declared by the Indian Ministry of Environment, Forest and Climate Change. The aerial distance between these two study sites is about 45 km (Fig. 1). As of the 2011 India census, S1-GH and S2-LH had populations of 583 and 17475, respectively. Besides the local populations, millions of pilgrims and tourists travel along with this Himalayan study site, especially during the summer. In addition, these two study sites also serve as a major stopover on the road to Gangotri temple and the trek to the Gaumukh glacier. A detailed discussion about the S1-GH, surrounding vegetation types, and vehicular count is reported in our earlier publication (Soni et al. 2019). Moreover, local wind pattern and atmospheric boundary layer height during the sampling campaign are discussed in Sect. S1 and shown in Figs. S1 and S2 in the supplementary text.

The study sites in Northern India. On the left side of the figure, a map of India. On the right side, the measurement site map situated in the Uttarakhand state of India. The digital elevation model data was acquired from https://www.diva-gis.org/
Measurement of atmospheric aerosols
At both the study sites, the influence of pilgrimage activities is highest during the summer months, while pilgrimage activities are negligible during the winter months. In the present study, aerosol sampling campaigns were performed during the summer and winter periods. The infrastructure at the study sites was very limited if not absent, i.e., power supplies and shelters. This led to aerosol sampling for shorter, more intense time periods so that structures could be maintained; for site S1-GH, these sampling periods were in the months of November 2015 (5–20 November) and May 2016 (5–25 May); for site S2-LH, the sampling period took place during the summer only, May and June 2016 (25 May–5 June). November and May–June sampling campaigns were represented as winter and summer periods throughout the manuscript.
During the winter sampling campaign, PM samples were collected on four different Whatman filters having a diameter of 46.2 mm (two PTFE filters for PM10 and PM2.5, one glass fiber filter for PM10, and one quartz fiber filter for PM2.5). These four filters were placed on four separate ChemComb cartridges (Model no. 3500, Thermo Scientific, USA) mounted in a four-channel Partisol speciation sampler (Soni et al. 2020). A Partisol speciation sampler was operated at a constant flow rate of 10 L per minute (Thermo Scientific, USA). The PM samples were collected continuously for 24 h (6:00 to 6:00 Indian Standard Time) during the winter campaign.
Furthermore, due to technical issues with the motherboard of the Partisol speciation sampler, summer sampling campaigns were performed using a cascade impactor. PM samples were collected for the summer campaigns on eight different 81-mm glass micro-fiber filters (EPM 2000, Whatman, USA) placed in an eight-stage non-viable Andersen cascade impactor (Thermo Scientific, USA). The Andersen cascade impactor collects eight different sizes of bin of PM such as 0.43–0.65, 0.65–1.1, 1.1–2.1, 2.1–3.3, 3.3–4.7, 4.7–5.8, 5.8–9, and >9 μm. The Andersen cascade impactor was operated at a flow rate of 28.3 L per minute. As there is no separate stage in the cascade impactor for a cutoff diameter of 2.5 μm, we have considered the nearest stage for the cutoff diameter as 2.1 μm (stages 1 to 3) as fine PM (PM2.1). The PM on all the stages (stages 1 to 8) is considered PM≥10 (Panwar et al. 2020). For simplicity, PM≥10 measured during the summer campaigns is represented as PM10 throughout the manuscript. It is worth mentioning that the PM samples were collected continuously for 48 h (6:00 to 6:00 Indian Standard Time) during the summer campaigns to get sufficient deposition of eight different size fractions of PM.
All the filters were kept in a desiccator for about 24 h before and after sampling to remove moisture and weighed three times using a microbalance (Mettler Toledo-XS105DU, 0.1-μg sensitivity) at room temperature 21±5 °C. The average weight was calculated using the arithmetic mean of three recorded values of each filter. PM mass concentrations were calculated gravimetrically by taking the difference between post- and pre-weight of filters. All the filters were placed in airtight Petri dishes in the freezer (−4 °C) until further chemical analysis. A detailed description of filter conditioning is reported in a companion paper (Soni et al. 2020).
Moreover, real-time BC mass concentrations were also measured continuously using an Aethalometer AE-33 (Magee Scientific, USA) during the sampling campaigns. In the present study, per minute BC data were utilized for further analysis. A detailed description of BC measurements and data treatment is reported in our earlier paper (Soni et al. 2019).
Chemical composition measurement
For the water-soluble chemical composition measurement, the PM containing quartz and glass fiber filters having a diameter of 46.2 mm were cut into halves and extracted in 5 ml of Milli-Q water (18.2 MΩ) followed by shaking with a mechanical shaker (60 min.). The 81-mm glass fiber filters were soaked in 10 ml of Milli-Q water (18.2 MΩ), also followed by 60 min of mechanical shaking. For both quartz and glass fiber filters, the extract was filtered through a 0.45-μm syringe PTFE filter (PALL Corporation, IC Acrodisc) and analyzed with ion chromatography (Dionex ICS-2000) for the measurement of fifteen inorganic and organic ion species (cations, NH4+, methylamine, ethylamine, K+, dimethylamine, diethylamine, trimethylamine, Mg2+, and Ca2+; anions, acetate, formate, MSA, NO3−, SO42−, and oxalate). However, some organic cations such as methylamine, ethylamine, dimethylamine, diethylamine, and trimethylamine were observed below the detection limit; thus, they were excluded from further discussion. Ammonium measurements of the glass fiber filters were removed from the data matrix due to the high uncertainty associated with the filter media. Moreover, water-extracted samples were also analyzed in a total organic carbon (TOC) analyzer (Shimadzu TOC-5000A) for water-soluble organic carbon (WSOC) measurement. A detailed description of IC and TOC measurement procedures was reported elsewhere (Saarikoski et al. 2012; Montero-Martínez et al. 2014; Paglione et al. 2014; Sandrini et al. 2016).
For the acid-soluble chemical composition measurement, each of the PTFE filters was divided into two parts and subsequently heated in a microwave digester with HNO3 to HClO4 ratio of 4:1 (MARS 6, CEM, USA) (Geladi and Adams 1978; Prabhu and Shridhar 2019; Soni et al. 2020; Panwar et al. 2020). The extracted samples were diluted, filtered, and analyzed in an atomic absorption spectrometer (AAS) to measure six chemical elements (such as Cd, Cr, Cu, Fe, K, and Zn). A detail about the acid-soluble chemical composition measurement procedure is reported in our earlier publication (Soni et al. 2020). Due to the significant uncertainty in AAS results of glass fiber filters collected during the summer, these filters were not further analyzed for acid-soluble elements.
Every 5 days, one field blank was collected throughout the sampling campaigns. These field blanks and several laboratory blank filters were also extracted and analyzed for water-soluble and acid-soluble chemical compositions. The mass concentrations of chemical species were calculated based on the regression equation of the calibration curve (R2>0.99) using absorbance/area values of AAS and IC. For the detection limit calculation, blank Milli-Q water (18.2 MΩ) was analyzed several times in AAS and IC. The method of detection limit (MDL) of each chemical species was calculated by multiplying the standard deviation of chemical species in blank Milli-Q with t-statistics at a 95% confidence interval (Soni et al. 2020). The chemical species that did not respond to blank Milli-Q water then signal to noise ratio (S/N) were considered a detection limit. Finally, all the reported chemical data were subtracted from the field blank. The MDL value of each chemical element is reported in Table 1.
Optical properties and direct aerosol radiative forcing calculations
Aerosol optical parameters such as the aerosol optical depth (AOD), Ångström exponent, single-scattering albedo (SSA), and asymmetric parameter (AP) were estimated using the Optical Properties of Aerosols and Clouds (OPAC) model (Hess et al. 1998). The model allows users to select different aerosol types with different proportions and calculates spectrally resolved bulk aerosol optical properties, which then can be utilized as an input to a radiative transfer model. In this study, the ground-based aerosol chemical composition and BC mass concentration (soot) were used to decide the proportion of water-soluble, insoluble, mineral dust, and soot aerosol types and were input into the model to obtain optical properties from OPAC. Due to the unavailability of mineral dust, mass closure of mineral dust was estimated by data extracted from nearby sites (Arun et al. 2019). Mass closure of chemical composition data used for the direct aerosol radiative forcing calculations is shown in Table 2. The observed surface concentration cannot be directly used to calculate AOD since the AOD is a measure of columnar concentration. Hence, optical properties such as single-scattering albedo and asymmetry parameter (ASP) were taken from the OPAC model, while the AOD was utilized from the MODIS satellite from a period of ground-based observations. The approach has been used in several studies before (Gadhavi and Jayaraman 2004, 2010; Jayaraman et al. 2006; Satheesh and Srinivasan 2006; Ramachandran and Kedia 2010). In the present study, level-2 MODIS Terra C6.0 deep blue AOD product at 550 nm was used mainly because it showed good association (r≈0.90, p<0.05) with ground-based neighboring AERONET station (Kumar et al. 2018).
The spectral optical properties of composite aerosols obtained by the OPAC model, such as AOD, Ångström exponent, SSA, and ASP, were incorporated into the Santa Barbara DISORT Atmospheric Radiative Transfer (SBDART) model (Ricchiazzi et al. 1998) to obtain shortwave (0.3 to 3 μm) radiative fluxes at the surface and the top of the atmosphere (TOA). Along with optical properties, the MODIS-derived surface reflectance, column water vapor, and Ozone Monitoring Instrument (OMI) column ozone over the study sites were also essential parameters for the aerosol radiative forcing (ARF) calculations. The SBDART model uses the assumption of a plane-parallel atmosphere. A detail about the basic assumptions involved in the model calculation and related uncertainties is mentioned in Sect. S2 in the supplementary text. Furthermore, the detailed methodology of ARF calculations and associated uncertainties is reported in previous studies (Kedia et al. 2010; Ramachandran and Kedia 2010; Satheesh et al. 2010). A detailed description of the ARF calculation is illustrated in the flowchart (Fig. 2).

Flowchart showing the methodology of direct aerosol radiative forcing calculations
Results and discussion
Variation of atmospheric aerosols
The variability of PM10, fine PM (PM2.5 or PM2.1), and black carbon (BC) mass concentrations during the sampling periods is shown in Fig. 3. During the summer sampling period, average PM10, PM2.1, and BC were observed to be 189.95 ± 86.14, 68.38 ± 34.42, and 4.16 ± 3.67 μg m−3, respectively. These values were in contrast to the winter sampling campaign at S1-GH when the measured concentrations were observed to be 28.47 ± 8.72, 19.02 ± 4.76, and 1.85 ± 2.23 μg m−3, respectively. At the site S2-LH during the summer, the average PM10, PM2.1, and BC concentrations were observed to be 250.43 ± 80.44, 86.54 ± 26.18, and 5.85 ± 3.21 μg m−3, respectively (Fig. 3). At both study sites during the summer, PM mass concentrations were observed to be higher than the 24-h permissible limit stipulated by the Central Pollution Control Board of India (CPCB) for eco-sensitive zone (100 and 60 μg m−3 for PM10 and PM2.5, respectively) and World Health Organization (WHO) (50 and 25 μg m−3 for PM10 and PM2.5, respectively). In comparison, PM mass concentrations at S1-GH were observed to be within the permissible limits during the winter.

Box and whisker plot of PM10, fine PM (PM2.5 or PM2.1), and black carbon (BC) mass concentrations at the sampling sites during the summer and winter sampling campaigns. The horizontal solid lines inside the boxes indicate the median, and the solid squares indicate the mean. The top and bottom of the box indicate the 75th and 25th percentile, respectively, while the top and bottom extents of the whiskers indicate the standard deviation (1σ). The top and bottom asterisk symbols represent the maximum and minimum, respectively
Interestingly, PM mass concentrations were observed to be low during the winter period when the neighboring IGP region reels under extensive emissions of aerosols. In contrast, high PM concentration was observed during the summer. The variability in PM mass concentrations can be related to the difference in boundary layer conditions during summer and winter campaigns. During the summer, the boundary layer remains deeper, which might favor aerosol transportation from the IGP regions, whereas the boundary layer remains shallow during the winter, which might restrict the transportation (Fig. S1 in supplementary text). In addition, the high concentrations during the summer can also be related to an intensification of the local anthropogenic sources. Pilgrimage activities involve wood and coal burning for heating and cooking purposes, waste burning, vehicular emissions, and soil resuspension associated with vehicular movement. Also, it is worth mentioning that more than 4000 ha of forest cover were burned during the summer sampling campaigns across the state of Uttarakhand (Indian Express 2016). In contrast to summer, human influence is low during winter since the majority of the local people migrate to lower altitudes due to the extreme cold. Several earlier studies have reported similar seasonal trends of aerosol mass concentrations over the Himalayan regions (Nair et al. 2013; Sundriyal et al. 2018; Soni et al. 2019; Negi et al. 2019).
The fine to coarse mass ratio (PM2.5/PM10) study is an important parameter to identify the dominant size fraction of aerosols. In this study at site S1-GH, the fine to coarse mass ratio was lower in summer than in winter, i.e., 0.36 and 0.67, respectively. The most plausible reason for the lower summer ratio could be soil resuspension from unpaved road use and high vehicular movement associated with pilgrim activities. Besides, some earlier studies have reported that the coarse PM can also be transported from the western arid and semi-arid regions (Sikka 1997; Hegde et al. 2007; Kumar et al. 2014). However, the wind-rose diagram and air-mass back trajectory analysis during the sampling campaigns reveal the minimal influence of wind convergence from the western arid regions or the Thar desert (Fig. S2 (a) and S3 (a) in supplementary text). The results showed that the dominance of coarse particles during the summer was mainly localized rather than long-range transported mineral dust. A similar trend was also observed at the lesser Himalayan valley site (S2-LH).
Furthermore, the BC concentrations in the summer were lower at S1-GH than that at S2-LH as expected, but the BC fractions in PM10 were not that different between the two sites (it is roughly 2–3% at both sites). That gave clear evidence of active BC sources at the study sites because the BC mass fraction was not lowered by mixing regional pollution with background aerosols (dust). Moreover, BC mass fractions were the highest at S1-GH in winter (approximately 8%), when the station is substantially disconnected by the IGP due to shallow boundary layer, hence pointing to local combustion sources (Soni et al. 2019).
Aerosol chemical composition and possible sources
Water-soluble chemical composition
The average mass concentrations of the water-soluble ions along with the average cation-to-anion charge equivalent ratios (Σ+/Σ−) are shown in Table 3. During the summer campaign at S1-GH, the cation-to-anion charge equivalent ratio showed a noticeable cation deficit with Σ+/Σ− ratio of 0.83 and 0.63 for PM10 and PM2.1. In contrast, cation-to-anion charge equivalent ratios showed a noticeable anion deficit at S2-LH with Σ+/Σ− ratio of 1.39 and 1.52 for PM10 and PM2.1. Earlier studies have reported that the anion deficiency in aerosol samples is mainly due to a lack of bicarbonate measurements (Venkataraman et al. 2002).
The percentage contributions of measured cations and anions are shown in Fig. 4. At both the Himalayan study sites, the major PM species were observed to be from sulfate (SO42−), nitrate (NO3−), and ammonium (NH4+), which are considered the main components of secondary inorganic aerosol (SIA). These SIA components contributed approximately 56–72% of the total ions associated with PM10 and 59–78% of total ions associated with PM2.5. Usually, possible secondary inorganic compounds were ammonium sulfate ((NH4)2SO4) and ammonium nitrate (NH4NO3), which originated from the neutralization of acidic sulfuric acid and nitric acid with ammonia (Gupta and Mandariya 2013). Besides the ammonium ion, crustal ions Ca2+ and Mg2+ can also neutralize the acidic SO42− and NO3− ions (Possanzini et al. 1988).

Percentage contribution of water-soluble ionic composition associated with the PM10 and fine PM (PM2.5 or PM2.1) during the winter and summer sampling campaigns
The neutralization factor can be used to evaluate the neutralization capacity of NH4+, Ca2+, and Mg2+ with acidic SO42− and NO3−. In the present study, neutralization factors were calculated and shown in Table 4. A detail about the calculation of the neutralization factor is mentioned in Sect. S3 in the supplementary text. The neutralization factors for summer campaigns were not reported mainly due to significant uncertainty associated with ammonium ion measurement from the glass fiber filters. However, it is worth mentioning that the ammonium ions contributed significantly during the winter campaign (Table 3), giving indirect evidence that ammonium ions could be present significantly during the summer campaigns as well.
Nevertheless, a significant fraction of Ca2+ ion is present even at fine-mode PM (Fig. S4 in the supplementary text), pointing to diffuse crustal sources from local and distant emissions. That has also importance for condensation or fixing of nitrate in particulate form when the semivolatile ammonium nitrate salts tend to partition to the gas phase. During all the sampling campaigns at both the Himalayan study sites, the neutralization capacity of crustal Ca2+ with acidic anions was highest. At S1-GH, the total neutralization factor was observed below unity, which depicted that the aerosols were acidic. In contrast, the total neutralization factor was observed above the unity at S2-LH, which depicted that most acidic anions were neutralized.
As mentioned in an earlier section, local anthropogenic activities were assumed to be the most prominent sources of atmospheric aerosols over the high-altitude Himalayan study sites. Therefore, the mass concentrations of anthropogenic source marker ions (non-crustal_K+, NO3−, and SO42−) observed during the sampling campaigns are segregated and shown in Fig. 5. The nitrate was associated mainly with coarse PM10 particles during all the measurement campaigns, which are less efficient for the long-range transported aerosols. Thus, the prominent emission sources of nitrate precursors in this area are expected to be from local anthropogenic activities.

Contribution of anthropogenic water-soluble ions associated with the PM10 and fine PM (PM2.5 or PM2.1) during the winter and summer sampling campaigns. (a) and (b) represent the ionic compositions during the summer at S1-GH and S2-LH; (c) represents the ionic compositions during the winter at S1-GH
During the summer campaigns, the total concentrations of these anthropogenic ionic compounds were much more significant at S2-LH than that at S1-GH in PM10. However, the difference was less significant for PM2.5, especially for sulfate (half of the samples at S2-LH show sulfate concentrations in line with those at S1-GH). It is not easy to assess the origin of sulfate in PM2.5 particles because it is a typical secondary aerosol compound that can accumulate during transport. Previously, Arun et al. (2019) reported sulfate mass concentrations in the range of 0.17 to 0.50 μg m−3 at the neighboring background site in Western Himalayas (4080 m a.s.l.). In the present study at S1-GH, sulfate concentration in fine PM was approximately an order of magnitude higher than the background Western Himalayan site. The observed high sulfate fractions gave clear pointers that the study site was significantly influenced by the secondary aerosol formation associated with combustion sources in high Himalayan valleys. In addition, a significant correlation between sulfate and oxalate (r=0.60, p<0.05) associated with PM2.5 gave strong evidence of secondary aerosol formation. Oxalate is the most abundant dicarboxylic acid in organic aerosols and also an important constituent of secondary organic aerosols (SOA) (Kawamura and Ikushima 1993; Kawamura and Yasui 2005).
During the winter, the mass fractions of the anthropogenic ionic compounds (non-crustal_K+, NO3−, and SO42−) in PM tend to be higher at S1-GH with respect to the summer despite the small PM concentrations. The high fractions of these anthropogenic ions were observed during the year when transport from IGP was unlikely. The NO3− showed the partitioning to coarse particles during the winter campaign, which is similar to the summer campaigns. These results indicated the influence of local combustion sources in the high Himalayan valley even during the low-pilgrimage period. Our earlier study (Soni et al. 2019) also reported similar results; the local biomass/biofuel burning led to high BC mass concentrations, especially during the morning hours.
The significant fractions of WSOC associated with the PM10 (50%) and fine PM (33%) showed dominant contributions of organic aerosols during the winter. The plausible reasons for the high fractions of WSOC associated with PM10 could be contributions from the localized biomass burning emissions, as no other ions increase in coarse PM10 except for nitrate. The WSOC fraction is oxygenated organic compounds directly emitted from the primary combustion processes and/or are formed through secondary processes (Saxena and Hildemann 1996). Similar to summer campaign, sulfate concentration in PM2.5 was approximately an order of magnitude higher than the background Western Himalayan site (Arun et al. 2019), which gave clear pointers that the secondary aerosol formation significantly influenced the study site. Furthermore, a significant correlation between sulfate and SOA marker oxalate and WSOC associated with PM2.5 strengthens secondary aerosol formation evidence (r≥0.35, p<0.05).
During the winter sampling campaign, very low levels of acetate (CH3COO−), formate (HCOO−), and MSA (methane sulfonic acid) were also observed in the fine PM2.5 (7% of total ions). MSA is generally considered a tracer for marine aerosols (Andreae 1986). There is also evidence that a minor fraction of MSA can be emitted from forests (Andreae et al. 1990; Lukács et al. 2008). As the present study sites are far away from any marine aerosol sources, emitted MSA could be mainly of biogenic origin. Moreover, the acetate, formate, and MSA were significantly correlated with the SOA marker oxalate as well as WSOC (r≥0.49, p<0.05). These results indicated that the SOA formation could be due to the oxidation of biogenic volatile organic compounds emitted from the neighboring forested regions.
Throughout the campaigns, oxalate was observed to be in the range of 3–8% of the total water-soluble ionic composition. The highest concentration of oxalate was observed to be in size range of 2.1–3.3 μm (Fig. S4 in the supplementary text). Interestingly, oxalate observed in fine PM2.5 samples was also strongly correlated with the Mg2+ ion, especially during the winter period at S1-GH (r=0.83, p<0.05). The particle-phase oxalate can also exist as a metal oxalate complex, which ultimately makes oxalate species more insoluble or less hygroscopic than non-complexed oxalate species (Furukawa and Takahashi 2011).
Acid-soluble chemical elements
The average mass concentrations of the acid-soluble chemical elements are shown in Table 5, and their percentage contributions are represented in Fig. 6. During the winter period at site S1-GH, these acid-soluble chemical elements constitute approximately 13% and 16% of the total PM10 and PM2.5, respectively, depicting that most of these elements were associated with the finer PM2.5 particles. The trends in the average mass concentration of the chemical elements during the winter followed the order of Fe>Cu>K>Zn for PM10 and Cu>Fe>K>Zn for PM2.5. Both Cu and Fe are very high in concentrations and are ubiquitous in crustal and combustion sources (Mao et al. 2017). However, time trends of Cu and Fe did not correlate, which suggests contributions from different sources. The high Himalayan study region (S1-GH) is surrounded by the apple orchards, which is the primary economy of local residents besides tourism. Every year, apple orchard owners spray Cu-based fungicides (copper hydroxide or copper oxychloride) to control canker after harvesting of apple, especially during November. It is expected that the high fractions of Cu could be due to fungicide spray, as the measurement campaign was also focused on during November. Very few studies have reported the influence of Cu-based fungicides on orchard soil (Arias et al. 2005; Wang et al. 2015). However, the present study results gave a pointer that the Cu-based fungicides can lead to high Cu in the atmospheric particle phase. Acid-soluble K has multiple sources such as biomass burning, fossil fuel combustion, waste burning, dust, and sea salt (Lawson and Winchester 1979; Andreae 1983). Earlier, several authors have reported a K/K+ mass ratio for different emission sources. For instance, the ratio for biomass burning and dust should be about less than two (approximately 1.17–2.01) and greater than three (approximately 3.93–8.27), respectively (Yu et al. 2018). In the present study, the K/K+ mass ratio was observed to be 3.90 during the winter campaign at S1-GH. The high ratio indicated that the dominant fraction of K was mainly of geographic crustal origin.

Pie charts representing the percentage contribution of the acid-soluble elements. (a) represents acid-soluble elements associated with PM10, (b) represents acid-soluble associated with PM2.5
Direct aerosol radiative forcing
Down-welling and up-welling aerosol radiative fluxes at the surface, top of atmosphere (TOA), and atmosphere have been estimated through a combination of OPAC (Hess et al. 1998) and SBDART (Ricchiazzi et al. 1998) models. In brief, we used the OPAC model to get aerosol optical properties based on the observed chemistry of aerosol. Since often smaller aerosols contribute more in aerosol radiative forcing than bigger aerosols, we separately estimated optical properties for PM2.5 and PM10 as the relative contribution of aerosol types varied. The mean value of surface, TOA, and atmosphere radiative forcing is shown in Fig. 7.

Direct aerosol radiative forcing (ARF) at surface, TOA, and atmosphere during summer and winter. (a) represents the ARF of PM10, (b) the ARF of fine PM
At the greater Himalayan study site (S1-GH), the direct aerosol radiative forcing (ARF) values at the TOA were nearly zero for PM10 aerosol for both the sampling periods and PM2.5 in winter. The TOA ARF was calculated around +1.1 W m−2 for PM2.5 in summer. The high values of TOA ARF for PM2.5 in summer are due to a relatively higher percentage of black carbon particles (6%). The surface and atmospheric ARF exhibit significant changes from summer to winter. This change is mainly because of lower aerosol optical depth (AOD) in winter than that in summer. The MODIS-derived AOD during the sampling campaign is shown in Fig. S5 in supplementary text. Also, the difference in the solar insolation period contributes significantly. During winter over the Himalayas, the solar insolation periods are significantly shorter, allowing less absorption and scattering by aerosols. Overall, the estimated ARF was observed to be lower than that observed at the neighboring Himalayan Manora peak station (Srivastava et al. 2015).
Nearly zero TOA ARF and large negative surface ARF mean absorbing aerosol, which outweighs the scattering in causing the radiative forcing. The absorbing nature of aerosol contributes to additional heating of the atmosphere, reflecting as positive atmospheric radiative forcing. The atmospheric forcing of fine PM (PM2.5 or PM2.1) was high compared to PM10, and we attribute this mainly to a higher fraction of absorbing BC particles present in fine PM.
Conclusions
In the present study, size fractions of particulate matter (PM) and black carbon (BC) mass concentrations were measured at data-deficient high-altitude Himalayan valley sites (2600 m a.s.l. and 1200 m a.s.l.) during the winter and summer months of the years 2015–2016. The PM samples were analyzed for their chemical composition as a function of size to identify possible emission sources. Moreover, mass closure of the chemical compositions of PM was used to estimate direct aerosol radiative forcing (ARF).
The average mass concentration of PM10 and PM2.5 during the summer was much higher than the permissible limit set by the Central Pollution Control Board of India for eco-sensitive zone and the World Health Organization (WHO). Based on the chemical characterizations, attributed major emission sources were pilgrimage/touristic activities and forest fires in the valley during the summer. All these emission sources at the greater Himalayan study site (S1-GH) raised PM10, PM2.5, and BC mass concentrations by around 7, 4, and 2 times higher than the low-pilgrimage winter campaign (when human influence was negligible), respectively.
Interestingly, the chemical and air-mass back trajectory analysis revealed that local emission sources substantially influence the study sites, even though the region was not influenced by the seasonal pilgrimage movement and forest fires. While most studies in the Himalayas have addressed pollution in the high Himalayas in terms of transport from Indo-Gangetic Plains (IGP), our study provides clear evidence that local sources cannot be overlooked. The chemical analysis of PM also indicated secondary aerosol formation from the oxidation of biogenic volatile organic compounds emitted from the neighboring forested regions, especially during the winter. Surprisingly, particle-phase Cu was observed to be one of the highest fractions of acid-soluble elements; attributed primary reason is the use of Cu-based fungicides for apple orchards in the surrounding regions, which was instead noticed or studied.
For PM10 measured at S1-GH, direct aerosol radiative forcing (ARF) at the top of the atmosphere was estimated to be between −0.1 W m−2 (in winter) and +0.1 W m−2 (in summer), while the surface ARF was estimated to be between −1.8 W m−2 (in winter) and −5.3 W m−2 (in summer). The estimated ARF was observed to be lower than that observed at the neighboring Himalayan Manora peak station. For PM10 measured at S2-LH, direct ARF was estimated to be +1.6, −29.1, and +30.7 at the top of the atmosphere, surface, and atmosphere, respectively. From the direct ARF estimations, it is inferred that the aerosol causes significant absorption and hence heating in the atmosphere over these high altitude Himalayan study sites.
Although this study is restricted to the short sampling campaigns over the Himalayas due to the lack of constant power supply, shelters, and other resources, there are clear pointers that the localized emissions significantly influence the local air quality over the Western Himalayas, even though human influence and forest fires were negligible. The present study has attempted to identify all possible sources of atmospheric aerosols and their climatic impact (such as direct ARF) based on the chemical compositions at the data-deficient Western Himalayan region. Aerosol/air pollution monitoring networks over this region of the Himalayas are very scarce. Therefore, it is anticipated that this study will open the way for better longterm assessment and its impacts (regionally as well as globally) of primary as well as secondary aerosols in this region of Western Himalayas.
References
Andreae MO (1986) The ocean as a source of atmospheric sulfur compounds. In: Buat-Ménard P. (eds) The Role of Air-Sea Exchange in Geochemical Cycling. NATO ASI Series (Series C: Mathematical and Physical Sciences), vol 185. Springer, Dordrecht. https://doi.org/10.1007/978-94-009-4738-2_14
Andreae MO, Berresheim H, Bingemer H et al (1990) The atmospheric sulfur cycle over the Amazon Basin: 2. Wet Season. J Geophys Res. https://doi.org/10.1029/JD095iD10p16813
Andreae MO (1983) Soot carbon and excess fine potassium: long-range transport of combustion-derived aerosols. Science 220:10–13. https://doi.org/10.1126/science.220.4602.1148
Arias M, Lopez E, Soto B (2005) Copper distribution and fractionation in aggregate fractions from vineyard soils: comparison with zinc. Agrochimica 49:60–69
Arun BS, Aswini AR, Gogoi MM et al (2019) Physico-chemical and optical properties of aerosols at a background site (~4 km a.s.l.) in the western Himalayas. Atmos Environ 218:117017. https://doi.org/10.1016/j.atmosenv.2019.117017
Bianchi F, Junninen H, Bigi A et al (2021) Biogenic particles formed in the Himalaya as an important source of free tropospheric aerosols. Nat Geosci 14:4–9. https://doi.org/10.1038/s41561-020-00661-5
Bonasoni P, Cristofanelli P, Marinoni A et al (2012) Atmospheric pollution in the Hindu Kush–Himalaya region. Mt Res Dev 32:468–479. https://doi.org/10.1659/MRD-JOURNAL-D-12-00066.1
Decesari S, Facchini MC, Carbone C et al (2010) Chemical composition of PM10 and PM1 at the high-altitude Himalayan station Nepal Climate Observatory-Pyramid (NCO-P) (5079 m a.s.l.). Atmos Chem Phys 10:4583–4596. https://doi.org/10.5194/acp-10-4583-2010
Furukawa T, Takahashi Y (2011) Oxalate metal complexes in aerosol particles: implications for the hygroscopicity of oxalate-containing particles. Atmos Chem Phys 11:4289–4301. https://doi.org/10.5194/acp-11-4289-2011
Gadhavi H, Jayaraman A (2004) Aerosol characteristics and aerosol radiative forcing over Maitri, Antarctica. Curr Sci 86:296–304
Gadhavi H, Jayaraman A (2010) Absorbing aerosols: Contribution of biomass burning and implications for radiative forcing. Ann Geophys 28:103–111. https://doi.org/10.5194/angeo-28-103-2010
Geladi P, Adams F (1978) The determination of cadmium, copper, iron, lead and zinc in aerosols by atomic-absorption spectrometry. Anal Chim Acta 96:229–241. https://doi.org/10.1016/S0003-2670(01)83658-8
George IJ, Abbatt JPD (2010) Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat Chem 2:713–722. https://doi.org/10.1038/nchem.806
George C, Ammann M, D’Anna B et al (2015) Heterogeneous photochemistry in the atmosphere. Chem Rev 115:4218–4258
Gupta T, Mandariya A (2013) Sources of submicron aerosol during fog-dominated wintertime at Kanpur. Environ Sci Pollut Res 20:5615–5629. https://doi.org/10.1007/s11356-013-1580-6
Hegde P, Kawamura K (2012) Seasonal variations of water-soluble organic carbon, dicarboxylic acids, ketocarboxylic acids, and α-dicarbonyls in Central Himalayan aerosols. Atmos Chem Phys 12:6645–6665. https://doi.org/10.5194/acp-12-6645-2012
Hegde P, Pant P, Naja M et al (2007) South Asian dust episode in June 2006: aerosol observations in the central Himalayas. Geophys Res Lett 34:1–5. https://doi.org/10.1029/2007GL030692
Hess M, Koepke P, Schult I (1998) Optical properties of aerosols and clouds: the software package OPAC. Bull Am Meteorol Soc 79:831–844. https://doi.org/10.1175/1520-0477(1998)079<0831:OPOAAC>2.0.CO;2
Indian Express, 2016. https://indianexpress.com/article/india/india-news-india/uttarakhand-forest-fires-180-hectares-green-cover-2806814/. Accessed 19 Nov 2018
Izhar S, Goel A, Chakraborty A, Gupta T (2016) Annual trends in occurrence of submicron particles in ambient air and health risk posed by particle bound metals. Chemosphere 146:582–590. https://doi.org/10.1016/j.chemosphere.2015.12.039
Jacobson MZ (2000) A physically-based treatment of elemental carbon optics: Implications for global direct forcing of aerosols. Geophys Res Lett 27:217–220. https://doi.org/10.1029/1999GL010968
Jayaraman A, Gadhavi H, Ganguly D et al (2006) Spatial variations in aerosol characteristics and regional radiative forcing over India: measurements and modeling of 2004 road campaign experiment. Atmos Environ 40:6504–6515. https://doi.org/10.1016/j.atmosenv.2006.01.034
Kawamura K, Ikushima K (1993) Seasonal changes in the distribution of dicarboxylic acids in the urban atmosphere. Environ Sci Technol 27:2227–2235. https://doi.org/10.1021/es00047a033
Kawamura K, Yasui O (2005) Diurnal changes in the distribution of dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban Tokyo atmosphere. Atmos Environ 39:1945–1960. https://doi.org/10.1016/j.atmosenv.2004.12.014
Kedia S, Ramachandran S, Kumar A, Sarin MM (2010) Spatiotemporal gradients in aerosol radiative forcing and heating rate over Bay of Bengal and Arabian Sea derived on the basis of optical, physical, and chemical properties. J Geophys Res Atmos 115. https://doi.org/10.1029/2009JD013136
Kumar R, Barth MC, Pfister GG et al (2014) WRF-Chem simulations of a typical pre-monsoon dust storm in northern India: influences on aerosol optical properties and radiation budget. Atmos Chem Phys 14:2431–2446. https://doi.org/10.5194/acp-14-2431-2014
Kumar A, Singh N, Anshumali SR (2018) Evaluation and utilization of MODIS and CALIPSO aerosol retrievals over a complex terrain in Himalaya. Remote Sens Environ 206:139–155. https://doi.org/10.1016/j.rse.2017.12.019
Lawson R, Winchester JW (1979) From South American tropical rain forests. J Geophys Res 84:3723–3727
Lohmann U, Feichter J (2010) Global indirect aerosol effects: a review. Atmos Chem Phys 5:715–737. https://doi.org/10.5194/acpd-4-7561-2004
Lukács H, Gelencsér A, Hoffer A et al (2008) Quantitative assessment of organosulfates in size-segregated rural fine aerosol. Atmos Chem Phys Discuss 8:6825–6843. https://doi.org/10.5194/acpd-8-6825-2008
Mao J, Fan S, Horowitz LW (2017) Soluble Fe in aerosols sustained by gaseous HO2 uptake. Environ Sci Technol Lett 4:98–104. https://doi.org/10.1021/acs.estlett.7b00017
Montero-Martínez G, Rinaldi M, Gilardoni S et al (2014) On the water-soluble organic nitrogen concentration and mass size distribution during the fog season in the Po Valley, Italy. Sci Total Environ 485–486:103–109. https://doi.org/10.1016/j.scitotenv.2014.03.060
Myhre G, Myhre CEL, Samset BH, Storelvmo T (2013) Aerosols and their relation to global climate and climate sensitivity. In: Nature Education
Nair VS, Babu SS, Moorthy KK et al (2013) Black carbon aerosols over the Himalayas: Direct and surface albedo forcing. Tellus Ser B Chem Phys Meteorol 65:19738. https://doi.org/10.3402/tellusb.v65i0.19738
Nasir J, Zeb B, Sorooshian A et al (2019) Spatio-temporal variations of absorbing aerosols and their relationship with meteorology over four high altitude sites in glaciated region of Pakistan. J Atmos Solar-Terrestrial Phys 190:84–95. https://doi.org/10.1016/j.jastp.2019.05.010
Negi PS, Pandey CP, Singh N (2019) Black carbon aerosols in the ambient air of Gangotri Glacier valley of north-western Himalaya in India. Atmos Environ 214:116879. https://doi.org/10.1016/j.atmosenv.2019.116879
Paglione M, Kiendler-Scharr A, Mensah AA et al (2014) Identification of humic-like substances (HULIS) in oxygenated organic aerosols using NMR and AMS factor analyses and liquid chromatographic techniques. Atmos Chem Phys 14:25–45. https://doi.org/10.5194/acp-14-25-2014
Panda U, Boopathy R, Gadhavi HS et al (2021) Metals in coarse ambient aerosol as markers for source apportionment and their health risk assessment over an eastern coastal urban atmosphere in India. Environ Monit Assess 193:1–27. https://doi.org/10.1007/s10661-021-09057-3
Panwar P, Prabhu V, Soni A et al (2020) Sources and health risks of atmospheric particulate matter at Bhagwanpur, an industrial site along the Himalayan foothills. SN Appl Sci. https://doi.org/10.1007/s42452-020-2420-1
Penner JE, Andreae M, Annegarn H et al (2001) Aerosols, their direct and indirect effects. In: Climate Change 2001: The Scientific Basis Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change
Possanzini M, Buttini P, Di Palo V (1988) Characterization of a rural area in terms of dry and wet deposition. Sci Total Environ 74:111–120. https://doi.org/10.1016/0048-9697(88)90132-5
Prabhu V, Shridhar V (2019) Investigation of potential sources, transport pathway, and health risks associated with respirable suspended particulate matter in Dehradun city, situated in the foothills of the Himalayas. Atmos Pollut Res 10:187–196. https://doi.org/10.1016/j.apr.2018.07.009
Prabhu V, Prakash J, Soni A et al (2019) Atmospheric aerosols and inhalable particle number count during Diwali in Dehradun. City Environ Interact 100006. https://doi.org/10.1016/j.cacint.2019.100006
Prabhu V, Soni A, Madhwal S et al (2020) Black carbon and biomass burning associated high pollution episodes observed at Doon valley in the foothills of the Himalayas. Atmos Res:243. https://doi.org/10.1016/j.atmosres.2020.105001
Rajeev P, Kumar A, Kumar G et al (2021) Chemical characterization , source identification and health risk assessment of polycyclic aromatic hydrocarbons in ambient particulate matter over central Indo-Gangetic Plain. Urban Clim 35:100755. https://doi.org/10.1016/j.uclim.2020.100755
Ram K, Sarin MM (2011) Day-night variability of EC, OC, WSOC and inorganic ions in urban environment of Indo-Gangetic Plain: implications to secondary aerosol formation. Atmos Environ 45:460–468. https://doi.org/10.1016/j.atmosenv.2010.09.055
Ramachandran S, Kedia S (2010) Black carbon aerosols over an urban region: radiative forcing and climate impact. J Geophys Res Atmos. https://doi.org/10.1029/2009JD013560
Ramanathan V, Carmichael G (2008) Global and regional climate changes due to black carbon. Nat Geosci 1:221–227
Ramaswamy V, Boucher O, Haigh J et al (2001) Radiative forcing of climate change. In: Climate Change 2001: The Scientific Basis, Contribution of Working Group I to the Third Assessment. IPCC
Ricchiazzi P, Yang S, Gautier C, Sowle D (1998) SBDART: a research and teaching software tool for plane-parallel radiative transfer in the earth’s atmosphere. Bull Am Meteorol Soc 79:2101–2114. https://doi.org/10.1175/1520-0477(1998)079<2101:SARATS>2.0.CO;2
Saarikoski S, Carbone S, Decesari S et al (2012) Chemical characterization of springtime submicrometer aerosol in Po Valley, Italy. Atmos Chem Phys 12:8401–8421. https://doi.org/10.5194/acp-12-8401-2012
Sandrini S, Van Pinxteren D, Giulianelli L et al (2016) Size-resolved aerosol composition at an urban and a rural site in the Po Valley in summertime: implications for secondary aerosol formation. Atmos Chem Phys 16:10879–10897. https://doi.org/10.5194/acp-16-10879-2016
Satheesh SK, Srinivasan J (2006) A method to estimate aerosol radiative forcing from spectral optical depths. J Atmos Sci 63:1082–1092. https://doi.org/10.1175/JAS3663.1
Satheesh SK, Vinoj V, Krishna Moorthy K (2010) Radiative effects of aerosols at an urban location in southern India: observations versus model. Atmos Environ 44:5295–5304. https://doi.org/10.1016/j.atmosenv.2010.07.020
Saxena P, Hildemann LM (1996) Water-soluble organics in atmospheric particles: a critical review of the literature and application of thermodynamics to identify candidate compounds. J Atmos Chem 24:57–109. https://doi.org/10.1007/BF00053823
Seinfeld JH, Pandis SN (2016) Atmospheric chemistry and physics from air pollution to climate change, Second edn
Sikka DR (1997) Desert climate and its dynamics. Curr Sci 72:35–46
Soni A, Decesari S, Shridhar V et al (2019) Investigation of potential source regions of atmospheric black carbon in the data deficit region of the western Himalayas and its foothills. Atmos Pollut Res 10:1832–1842. https://doi.org/10.1016/J.APR.2019.07.015
Soni A, Kumar U, Prabhu V, Shridhar V (2020) Characterization, source apportionment and carcinogenic risk assessment of atmospheric particulate matter at Dehradun, situated in the foothills of Himalayas. J Atmos Solar-Terrestrial Phys 199:105205. https://doi.org/10.1016/j.jastp.2020.105205
Srivastava AK, Ram K, Singh S, et al (2015) Aerosol optical properties and radiative effects over Manora Peak in the Himalayan foothills: seasonal variability and role of transported aerosols. Sci Total Environ 502:287–295. https://doi.org/10.1016/j.scitotenv.2014.09.015
Stocker TF, Qin D, Plattner GK, et al (2013) Climate change 2013 the physical science basis: Working Group I contribution to the fifth assessment report of the intergovernmental panel on climate change
Sundriyal S, Shridhar V, Madhwal S et al (2018) Impacts of tourism development on the physical environment of Mussoorie, a hill station in the lower Himalayan range of India. J Mt Sci 15:2276–2291. https://doi.org/10.1007/s11629-017-4786-0
Venkataraman C, Reddy CK, Josson S, Reddy MS (2002) Aerosol size and chemical characteristics at Mumbai, India, during the INDOEX-IFP (1999). Atmos Environ 36:1979–1991. https://doi.org/10.1016/S1352-2310(02)00167-X
Wang QY, Liu JS, Wang Y, Yu HW (2015) Accumulations of copper in apple orchard soils: distribution and availability in soil aggregate fractions. J Soils Sediments 15:1075–1082. https://doi.org/10.1007/s11368-015-1065-y
Yu H, Remer LA, Chin M et al (2012) Aerosols from overseas rival domestic emissions over North America. Science. https://doi.org/10.1126/science.1217576
Yu J, Yan C, Liu Y et al (2018) Potassium: a tracer for biomass burning in Beijing? Aerosol Air Qual Res 18:2447–2459. https://doi.org/10.4209/aaqr.2017.11.0536
Zhang T, Li T, Yue X, Yang X (2017) Impacts of aerosol pollutant mitigation on lowland rice yields in China. Environ Res Lett 12:104003. https://doi.org/10.1088/1748-9326/aa80f0
Acknowledgements
The authors express sincere thanks to anonymous reviewers for critical evaluation and constructive suggestions to improve the manuscript. We appreciate Prof. Gerhard Lammel for his kind editorial handling.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
University Grant Commission (UGC) provided funding for the instruments and fellowship to Ashish Soni through UGC major project (ENVI-2013-42/419). Ashish Soni is also thankful to the Ministry of Human Resource Development, India, and Ministry of Foreign Affairs and International Cooperation, Italy, for providing scholarship for research mobility in Italy (Protocol No. 1198).
Author information
Authors and Affiliations
Contributions
AS, VS, and UK designed the manuscript. AS prepared outline of the manuscript. AS, SD, HG, and MP did the data curation; AS, SD, HG, MP, DO, and FV helped with analysis and editing; AS, SD, HG, MP, and DO finally reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: Gerhard Lammel
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 1498 kb)
Rights and permissions
About this article

Cite this article
Soni, A., Decesari, S., Gadhavi, H. et al. Chemical composition and radiative forcing of atmospheric aerosols over the high-altitude Western Himalayas of India. Environ Sci Pollut Res 29, 1961–1974 (2022). https://doi.org/10.1007/s11356-021-15609-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-021-15609-4