
Abstract
Air pollution is an important cause of non-communicable diseases globally with particulate matter (PM) as one of the main air pollutants. PM is composed of microscopic particles that contain a mixture of chemicals and biological elements that can be harmful to human health. The aerodynamic diameter of PM facilitates their deposition when inhaled. For instance, coarse PM having a diameter of < 10 μm is deposited mainly in the large conducting airways, but PM of < 2.5 μm can cross the alveolar-capillary barrier, traveling to other organs within the body. Epidemiological studies have shown the association between PM exposure and risk of disease, namely those of the respiratory system such as lung cancer, asthma, and chronic obstructive pulmonary disease (COPD). However, cardiovascular and neurological diseases have also been reported, including hypertension, atherosclerosis, acute myocardial infarction, stroke, loss of cognitive function, anxiety, and Parkinson’s and Alzheimer’s diseases. Inflammation is a common hallmark in the pathogenesis of many of these diseases associated with exposure to a variety of air pollutants, including PM. This review focuses on the main effects of PM on human health, with an emphasis on the role of inflammation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Air pollution is a public health problem many cities face worldwide. It is characterized by a conglomeration of harmful substances present in the atmosphere (World Health Organization 2013, 2016). Population growth, associated with the development of industry and an increase in fossil fuel consumption, and inappropriate agricultural activities are factors that increase the generation of pollutants in the air, affecting human health (World Health Organization 2006). Among the different types of air pollutants, particulate matter (PM) causes the most significant harmful effects (Thompson 2018).
PM is composed of solid and liquid particles produced by diesel usage, road and agricultural dust, and industrial activities that are emitted directly into the air. PM has a wide range of morphological, chemical, physical, and thermodynamic properties. According to its aerodynamic diameter, it is usually classified as PM10 (< 10 μm, coarse particles), PM2.5 (< 2.5 μm, fine particles), and PM0.1 (< 0.1 μm, ultrafine particulates) (César 2012; World Health Organization 2016; Yang et al. 2020).
PM remains suspended in the air due to its size, density, and thermic conditions, and to wind speed. These suspended particles are a complex mixture of chemical and biological elements, such as heavy metals, carbonaceous materials, persistent organic pollutants (POPs), volatile compounds, and polycyclic aromatic hydrocarbons (PAHs). PMs interact with each other through phytochemical properties which leads to complex mixtures (World Health Organization 2013). Organic compounds present in PM, such as plant pollen, spores, and microorganisms (mold and bacteria) or microbial components, for example, lipopolysaccharides (LPS), play an essential role in inflammation (Lee et al. 2015; Sayan and Mossman 2015; Uh et al. 2017).
According to the World Health Organization (WHO), 90% of individuals worldwide, especially those in urban areas, inhale air contaminated with high levels of pollutants, and they estimate that approximately 7 million people die each year due to particles in polluted air (World Health Organization 2013, 2014). Due to the genotoxic, mutagenic, and carcinogenic effects of PM, air pollution exposure is a risk factor for non-communicable diseases, including heart disease, stroke, chronic obstructive pulmonary disease (COPD), and lung cancer (Risom et al. 2005; Brook et al. 2010; Lim et al. 2012) (Fig. 1). In low-income countries, air pollution is a significant problem and is generated mainly by wood burning and the use of low-quality fossil fuels, such as diesel (World Health Organization 2013; Newell et al. 2017).

Main mechanisms of cell injury induced by PM
The WHO estimates that PM exposure causes around 1 out of 10 deaths worldwide. As a strategy to reduce the impact of PM, the WHO has set a limit for annual average outdoor air pollution of PM2.5 and PM10 at 10 and 20 μg/m3, respectively. The WHO air quality guidelines estimate that reducing the annual average of PM2.5 concentrations from 35 μg/m3 to the recommended levels of 10 μg/m3 could decrease air pollution-related deaths to approximately 15% (World Health Organization 2006, 2013). However, even in cities where PM concentrations comply with the WHO guidelines, exposure to low levels of PM still reduces life expectancy by 8.6 months (World Health Organization 2013). This review aims to detail current research related to the harmful effects of PM, with an emphasis on the inflammation.
PM contains different chemical and biological substances that can upregulate the expression of several inflammatory pathways, cytokines, and genes that cause inflammatory-related injury. PM promotes the generation of ROS which can cause DNA damage and cell death. PM also suppresses DNA repair and promotes the replication of damaged DNA fragments and, therefore, triggers carcinogenesis.
Effects of PM on health: epidemiological evidence
PM exposure can affect multiple organ systems including the respiratory, cardiovascular, and central nervous systems (Table 1).
In the respiratory system, evidence suggests a link between PM2.5 exposure and the exacerbation of pre-existing cardiopulmonary diseases leading to increased morbidity and mortality (Lee et al. 2014; Morantes-Caballero et al. 2019). For every 10 μg/m3 of daily exposure to PM2.5, studies have shown that risk of mortality increases by 1%. The number of hospital admissions for non-communicable respiratory and cardiovascular diseases has also been shown to increase with increasing concentrations of PM (Lee et al. 2014; Xu et al. 2017; Hwang et al. 2017; Zhang et al. 2019). Furthermore, in a meta-analysis conducted by Li et al., an increase of 10 μg/m3 in daily PM2.5 exposure was associated with a 3.1% increase in COPD hospitalizations and a 2.5% increase in COPD-related mortality (Li et al. 2016). Others have also reported similar findings (Tsai et al. 2013; Morantes-Caballero et al. 2019). Moreover, Hwang et al. and Morantes et al. reported that acute exacerbation of COPD admissions was significantly associated with high PM2.5 concentrations. These patients required greater use of antibiotics and corticosteroids (Hwang et al. 2017; Morantes-Caballero et al. 2019).
Data from several large population cohorts have indicated that PM induces a strong systemic inflammatory response, increasing hospital admission rates due to pulmonary diseases such as asthma (Cheng et al. 2015; Kim et al. 2019), pneumonia (Darrow et al. 2014; Croft et al. 2018), influenza (Horne et al. 2018; Croft et al. 2018), and bronchiolitis (Ségala et al. 2008; Carugno et al. 2018) by up to 4, 3, 3.9, and 7%, respectively. PM exposure may also decrease lung function (up to 5.7%) (McCreanor et al. 2007). Of note, an increase in biomarkers of neutrophilic inflammation is frequent among patients exposed to increased PM2.5 (McCreanor et al. 2007). Likewise, PM2.5 was independently associated with increased risk of intensive care unit admission due to pneumonia (Zhang et al. 2017).
A study conducted by the American Cancer Society which evaluated a large cohort of non-smokers from 1982 to 2008 found that lung cancer mortality rates increased by 15–27% for every 10 μg/m3 increase in PM2.5 concentration (Turner et al. 2011). In contrast, a decrease of 10 μg/m in PM2.5 concentrations was associated with an increase in mean life expectancy of 0.35 years (Correia et al. 2013).
Several epidemiological studies have shown that exposure to PM is associated with increased cardiovascular morbidity and mortality and that these effects are exacerbated by individuals with pre-existing compromised cardiovascular function (Kim et al. 2020). Exposure to PM2.5 even at low concentrations results in a substantial decrease in life expectancy. Chronic exposure to PM early in life is directly linked to the development of significant cardiovascular alterations, including hypertension, heart failure, ischemic heart disease, cerebrovascular disease, prolonged inflammatory and thrombotic markers, heart rhythm disturbances (i.e., ventricular tachycardia and atrial fibrillation), obesity, metabolic disorders, and increased mortality due to cardiovascular disease (Brook et al. 2010; Zhang et al. 2019). Furthermore, PM is related to an autonomic imbalance and increases blood pressure between 1 and 4 mmHg for each 10 μg/m3 of PM (Fuks et al. 2014; Chen et al. 2017). In general, there is a 10% risk of cardiovascular mortality for each increment of 10 μg/m3 in long-term PM2.5 exposure (Cai et al. 2018; Kim et al. 2020).
Current evidence suggests that an increase of 10 μg/m3 in PM2.5 and PM10 concentrations was associated with a 3.8 and 2.7% increase in the risk of atrial fibrillation incidence, respectively (Liu et al. 2018). PM2.5 is also associated with a high incidence of episodes of ventricular tachycardia and ventricular fibrillation (Folino et al. 2017). In addition, PM2.5 exposure is associated with an increased likelihood of having severe coronary atherosclerosis (Yang et al. 2019). It is also significantly associated with an increased risk of myocardial infarction (Mustafic et al. 2012; Kim et al. 2019). These results suggest that PM exposure may be a modifiable risk factor for cardiovascular disease, especially in susceptible populations which have pre-existing diseases.
Several studies have shown significant associations between PM2.5 exposure and higher odds of hypertension (Dong et al. 2013; Cai et al. 2016), not only in adults but also in children and adolescents (Zhang et al. 2019). Moreover, PM2.5 exposure in pregnancy is associated with increased probability of early-onset preeclampsia. Meta-analyses have also shown increased risks of all hypertensive disorders during pregnancy (Mobasher et al. 2013; Pedersen et al. 2014).
Infants exposed to elevated PM concentrations during early life may exhibit more rapid postnatal weight gain and reduced fetal growth if pregnant mothers are exposed to increased PM2.5 (Fleisch et al. 2015). Pregnant women in the first and second trimester have higher probability of having gestation diabetes mellitus. Higher fasting blood glucose (FBG) levels in children were associated with exposure to PM2.5 and PM10 (Choe et al. 2019; Cai et al. 2019). In addition, PM10 is associated with the development of type 1 diabetes mellitus in children less than 5 years old (Hathout et al. 2002) and is also associated with elevated triglycerides, apolipoprotein B, and hemoglobin A1c, and reduced high-density lipoprotein cholesterol (Chuang et al. 2010). PM2.5 exposure is also associated with lipoprotein increases, specifically, low-density lipoprotein cholesterol, triglycerides, apolipoprotein B, and total cholesterol (McGuinn et al. 2019). This highlights the role that PM exposure plays in the development of multiple indicators of cardiovascular risk.
In the central nervous system, PM, especially PM2.5, can cross the blood–brain barrier and produce neuroinflammation, a process which is related to the development of several cognitive alterations (Brockmeyer and D’Angiulli 2016). For instance, exposure to PM may result in delayed neurodevelopment in early childhood. During the first 6 months of life, prenatal PM exposure is associated with reduced developmental, mental, and psychomotor indices (Kim et al. 2014). Moreover, evidence consistently shows that exposure to PM causes poor age-related cognitive performance (Cipriani et al. 2018). Living in areas with high levels of PM has been linked to markers of neuroinflammation and neuropathology associated with neurodegenerative conditions such as Alzheimer’s disease–like brain pathologies (Levesque et al. 2011; Babadjouni et al. 2017).
Likewise, in a large cohort of patients older than 65 years of age, long-term exposure to PM2.5 was associated with increased incidence of Alzheimer’s disease (AD) (Jung et al. 2015). High levels of PM exposure among women > 65 years old were associated with a significant reduction in white matter on structural brain magnetic resonance imaging (MRI) (Babadjouni et al. 2017). PM is also associated with exacerbations of AD and Parkinson’s diseases (PD) (Zanobetti et al. 2014; Jung et al. 2015) which emphasizes the role that PM exposure plays in exacerbating cognitive dysfunction and enhancing progression of neurodegenerative processes underlying AD and PD.
Studies looking at MRI in children exposed to high levels of PM saw prefrontal white matter hyperintense lesions, which could be related to reduced cognitive dysfunction (Calderón-Garcidueñas et al. 2008). In addition, PM10 levels have been positively correlated with attention deficit hyperactivity disorder (ADHD) in children, and there is a significant association between PM2.5 and poor results on neurobehavioral tests (Siddique et al. 2011). In contrast, exposure to PM2.5 was associated with increased anxiety and an increased risk of depression; associations which are consistent with increased odds of medication use following high PM exposure (Pun et al. 2017; Vert et al. 2017). Collectively, this shows the functional and structural central nervous system changes documented in pediatric and elderly cohorts exposed to PM.
Inflammatory mechanisms associated with PM
Respiratory system
Depending on their size, PM particles can reach different sites within the respiratory tract. For instance, PM10 particles can enter the upper airways and trigger allergenic and irritating responses. Conversely, finer particles, such as PM2.5 and PM0.1, can enter the terminal bronchioles and alveoli, and are small enough to enter the blood via the blood–air barrier which can affect other organ systems as well (Xing et al. 2016; Yang et al. 2020).
PM particles, especially PM2.5, can damage the respiratory system by reducing airway epithelial defense and by altering the immune response. Airway epithelial cells are the first line of defense against airborne irritants, pollutants, and infectious agents that serve as a barrier, with a role in mucociliary clearance, and antimicrobial proteins and peptides secretion. However, studies have shown that PM2.5 exposure can affect mucociliary movement and increase mucus production (Zheng et al. 2013; He et al. 2017) and is associated with the downregulation of antimicrobial peptides, such as beta-defensins, creating a favorable environment for invading bacteria (Park et al. 2011; Migliaccio et al. 2013).
Another essential cell that plays a crucial role in pulmonary defense includes alveolar macrophages (AM), which aid in clearing and processing inhaled PM. Studies have reported that PM2.5 can interfere with the phenotype and function of AM in vitro and in vivo (Park et al. 2011; Migliaccio et al. 2013). When AM are exposed to PM, a Th1 type-inflammatory response ensues which leads to a massive release of cytokines (IL-12, IFN-γ). PM impairs phagocytic capacity mainly via the presence of PAHs which decrease antigen presentation, and by causing AM to switch to a Th2-dominant profile (IL-4, IL-10, and IL-13). Moreover, PM impairs the function of other important immune cells, such as polymorphonuclear neutrophils (Sigaud et al. 2007), natural killer cells (Zhao et al. 2014), and lymphocytes (Wang et al. 2017; Chen et al. 2018). Collectively, these findings demonstrate an increased risk of infection following PM exposure (Miyata and van Eeden 2011; Zheng et al. 2013).
PM has been shown to cause a significant inflammatory response in in vitro and in vivo models, initiated by AM and airway epithelial cells. These cells produce pro-inflammatory mediators after phagocytosing PM that contributes to the lung immune response, leading to oxidative stress and systemic inflammation (Yang et al. 2016; Wang et al. 2017). PM increases the production of ROS which can result in cellular and tissue damage, mainly through activation of TLR4 (Toll-like receptor-4) via DAMPs and PAMPs (Lakey et al. 2016; Münzel et al. 2018).
PM exposure can lead to cell death through several pathways which depends on the amount and time of exposure. For instance, necrosis occurs following intense exposure and is mediated by the complex Receptor-interacting protein 1 and 3 (RIP1, RIP3), proteins which increase ROS production, promote DNA damage, and induce poly-ADP-ribose-polymerase-1(PARP-1) activation and protease. In particular, it interferes with mitochondrial function by decreasing ATP levels (Peixoto et al. 2017). Conversely, moderate exposure is related to autophagy induced by stress signals that inhibit mTOR, inducing III PI3K-Beclin1 autophagic complexes (Sachdeva et al. 2019). Finally, low exposure to PM is associated with apoptosis through activation of caspase-3 by extrinsic (death ligand) and intrinsic (mitochondrial) pathways. Inside the cell, p53 expression is increased due to DNA damage, which induces—in mitochondria—the release of cytochrome-c, which triggers apoptosome formation (Pope et al. 2016) (Fig. 2).

Main effects of particulate material in the respiratory system. a Inflammation: PM activates the inflammasome complex. b Necrosis via PARP-1 and proteases activation (calpains), that interfere with mitochondrial function. c Apoptosis by intrinsic and extrinsic pathways leads to the activation of caspase-3. d Autophagy induced by stress signals—that inhibit mTOR signaling—triggers the formation of the autophagosome
Evidence from in vitro and in vivo studies have found that PM induces high levels of several inflammatory markers, such as IL-1α, IL-1β, IL-6, IL-8, IL-17, IL-18, MIP-3α, MIP-1α, MIP-1β, TNF-α, granulocyte/macrophage colony-stimulating factor (GM-CSF), and Cox-2, among others, which is likely due to the composition of these particles (Øvrevik et al. 2009; Farina et al. 2013; He et al. 2017). PM contains endotoxins or LPS, which are potent activators of the NLRP3 inflammasome (Hernández and Urcuqui 2012). The multiprotein inflammasome complex activates caspase-1, which is needed for the release of mature IL-1β and IL-18, pro-inflammatory cytokines which participate in local and systemic inflammation (Sayan and Mossman 2015; Uh et al. 2017). However, it is difficult to determine the exact mechanism of activation of the NLRP3 inflammasome, considering the varied chemical and biological composition of PM particles. PM10 activates the NLRP3 inflammasome in airway epithelial cells, recruits inflammatory cells to the airways, and upregulates heme-oxygenase-1, increasing ROS production and causing the release of TNF-α, IL-1, IL-6, and IL-8, IL-17, IL-1β, keratinocyte-derived chemokine (KC), CC chemokine ligand-20 (CCL20) and GM-CSF (Bengalli et al. 2013; Farina et al. 2013). PM2.5 also triggers the NLRP3 inflammasome and induces systemic inflammation, which is associated with increased circulating growth factors and cytokines, such as TNF-α, MCP-1, MIP-1α and MIP-1β, IP-10, IL-1β, and IL-18 (Pope et al. 2016).
Additionally, several in vivo studies have found that short-term exposure to PM2.5 induces acute lung inflammation characterized by an increase in inflammatory cells, epithelial permeability, and oxidative stress, related to an increase in the activation of NLRP3 inflammasome and P2X7 receptor in lung tissue (Muñoz-Planillo et al. 2013; Sayan and Mossman 2015; Wang et al. 2017). Morphological lesions and changes in lung tissue during short-term exposure of PM have also been reported and are characterized by alveolar collapse, neutrophilic inflammation, and an increase in TNF-α production (Xing et al. 2016; Wang et al. 2017). However, the precise details regarding the pathophysiology of PM on acute lung inflammation remain poorly understood.
PM2.5 can activate airway epithelial cells and AM to release the growth factor, TGF-β1, which stimulates fibroblast proliferation and epithelial-mesenchymal transition, a process whereby epithelial cells transition to a motile mesenchymal phenotype which is a marker of epithelial plasticity. This process is associated with an increase in pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, which perpetuates inflammation and can lead to elastin degradation and alveolar destruction (Sayan and Mossman 2015; He et al. 2017). Moreover, PM2.5 exposure is also associated with a significant reduction in lung function (McCreanor et al. 2007; He et al. 2017). Taken together, the changes in the airway produced by the PM eventually cause functional changes in the lungs, triggering long-term adverse effects like COPD (He et al. 2017; Morantes-Caballero et al. 2019).
It has been reported that PM induces a wide range of immunological effects on the respiratory system and increases susceptibility to bacterial and viral infections by increasing inflammation which can have deleterious effects on AM function. Furthermore, when PM2.5 enters the alveoli, it causes a strong inflammatory response that can exacerbates lung infections. Exposure to PM is associated with accelerated bacterial/viral replication and a more severe respiratory infection as seen with Pseudomonas aeruginosa (Chen et al. 2018; Woo et al. 2018), Staphylococcus aureus (Zhao et al. 2014), Streptococcus pneumonia (Zhou and Kobzik 2007), Mycobacterium tuberculosis (Rivas-Santiago et al. 2015), Klebsiella pneumoniae (Zheng et al. 2013), human polyomaviruses (Dolci et al. 2018), adenovirus (Fujii et al. 2003), influenza virus (Jaspers et al. 2005; Mebratu et al. 2016), respiratory syncytial virus (RSV) (Vandini et al. 2015; Mebratu et al. 2016), and rhinovirus (Capistrano et al. 2016). PM exposure has also been associated with increased respiratory symptoms, inflammation, vascular injury markers, and host susceptibility to Pneumocystis jirovecii infection in people living with human immunodeficiency virus (HIV) (Blount et al. 2013; Kim et al. 2018).
PM adversely affects viral immune responses. In a recent clinical trial which evaluated the nasal inflammatory response during influenza infection found that IFN-γ-induced protein 10 (IP-10), an essential protein in the antiviral response (Rebuli et al. 2019), was decreased following exposure to PM. Additionally, several in vitro assays have shown that PM exposure in human respiratory epithelial cells increases the susceptibility to influenza infection by promoting viral attachment and entry. Additionally, PM affects NLRP3 inflammasome and antiviral responses by affecting ROS (Jaspers et al. 2005; Hirota et al. 2015b), through depletion of glutathione in dendritic cells, which downregulates IL-12 production and increases IL-4, favoring a Th2 phenotype (Gowdy et al. 2010).
Additionally, RSV-infected mice that were exposed to PM had increased lung inflammation and morphologic changes within the airways and had reduced levels of immunomodulatory proteins, like Clara cell secretory protein (CCSP) and surfactant protein A (SP-A), proteins which help control RSV infections (Harrod et al. 2003). Likewise, in human lung fibroblasts infected with rhinovirus and exposed to air pollutants, an increase in IL-6 and IL-8 production was seen. These cytokines are known to be positively correlated with respiratory symptoms, which demonstrates that air pollutants can modulate virus-induced inflammation during infection and can alter disease severity (Capistrano et al. 2016). These findings show that there may be a possible synergistic effect between PM and respiratory viral infections which can exacerbate inflammation.
Cardiovascular system
The main pathophysiological mechanisms linked to PM exposure in the cardiovascular system are systemic inflammation, impaired coagulation, impaired autonomic responses, and alteration in vascular cells, which collectively increase the risk of cardiovascular disease (Fig. 3). However, the specific mechanisms by which PM exposures promote cardiovascular disease are not yet entirely understood (Lee et al. 2014; Kim et al. 2020).

Effects of PM on the cardiovascular system. PM may cross the lung-blood barrier and reach into the circulation through two routes: (1) after ingestion by alveolar macrophages, but this way is prolonged, and the new evidence suggests that (2) it can cross the barrier by itself, although this depends by several factors including particle size, charge, and chemical composition. Once in the circulation, PM might interact with the vascular endothelium or have direct effects on atherosclerotic plaques, it might cause local oxidative stress and systemic inflammatory outputs. PM also has the potential for altering hemostasis and cardiovascular integrity
According to the American Heart Association, the inflammatory response to PM increases acute phase reactants (i.e., C-reactive protein, fibrinogen, and D-dimer), blood coagulability, and endothelium dysfunction in healthy adults. This systemic response has been linked to cytokines, such as TNF- α, GM-CSF, IL-1β, MCP-1, IL-8, MIP-1α/MIP-1β, and IL-6 (Brook et al. 2010). These pro-inflammatory mediators may contribute to acute exacerbation of cardiovascular diseases due to the destabilization of atherosclerotic plaques, increasing their likelihood for rupture and thrombosis during the inflammatory response (Pope et al. 2016). Moreover, PM exposure and inflammatory mediators activate circulating leukocytes and the vascular endothelium and promote cell adhesion and migration, increasing vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), contributing to atherosclerogenesis (Suwa et al. 2002).
The NLRP3 inflammasome, and its effect on IL-1β, plays an essential role in systemic inflammation regulation. Different stimuli associated with PM exposure, such as the presence of LPS, PAHs, K+ Efflux, ROS, lysosomal damage, and TLR4 stimulation, among others, can activate the inflammasome (Bengalli et al. 2013; Muñoz-Planillo et al. 2013; Ferguson et al. 2013; Hirota et al. 2015a; Uh et al. 2017; Kelley et al. 2019). In a murine model, PM was shown to activate the NLRP3 inflammasome through a caspase-1 independent pathway (Provoost et al. 2011). Regardless of the mechanisms by which NLRP3 is activated, pro-inflammatory products derived from the inflammasome are essential in cardiovascular disease.
Exposure to PM also results in impaired endogenous fibrinolysis and increased thrombus formation. PM increases coagulation by reducing the activated partial thromboplastin time (aPTT) and the prothrombin time (PT) (Pope et al. 2016). Likewise, in vivo assays showed that mice exposed to PM exhibited a TNF-α-dependent increase in plasminogen activator inhibitor-1 (PAI-1) and an IL-6-dependent activation of coagulation (Mutlu et al. 2007). PAI-1 inhibits urokinase-type plasminogen activator and tissue-type plasminogen activator, which play an important role in the activation of plasmin from plasminogen (Budinger et al. 2011). Elevated levels of PAI-1 have been recognized as a risk factor for the development of ischemic events (Budinger et al. 2011). Mice exposed to PM also displayed increased production of IL-6 and tissue factor and had increased levels of thrombin–antithrombin complex in their plasma. IL-6, produced by AM following PM exposure, has been associated with increased generation of intravascular thrombin and acceleration of arterial thrombosis through increased expression of fibrinogen, factor VIII, and von Willebrand factor, and increased activity of factor II and X. IL-6 also reduces the transcription of thrombosis inhibitors including antithrombin and protein S while promoting activation of STAT3 in bone marrow progenitor cells, resulting in neutrophilia and thrombocytosis (Jenkins et al. 2007; Mutlu et al. 2007; Münzel et al. 2018).
PM2.5 exposure induces systemic microvascular dysfunction, arteriolar constrictions, and significantly reduces vasodilation (Nurkiewicz et al. 2008). Additionally, PM2.5 exposure potentiates hypertension by increasing angiotensin II concentrations, which promotes cardiac remodeling through an RhoA/Rho kinase-dependent mechanism, inducing cardiac hypertrophy and collagen deposition (Ying et al. 2009). The biological components of PM, namely LPS and β-1,3-D-Glucan, are associated with increased blood pressure and vascular endothelial growth factor (VEGF) production. In healthy adults exposed to PM, an increase in VEGF in response to acute endothelial injury was observed (Zhong et al. 2015b). VEGF is a potent angiogenic growth factor that induces endothelial proliferation and capillary formation and plays an essential role in the regulation of angiogenesis in heart disease and tumors (Sun et al. 2018b).
Both short-term and chronic PM2.5 exposure is associated with increased levels of circulating ET-1, elevated mean pulmonary arterial pressure and arterial vasoconstriction (Calderón-Garcidueñas et al. 2007b; Langrish et al. 2009). PM also increases vascular sensitivity to ET-1 (Langrish et al. 2009). The perturbation in vascular homeostatsis is exacerbated by reduced acetylcholine-induced dilation in coronary arteries in a nitric oxide synthase (NOS)-dependent manner. PM induces endothelial NOS dysfunction in coronary arterioles that are associated with increased ROS, limiting the availability of BH4, an essential cofactor for NOS (Cherng et al. 2011).
Furthermore, PM2.5 exposure causes epigenetic alterations such as abnormal DNA methylation patterns, which alters gene expression, and activates inflammatory and vascular responses. Recent evidence suggests that PM reduces TLR4-methylation, which was associated with a significant increase in blood pressure. Experimental models have shown that TLR4, which is activated by PM-associated LPS, contributes to inflammation, oxidative stress, and cardiovascular responses, thereby promoting the release of thromboxane A2, a potent vasoconstrictor (Bellavia et al. 2013). PM also decreases methylation of the long-interspersed nucleotide element-1 (LINE-1) and the short-interspersed nucleotide element Alu and NOS genes, and increases TLR2 methylation (Baccarelli et al. 2009). Alu and LINE-1 sequences are present in several genes related to cardiovascular diseases, and their hypomethylation has been linked to ischemic heart disease, stroke, and hypertension (Muka et al. 2016; Sun et al. 2018a). Low NOS gene methylation also associated with higher endogenous thrombin potential and could be associated with alterations in vascular tone (Tarantini et al. 2009; Cherng et al. 2011). Similarly, PM2.5 exposure is associated with high TLR2 methylation, which suppresses TLR2 expression and increases the individual’s susceptibility to adverse cardiac effects (Zhong et al. 2015a).
Although the specific mechanisms by which PM exposure promotes cardiovascular events are not entirely understood, PM2.5 adversely affects cardiovascular health increasing the production of pro-inflammatory cytokines, growth factors, cell adhesion molecules, causing epigenetic alterations leading to microvascular dysfunction, endothelial injury, impaired coagulation, and altering the autonomic response. These alterations might potentially initiate and promote atherosclerotic lesions and trigger cardiovascular and cerebrovascular events.
Central nervous system
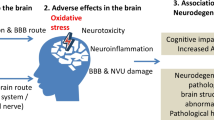
There are several mechanisms by which PM are believed to affect the central nervous system (CNS); the most accepted theories indicate that the inflammatory response to direct contact of PM, and the CNS and systemic inflammation can affect different brain structures (Cipriani et al. 2018). After PM reaches the circulatory system, it generates a systemic inflammatory response, negatively affecting the CNS. This response is evidenced by the presence of pro-inflammatory mediators in blood and cerebrospinal fluid (Brockmeyer and D’Angiulli 2016) which contributes to neuroinflammation and loss of neuronal tissue in diffuse areas of the brain, such as the olfactory bulb, frontal cortex, and hippocampus. One of the main mechanisms by which inflammation causes damage to the brain is through the disruption of the blood–brain barrier which permits monocyte infiltration, activation of microglia, and cytokine activity (Calderón-Garcidueñas et al. 2008, 2012) (Fig. 4). PM also induces brain inflammation in the olfactory system because PM can deposit in the olfactory epithelium and filter soluble metals and toxins into the nasal mucosa which can then be transported into the olfactory bulbs. Once inside, toxins can trigger the local immune response, or infiltrate upstream brain regions causing inflammation (Ljubimova et al. 2018).

Summary of the main mechanism of neuroinflammation produced by particulate material. PM might translocate to the brain parenchyma through olfactory nerve or translocate from systemic circulation and produce inflammation, with it is a consequent disruption of the blood–brain barrier; this increases monocyte infiltration, activation of microglia, and ROS production. Also, there are indirect pathways that trigger neuroinflammation, such as pro-inflammatory cytokines from the nasal epithelium, which are transferred to the brain, and systemic circulating cytokines released after pulmonary and endothelium damage. Similarly, the circulating stress hormones may regulate the brain’s pro-inflammatory environment
PM may enter the body through the lungs, and it has been demonstrated that a portion of PM translocate to the blood from the alveoli reaching extrapulmonary sources, including the blood–brain barrier and the brain parenchyma, leading to inflammation (Cipriani et al. 2018) (Fig. 3).
Air pollution, especially PM, can affect the CNS through a variety of molecular, cellular, and inflammatory pathways that can injure brain structures and lead to neurological diseases (Babadjouni et al. 2017). Studies which evaluated the presence of demyelinated neurons or white matter hyperintensities through the use of neuroimaging like MRI found that these brain structure alterations were related to PM2.5 exposure and were mainly associated with inflammatory mechanisms, i.e., the presence of TNF-α, COX2, and IL-1β expression (Calderón-Garcidueñas et al. 2007a, 2008; Chen et al. 2015).
Neural damage contributes to cognitive dysfunction. Moreover, PM2.5 is associated with lower total cerebral brain volume, a marker of age-associated brain atrophy, and with higher probabilities of brain infarcts, which are associated with high levels of ET-1 and results in a negative impact on the neuronal and circulatory architecture of the brain. It also increases blood pressure, affecting brain function (Calderón-Garcidueñas et al. 2007b). Although initially these changes appear asymptomatic, small infarcts, typically located deep within the brain, have been associated with neurological abnormalities, more reduced cognitive function, and the onset of dementia which are thought to reflect small vessel disease (Cipriani et al. 2018).
Protein characteristics of Alzheimer’s disease (i.e., amyloid peptide (Aβ42), alpha-synuclein, hyperphosphorylated tau, and the downregulation of the prion-related protein [PrP(C)]), which have a vital role in neuroprotection, neurodegeneration, and mood disorder states, are associated with PM exposure (Levesque et al. 2011; Hullmann et al. 2017). Children exposed to high levels of air pollution have been found to be APOE4 (a well-known genetic risk factor for Alzheimer’s disease) carriers and had higher hyperphosphorylated tau and amyloid peptides compared to APOE3 carriers (Calderón-Garcidueñas et al. 2012). Additionally, PM2.5 impairs synaptic and cognitive function and is associated with a role in β-secretase (BACE1)-related induction of NF-κB signaling-mediated miR-574-5p downregulation. BACE1 is an essential enzyme for amyloid β peptide synthesis, which is involved in Alzheimer’s disease (Ku et al. 2017). This finding suggests that genetic and post-transcriptional factors may play a role in people exposed to PM, by increasing the risk of Alzheimer’s disease, and contributing to rising neuroinflammation and loss of communication between neurons.
One of the mechanisms of immune defense in the CNS is the activation of microglia, innate immune cells which reside in the brain. Microglial cells respond to several types of stimuli, including disease markers, environmental toxins, and pro-inflammatory signals, to regulate the inflammatory environment within the brain However, when overstimulated, microglial activation can lead to an increase in TNF-α, IL-1β, IFN-γ, and ROS production, which when left uncontrolled can be neurotoxic (Roqué et al. 2016; Allen et al. 2017). PM activates microglia via the MAC1 receptor which, in turn, causes ROS production through the NOX2 pathway (Levesque et al. 2013). Microglia can also internalize PM through scavenger receptors resulting in an increase in glutamate, an excitatory neurotransmitter that, at high extracellular levels, induces neuronal injury (Allen et al. 2017). Likewise, PM2.5 exposure significantly reduced murine cognitive learning abilities and increased the presence of glutamate and other toxic substances (i.e., lead, manganese, and aluminum) in hippocampal tissues. Moreover, PM2.5 led to a decrease in N-methyl-d-aspartate glutamate receptor (NMDAR) expression while increasing metabotropic glutamate receptor type 1 (mGluR1) expression, causing excitatory neurotoxicity through the generation of glutaminase-mediated glutamate, as well as neuronal swelling and necrosis (Li et al. 2018). The neuronal and white matter damage produced by glutamate is associated with the pathogenesis of PD and AD and other neuronal diseases (Liu et al. 2015). PM2.5 also induces astrocyte activation and alters striatal dopaminergic transmission by decreasing dopamine D2-like receptors (D2Rs) (de los Andrade-Oliva et al. 2018). Dopamine and glutamate are overlapping neurotransmitter systems critical for cognitive function mediation (Allen et al. 2017).
PM might also induce cognitive impairment by dysregulating FBG function, an independent risk factor for dementia and cardiovascular and metabolic diseases. PM exposure is associated with elevated FBG levels in non-diabetic individuals. The evidence suggests that PM2.5 exposure upregulates inflammatory gene expression, for example, ICAM-1, increasing CNS inflammation (Peng et al. 2016). Regions of the brain associated with behavior and cognitive performance are affected by neuroinflammation, endothelial barrier changes, blood vessel rearrangement, and blood–brain barrier permeability (Brockmeyer and D’Angiulli 2016). Overall, these findings suggest that PM exposure increases inflammation and can have deleterious effects on myelination contributing to neuroinflammation and cell loss.
A recent clinical trial suggested that the CNS reacts to changes in PM exposure through activation of the hypothalamus–pituitary–adrenal (HPA) axis, evidenced by increased HPA axis hormones (ACTH, CRH, and cortisol and cortisone). In addition, catecholamines (i.e., norepinephrine and epinephrine), released by the sympathetic–adrenal–medullary axis, also increase in the presence of PM (Li et al. 2017). These hormones and pro-inflammatory mediators have a systemic effect—increasing the risk of developing respiratory, cardiovascular, metabolic, and neurological sequelae, highlighting the complex pathophysiology of PM exposure.
Conclusion
Evidence suggests that inflammation contributes to the pathogenesis of cardiovascular and respiratory diseases, which are attributable to PM exposure. However, PM may negatively affect other structures as well, such as the CNS, and contribute to adverse neurological outcomes. Research has elucidated the countless risk factors and mechanisms of harm associated with PM exposure. Implementation of appropriate interventions and the establishment of conventions and protocols for the prevention and control of air pollution has become a critical world health priority. The significant consequences of PM exposure on human health require the creation and implementation of more environmental and social policies in order to reduce exposure and minimize risks associated with air pollutants. Further studies are required to provide more understanding on how best to reduce the adverse effects of PM exposure on health.
References
Allen JL, Klocke C, Morris-Schaffer K, Conrad K, Sobolewski M, Cory-Slechta DA (2017) Cognitive effects of air pollution exposures and potential mechanistic underpinnings. Curr Environ Health Rep 4:180–191. https://doi.org/10.1007/s40572-017-0134-3
Babadjouni RM, Hodis DM, Radwanski R, Durazo R, Patel A, Liu Q, Mack WJ (2017) Clinical effects of air pollution on the central nervous system; a review. J Clin Neurosci 43:16–24. https://doi.org/10.1016/j.jocn.2017.04.028
Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J (2009) Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med 179:572–578. https://doi.org/10.1164/rccm.200807-1097OC
Bellavia A, Urch B, Speck M, Brook RD, Scott JA, Albetti B, Behbod B, North M, Valeri L, Bertazzi PA, Silverman F, Gold D, A. Baccarelli A (2013) DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc 2:e000212. https://doi.org/10.1161/JAHA.113.000212
Bengalli R, Molteni E, Longhin E, Refsnes M, Camatini M, Gualtieri M (2013) Release of IL-1 β triggered by Milan summer PM10: molecular pathways involved in the cytokine release. Biomed Res Int 2013:158093–158099. https://doi.org/10.1155/2013/158093
Blount RJ, Djawe K, Daly KR, Jarlsberg LG, Fong S, Balmes J, Miller RF, Walzer PD, Huang L, on behalf of the International HIV-associated Opportunistic Pneumonias (IHOP) Study (2013) Ambient air pollution associated with suppressed serologic responses to Pneumocystis jirovecii in a prospective cohort of HIV-infected patients with Pneumocystis pneumonia. PLoS ONE:8. https://doi.org/10.1371/journal.pone.0080795
Brockmeyer S, D’Angiulli A (2016) How air pollution alters brain development: the role of neuroinflammation. Transl Neurosci 7:24–30. https://doi.org/10.1515/tnsci-2016-0005
Brook RD, Rajagopalan S, Pope CA et al (2010) Particulate matter air pollution and cardiovascular disease. Circulation 121:2331–2378. https://doi.org/10.1161/CIR.0b013e3181dbece1
Budinger GRS, McKell JL, Urich D et al (2011) Particulate matter-induced lung inflammation increases systemic levels of PAI-1 and activates coagulation through distinct mechanisms. PLoS One 6:e18525. https://doi.org/10.1371/journal.pone.0018525
Cai Y, Zhang B, Ke W, Feng B, Lin H, Xiao J, Zeng W, Li X, Tao J, Yang Z, Ma W, Liu T (2016) Associations of short-term and long-term exposure to ambient air pollutants with hypertension: a systematic review and meta-analysis. Hypertension 68:62–70. https://doi.org/10.1161/HYPERTENSIONAHA.116.07218
Cai J, Yu S, Pei Y, Peng C, Liao Y, Liu N, Ji J, Cheng J (2018) Association between airborne fine particulate matter and residents’ cardiovascular diseases, ischemic heart disease and cerebral vascular disease mortality in areas with lighter air pollution in China. Int J Environ Res Public Health 15:1918. https://doi.org/10.3390/ijerph15091918
Cai L, Wang S, Gao P, Shen X, Jalaludin B, Bloom MS, Wang Q, Bao J, Zeng X, Gui Z, Chen Y, Huang C (2019) Effects of ambient particulate matter on fasting blood glucose among primary school children in Guangzhou, China. Environ Res 176:108541. https://doi.org/10.1016/j.envres.2019.108541
Calderón-Garcidueñas L, Reed W, Maronpot RR, Henriquez-Roldán C, Delgado-Chavez R, Calderón-Garcidueñas A, Dragustinovis I, Franco-Lira M, Aragón-Flores M, Solt AC, Altenburg M, Torres-Jardón R, Swenberg JA (2004) Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol 32:650–658. https://doi.org/10.1080/01926230490520232
Calderón-Garcidueñas L, Franco-Lira M, Torres-Jardón R, Henriquez-Roldán C, Barragán-Mejía G, Valencia-Salazar G, Gonzaléz-Maciel A, Reynoso-Robles R, Villarreal-Calderón R, Reed W (2007a) Pediatric respiratory and systemic effects of chronic air pollution exposure: nose, lung, heart, and brain pathology. Toxicol Pathol 35:154–162. https://doi.org/10.1080/01926230601059985
Calderón-Garcidueñas L, Vincent R, Mora-Tiscareño A, Franco-Lira M, Henríquez-Roldán C, Barragán-Mejía G, Garrido-García L, Camacho-Reyes L, Valencia-Salazar G, Paredes R, Romero L, Osnaya H, Villarreal-Calderón R, Torres-Jardón R, Hazucha MJ, Reed W (2007b) Elevated plasma endothelin-1 and pulmonary arterial pressure in children exposed to air pollution. Environ Health Perspect 115:1248–1253. https://doi.org/10.1289/ehp.9641
Calderón-Garcidueñas L, Mora-Tiscareño A, Ontiveros E, Gómez-Garza G, Barragán-Mejía G, Broadway J, Chapman S, Valencia-Salazar G, Jewells V, Maronpot RR, Henríquez-Roldán C, Pérez-Guillé B, Torres-Jardón R, Herrit L, Brooks D, Osnaya-Brizuela N, Monroy ME, González-Maciel A, Reynoso-Robles R, Villarreal-Calderon R, Solt AC, Engle RW (2008) Air pollution, cognitive deficits and brain abnormalities: a pilot study with children and dogs. Brain Cogn 68:117–127. https://doi.org/10.1016/j.bandc.2008.04.008
Calderón-Garcidueñas L, Kavanaugh M, Block M, D'Angiulli A, Delgado-Chávez R, Torres-Jardón R, González-Maciel A, Reynoso-Robles R, Osnaya N, Villarreal-Calderon R, Guo R, Hua Z, Zhu H, Perry G, Diaz P (2012) Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J Alzheimers Dis 28:93–107. https://doi.org/10.3233/JAD-2011-110722
Capistrano S, Zakarya R, Chen H, Oliver B (2016) Biomass smoke exposure enhances rhinovirus-induced inflammation in primary lung fibroblasts. Int J Mol Sci 17:1403. https://doi.org/10.3390/ijms17091403
Carugno M, Dentali F, Mathieu G, Fontanella A, Mariani J, Bordini L, Milani GP, Consonni D, Bonzini M, Bollati V, Pesatori AC (2018) PM10 exposure is associated with increased hospitalizations for respiratory syncytial virus bronchiolitis among infants in Lombardy, Italy. Environ Res 166:452–457. https://doi.org/10.1016/j.envres.2018.06.016
César AA (2012) Diagnosis and control of particulate matter: total suspended particles PM10 breathable fraction. Luna Azul 195–213
Chen JC, Wang X, Wellenius GA, Serre ML, Driscoll I, Casanova R, McArdle JJ, Manson JAE, Chui HC, Espeland MA (2015) Ambient air pollution and neurotoxicity on brain structure: evidence from women’s health initiative memory study. Ann Neurol 78:466–476. https://doi.org/10.1002/ana.24460
Chen S-Y, Chan C-C, Su T-C (2017) Particulate and gaseous pollutants on inflammation, thrombosis, and autonomic imbalance in subjects at risk for cardiovascular disease. Environ Pollut 223:403–408. https://doi.org/10.1016/j.envpol.2017.01.037
Chen X, Liu J, Zhou J, Wang J, Chen C, Song Y, Pan J (2018) Urban particulate matter (PM) suppresses airway antibacterial defence 19:5. https://doi.org/10.1186/s12931-017-0700-0
Cheng M-H, Chiu H-F, Yang C-Y (2015) Coarse particulate air pollution associated with increased risk of hospital admissions for respiratory diseases in a Tropical City, Kaohsiung, Taiwan. Int J Environ Res Public Health 12:13053–13068. https://doi.org/10.3390/ijerph121013053
Cheng H, Saffari A, Sioutas C, Forman HJ, Morgan TE, Finch CE (2016) Nanoscale particulate matter from urban traffic rapidly induces oxidative stress and inflammation in olfactory epithelium with concomitant effects on brain. Environ Health Perspect 124:1537–1546. https://doi.org/10.1289/EHP134
Cherng TW, Paffett ML, Jackson-Weaver O, Campen MJ, Walker BR, Kanagy NL (2011) Mechanisms of diesel-induced endothelial nitric oxide synthase dysfunction in coronary arterioles. Environ Health Perspect 119:98–103. https://doi.org/10.1289/ehp.1002286
Choe SA, Eliot MN, Savitz DA, Wellenius GA (2019) Ambient air pollution during pregnancy and risk of gestational diabetes in New York City. Environ Res 175:414–420. https://doi.org/10.1016/j.envres.2019.04.030
Chuang KJ, Yan YH, Cheng TJ (2010) Effect of air pollution on blood pressure, blood lipids, and blood sugar: a population-based approach. J Occup Environ Med 52:258–262. https://doi.org/10.1097/JOM.0b013e3181ceff7a
Cipriani G, Danti S, Carlesi C, Borin G (2018) Danger in the air: air pollution and cognitive dysfunction. Am J Alzheimers Dis Dementias 33:333–341. https://doi.org/10.1177/1533317518777859
Correia AW, Pope CA III, Dockery DW, Wang Y, Ezzati M, Dominici F (2013) The effect of air pollution control on life expectancy in the United States: an analysis of 545 US counties for the period 2000 to 2007. Epidemiology Camb Mass 24:23–31. https://doi.org/10.1097/EDE.0B013E3182770237
Croft DP, Zhang W, Lin S et al (2018) The association between respiratory infection and air pollution in the setting of air quality policy and economic change. Ann Am Thorac Soc 16:AnnalsATS.201810-691OC. https://doi.org/10.1513/AnnalsATS.201810-691OC
Darrow LA, Klein M, Flanders WD, Mulholland JA, Tolbert PE, Strickland MJ (2014) Air pollution and acute respiratory infections among children 0-4 years of age: an 18-year time-series study. Am J Epidemiol 180:968–977. https://doi.org/10.1093/aje/kwu234
de los Andrade-Oliva MA, Aztatzi-Aguilar OG, García-Sierra F et al (2018) Effect of in vivo exposure to ambient fine particles (PM2.5) on the density of dopamine D2-like receptors and dopamine-induced [35S]-GTPγS binding in rat prefrontal cortex and striatum membranes. Environ Toxicol Pharmacol 60:58–65. https://doi.org/10.1016/j.etap.2018.04.001
Dolci M, Favero C, Bollati V, Campo L, Cattaneo A, Bonzini M, Villani S, Ticozzi R, Ferrante P, Delbue S (2018) Particulate matter exposure increases JC polyomavirus replication in the human host. Environ Pollut 241:234–239. https://doi.org/10.1016/j.envpol.2018.05.044
Dong G-H, Min QZ, Xaverius PK et al (2013) Association between long-term air pollution and increased blood pressure and hypertension in China. Hypertension 61:578–584. https://doi.org/10.1161/HYPERTENSIONAHA.111.00003
Dvonch JT, Kannan S, Schulz AJ, Keeler GJ, Mentz G, House J, Benjamin A, Max P, Bard RL, Brook RD (2009) Acute effects of ambient particulate matter on blood pressure: differential effects across urban communities. Hypertens Dallas Tex 1979 53:853–859. https://doi.org/10.1161/HYPERTENSIONAHA.108.123877
Farina F, Sancini G, Battaglia C, Tinaglia V, Mantecca P, Camatini M, Palestini P (2013) Milano summer particulate matter (PM10) triggers lung inflammation and extra pulmonary adverse events in mice. PLoS One 8:e56636. https://doi.org/10.1371/journal.pone.0056636
Ferguson MD, Migliaccio C, Ward T (2013) Comparison of how ambient PMc and PM2.5 influence the inflammatory potential. Inhal Toxicol 25:766–773. https://doi.org/10.3109/08958378.2013.847993
Finch J, Conklin DJ (2016) Air pollution-induced vascular dysfunction: potential role of endothelin-1 (ET-1) system. Cardiovasc Toxicol 16:260–275. https://doi.org/10.1007/s12012-015-9334-y
Fleisch AF, Rifas-Shiman SL, Koutrakis P, Schwartz JD, Kloog I, Melly S, Coull BA, Zanobetti A, Gillman MW, Gold DR, Oken E (2015) Prenatal exposure to traffic pollution: associations with reduced fetal growth and rapid infant weight gain. Epidemiology 26:43–50. https://doi.org/10.1097/EDE.0000000000000203
Folino F, Buja G, Zanotto G, Marras E, Allocca G, Vaccari D, Gasparini G, Bertaglia E, Zoppo F, Calzolari V, Suh RN, Ignatiuk B, Lanera C, Benassi A, Gregori D, Iliceto S (2017) Association between air pollution and ventricular arrhythmias in high-risk patients (ARIA study): a multicentre longitudinal study. Lancet Planet Health 1:e58–e64. https://doi.org/10.1016/S2542-5196(17)30020-7
Fujii T, Hogg JC, Keicho N et al (2003) Adenoviral E1A modulates inflammatory mediator expression by lung epithelial cells exposed to PM10. Am J Physiol Lung Cell Mol Physiol 284. https://doi.org/10.1152/ajplung.00197.2002
Fuks KB, Weinmayr G, Foraster M, Dratva J, Hampel R, Houthuijs D, Oftedal B, Oudin A, Panasevich S, Penell J, Sommar JN, Sørensen M, Tiittanen P, Wolf K, Xun WW, Aguilera I, Basagaña X, Beelen R, Bots ML, Brunekreef B, Bueno-de-Mesquita HB, Caracciolo B, Cirach M, de Faire U, de Nazelle A, Eeftens M, Elosua R, Erbel R, Forsberg B, Fratiglioni L, Gaspoz JM, Hilding A, Jula A, Korek M, Krämer U, Künzli N, Lanki T, Leander K, Magnusson PKE, Marrugat J, Nieuwenhuijsen MJ, Östenson CG, Pedersen NL, Pershagen G, Phuleria HC, Probst-Hensch NM, Raaschou-Nielsen O, Schaffner E, Schikowski T, Schindler C, Schwarze PE, Søgaard AJ, Sugiri D, Swart WJR, Tsai MY, Turunen AW, Vineis P, Peters A, Hoffmann B (2014) Arterial blood pressure and long-term exposure to traffic-related air pollution: an analysis in the European Study of Cohorts for Air Pollution Effects (ESCAPE). Environ Health Perspect 122:896–905. https://doi.org/10.1289/ehp.1307725
Gowdy KM, Krantz QT, King C, Boykin E, Jaspers I, Linak WP, Gilmour MI (2010) Role of oxidative stress on diesel-enhanced influenza infection in mice. Part Fibre Toxicol 7:34. https://doi.org/10.1186/1743-8977-7-34
Harris MH, Gold DR, Rifas-Shiman SL, Melly SJ, Zanobetti A, Coull BA, Schwartz JD, Gryparis A, Kloog I, Koutrakis P, Bellinger DC, Belfort MB, Webster TF, White RF, Sagiv SK, Oken E (2016) Prenatal and childhood traffic-related air pollution exposure and childhood executive function and behavior. Neurotoxicol Teratol 57:60–70. https://doi.org/10.1016/j.ntt.2016.06.008
Harrod KS, Jaramillo RJ, Rosenberger CL, Wang SZ, Berger JA, McDonald JD, Reed MD (2003) Increased susceptibility to RSV infection by exposure to inhaled diesel engine emissions. Am J Respir Cell Mol Biol 28:451–463. https://doi.org/10.1165/rcmb.2002-0100OC
Hathout EH, Beeson WL, Nahab F, Rabadi A, Thomas W, Mace JW (2002) Role of exposure to air pollutants in the development of type 1 diabetes before and after 5 yr of age. Pediatr Diabetes 3:184–188. https://doi.org/10.1034/j.1399-5448.2002.30403.x
He F, Liao B, Pu J, Li C, Zheng M, Huang L, Zhou Y, Zhao D, Li B, Ran P (2017) Exposure to ambient particulate matter induced COPD in a rat model and a description of the underlying mechanism. Sci Rep 7:45666. https://doi.org/10.1038/srep45666
Hernández J, Urcuqui S (2012) Activación y regulación del inflamasoma NLRP3 en las enfermedades infecciosas. Iatreia 25:380–390
Hiraiwa K, van Eeden SF (2013) Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat Inflamm 2013:619523–619510. https://doi.org/10.1155/2013/619523
Hirota JA, Gold MJ, Hiebert PR, Parkinson LG, Wee T, Smith D, Hansbro PM, Carlsten C, VanEeden S, Sin DD, McNagny KM, Knight DA (2015a) The nucleotide-binding domain, leucine-rich repeat protein 3 inflammasome/IL-1 receptor I axis mediates innate, but not adaptive, immune responses after exposure to particulate matter under 10 μm. Am J Respir Cell Mol Biol 52:96–105. https://doi.org/10.1165/rcmb.2014-0158OC
Hirota JA, Marchant DJ, Singhera GK, Moheimani F, Dorscheid DR, Carlsten C, Sin D, Knight D (2015b) Urban particulate matter increases human airway epithelial cell IL-1β secretion following scratch wounding and H1N1 influenza a exposure in vitro. Exp Lung Res 41:353–362. https://doi.org/10.3109/01902148.2015.1040528
Horne BD, Joy EA, Hofmann MG, Gesteland PH, Cannon JB, Lefler JS, Blagev DP, Korgenski EK, Torosyan N, Hansen GI, Kartchner D, Pope CA III (2018) Short-term elevation of fine particulate matter air pollution and acute lower respiratory infection. Am J Respir Crit Care Med 198:759–766. https://doi.org/10.1164/rccm.201709-1883OC
Hullmann M, Albrecht C, van Berlo D, Gerlofs-Nijland ME, Wahle T, Boots AW, Krutmann J, Cassee FR, Bayer TA, Schins RPF (2017) Diesel engine exhaust accelerates plaque formation in a mouse model of Alzheimer’s disease. Part Fibre Toxicol 14:35. https://doi.org/10.1186/s12989-017-0213-5
Hwang S-L, Lin Y-C, Guo S-E, Chou CT, Lin CM, Chi MC (2017) Fine particulate matter on hospital admissions for acute exacerbation of chronic obstructive pulmonary disease in southwestern Taiwan during 2006–2012. Int J Environ Health Res 27:95–105. https://doi.org/10.1080/09603123.2017.1278748
Jaspers I, Ciencewicki JM, Zhang W, Brighton LE, Carson JL, Beck MA, Madden MC (2005) Diesel exhaust enhances influenza virus infections in respiratory epithelial cells. Toxicol Sci 85:990–1002. https://doi.org/10.1093/toxsci/kfi141
Jenkins BJ, Roberts AW, Greenhill CJ, Najdovska M, Lundgren-May T, Robb L, Grail D, Ernst M (2007) Pathologic consequences of STAT3 hyperactivation by IL-6 and IL-11 during hematopoiesis and lymphopoiesis. Blood 109:2380–2388. https://doi.org/10.1182/blood-2006-08-040352
Jung C-R, Lin Y-T, Hwang B-F (2015) Ozone, particulate matter, and newly diagnosed Alzheimer’s disease: a population-based cohort study in Taiwan. J Alzheimers Dis 44:573–584. https://doi.org/10.3233/JAD-140855
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20
Kim E, Park H, Hong Y-C, Ha M, Kim Y, Kim BN, Kim Y, Roh YM, Lee BE, Ryu JM, Kim BM, Ha EH (2014) Prenatal exposure to PM10 and NO2 and children’s neurodevelopment from birth to 24months of age: mothers and Children’s Environmental Health (MOCEH) study. Sci Total Environ 481:439–445. https://doi.org/10.1016/j.scitotenv.2014.01.107
Kim C, Jary H, Mortimer K, Schweitzer KS, Curran-Everett D, Gordon S, Petrache I (2018) Effects of household air pollution in Malawi and human immunodeficiency virus status on respiratory symptoms and inflammation, injury, and repair markers. Ann Am Thorac Soc 15:S132–S133. https://doi.org/10.1513/AnnalsATS.201707-614MG
Kim JS, Chen Z, Alderete TL, Toledo-Corral C, Lurmann F, Berhane K, Gilliland FD (2019) Associations of air pollution, obesity and cardiometabolic health in young adults: the meta-AIR study. Environ Int 133:105180. https://doi.org/10.1016/j.envint.2019.105180
Kim JB, Prunicki M, Haddad F, Dant C, Sampath V, Patel R, Smith E, Akdis C, Balmes J, Snyder MP, Wu JC, Nadeau KC (2020) Cumulative lifetime burden of cardiovascular disease from early exposure to air pollution. J Am Heart Assoc 9:e014944. https://doi.org/10.1161/JAHA.119.014944
Ku T, Li B, Gao R et al (2017) NF-ΚB-regulated microRNA-574-5p underlies synaptic and cognitive impairment in response to atmospheric PM2.5 aspiration. Part Fibre Toxicol 14. https://doi.org/10.1186/s12989-017-0215-3
Lakey PSJ, Berkemeier T, Tong H, Arangio AM, Lucas K, Pöschl U, Shiraiwa M (2016) Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci Rep 6:32916. https://doi.org/10.1038/srep32916
Langrish JP, Lundback M, Mills NL et al (2009) Contribution of Endothelin 1 to the vascular effects of diesel exhaust inhalation in humans. Hypertension 54:910–915. https://doi.org/10.1161/HYPERTENSIONAHA.109.135947
Lee B-J, Kim B, Lee K (2014) Air pollution exposure and cardiovascular disease. Toxicol Res 30:71–75. https://doi.org/10.5487/TR.2014.30.2.071
Lee A, Kinney P, Chillrud S, Jack D (2015) A systematic review of innate Immunomodulatory effects of household air pollution secondary to the burning of biomass fuels. Ann Glob Health 81:368. https://doi.org/10.1016/j.aogh.2015.08.006
Levesque S, Surace MJ, McDonald J, Block ML (2011) Air pollution & the brain: subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J Neuroinflammation 8:105. https://doi.org/10.1186/1742-2094-8-105
Levesque S, Taetzsch T, Lull ME, Johnson JA, McGraw C, Block ML (2013) The role of MAC1 in diesel exhaust particle-induced microglial activation and loss of dopaminergic neuron function. J Neurochem 125:756–765. https://doi.org/10.1111/jnc.12231
Li N, Harkema JR, Lewandowski RP, Wang M, Bramble LA, Gookin GR, Ning Z, Kleinman MT, Sioutas C, Nel AE (2010) Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol 299:L374–L383. https://doi.org/10.1152/ajplung.00115.2010
Li M-H, Fan L-C, Mao B, Yang JW, Choi AMK, Cao WJ, Xu JF (2016) Short-term exposure to ambient fine particulate matter increases hospitalizations and mortality in COPD. Chest 149:447–458. https://doi.org/10.1378/chest.15-0513
Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, Hao K, Kinney PL, Chen H, Kan H (2017) Particulate matter exposure and stress hormone levels: a randomized, double-blind, crossover trial of air purification. Circulation 136:618–627. https://doi.org/10.1161/CIRCULATIONAHA.116.026796
Li Q, Zheng J, Xu S, Zhang J, Cao Y, Qin Z, Liu X, Jiang C (2018) The neurotoxicity induced by PM2.5 might be strongly related to changes of the hippocampal tissue structure and neurotransmitter levels. Toxicol Res 7:1144–1152. https://doi.org/10.1039/c8tx00093j
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, AlMazroa MA, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng ATA, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FGR, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD III, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Memish ZA, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Hanafiah KM, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CDH, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA III, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJC, Steenland K, Stöckl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJL, Ezzati M (2012) A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380:2224–2260. https://doi.org/10.1016/S0140-6736(12)61766-8
Liu F, Huang Y, Zhang F, Chen Q, Wu B, Rui W, Zheng JC, Ding W (2015) Macrophages treated with particulate matter PM2.5 induce selective neurotoxicity through glutaminase-mediated glutamate generation. J Neurochem 134:315–326. https://doi.org/10.1111/jnc.13135
Liu X, Kong D, Liu Y, Fu J, Gao P, Chen T, Fang Q, Cheng K', Fan Z (2018) Effects of the short-term exposure to ambient air pollution on atrial fibrillation. Pacing Clin Electrophysiol 41:1441–1446. https://doi.org/10.1111/pace.13500
Ljubimova JY, Braubach O, Patil R et al (2018) Coarse particulate matter (PM2.5–10) in Los Angeles Basin air induces expression of inflammation and cancer biomarkers in rat brains. Sci Rep 8:5708. https://doi.org/10.1038/s41598-018-23885-3
McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, Harrington R, Svartengren M, Han IK, Ohman-Strickland P, Chung KF, Zhang J (2007) Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med 357:2348–2358. https://doi.org/10.1056/NEJMoa071535
McGuinn LA, Schneider A, McGarrah RW et al (2019) Association of long-term PM 2.5 exposure with traditional and novel lipid measures related to cardiovascular disease risk. Environ Int 122:193–200. https://doi.org/10.1016/j.envint.2018.11.001
Mebratu YA, Smith KR, Agga GE, Tesfaigzi Y (2016) Inflammation and emphysema in cigarette smoke-exposed mice when instilled with poly (I:C) or infected with influenza a or respiratory syncytial viruses. Respir Res 17:75. https://doi.org/10.1186/s12931-016-0392-x
Migliaccio CT, Kobos E, King QO, Porter V, Jessop F, Ward T (2013) Adverse effects of wood smoke PM2.5 exposure on macrophage functions. Inhal Toxicol 25:67–76. https://doi.org/10.3109/08958378.2012.756086
Miyata R, van Eeden SF (2011) The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol 257:209–226. https://doi.org/10.1016/j.taap.2011.09.007
Mobasher Z, Salam MT, Goodwin TM, Lurmann F, Ingles SA, Wilson ML (2013) Associations between ambient air pollution and hypertensive disorders of pregnancy. Environ Res 123:9–16. https://doi.org/10.1016/j.envres.2013.01.006
Morantes-Caballero JA, Alberto H, Rodriguez F (2019) Effects of air pollution on acute exacerbation of chronic obstructive pulmonary disease: a descriptive retrospective study (pol-AECOPD). Int J COPD 14:1549–1557. https://doi.org/10.2147/COPD.S192047
Muka T, Koromani F, Portilla E, O'Connor A, Bramer WM, Troup J, Chowdhury R, Dehghan A, Franco OH (2016) The role of epigenetic modifications in cardiovascular disease: a systematic review. Int J Cardiol 212:174–183. https://doi.org/10.1016/j.ijcard.2016.03.062
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153. https://doi.org/10.1016/J.IMMUNI.2013.05.016
Münzel T, Gori T, Al-Kindi S et al (2018) Effects of gaseous and solid constituents of air pollution on endothelial function. Eur Heart J 39:3543–3550. https://doi.org/10.1093/eurheartj/ehy481
Mustafic H, Jabre P, Caussin C et al (2012) Main air pollutants and myocardial infarction: a systematic review and meta-analysis. JAMA 307:713–721. https://doi.org/10.1001/jama.2012.126
Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, Chandel NS, Dean DA, Sznajder JI, Budinger GRS (2007) Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest 117:2952–2961. https://doi.org/10.1172/JCI30639
Nemmar A, Zia S, Subramaniyan D, Fahim MA, Ali BH (2011) Exacerbation of thrombotic events by diesel exhaust particle in mouse model of hypertension. Toxicology 285:39–45. https://doi.org/10.1016/J.TOX.2011.03.018
Newell K, Kartsonaki C, Lam KBH, Kurmi OP (2017) Cardiorespiratory health effects of particulate ambient air pollution exposure in low-income and middle-income countries: a systematic review and meta-analysis. Lancet Planet Health 1:e368–e380. https://doi.org/10.1016/S2542-5196(17)30166-3
Nurkiewicz TR, Porter DW, Hubbs AF, Cumpston JL, Chen BT, Frazer DG, Castranova V (2008) Nanoparticle inhalation augments particle-dependent systemic microvascular dysfunction. Part Fibre Toxicol 5:1. https://doi.org/10.1186/1743-8977-5-1
Øvrevik J, Låg M, Holme JA, Schwarze PE, Refsnes M (2009) Cytokine and chemokine expression patterns in lung epithelial cells exposed to components characteristic of particulate air pollution. Toxicology 259:46–53. https://doi.org/10.1016/j.tox.2009.01.028
Park EJ, Roh J, Kim Y, Park K, Kim DS, Yu SD (2011) PM 2.5 collected in a residential area induced Th1-type inflammatory responses with oxidative stress in mice. Environ Res 111:348–355. https://doi.org/10.1016/j.envres.2010.11.001
Pedersen M, Stayner L, Slama R, Sørensen M, Figueras F, Nieuwenhuijsen MJ, Raaschou-Nielsen O, Dadvand P (2014) Ambient air pollution and pregnancy-induced hypertensive disorders: a systematic review and meta-analysis. Hypertension 64:494–500. https://doi.org/10.1161/HYPERTENSIONAHA.114.03545
Peixoto MS, de Oliveira Galvão MF, Batistuzzo de Medeiros SR (2017) Cell death pathways of particulate matter toxicity. Chemosphere 188:32–48. https://doi.org/10.1016/j.chemosphere.2017.08.076
Peng C, Bind M-AC, Colicino E, Kloog I, Byun HM, Cantone L, Trevisi L, Zhong J, Brennan K, Dereix AE, Vokonas PS, Coull BA, Schwartz JD, Baccarelli AA (2016) Particulate air pollution and fasting blood glucose in nondiabetic individuals: associations and epigenetic mediation in the normative aging study, 2000–2011. Environ Health Perspect 124:1715–1721. https://doi.org/10.1289/EHP183
Peters A, Dockery DW, Muller JE, Mittleman MA (2001) Increased particulate air pollution and the triggering of myocardial infarction. Circulation 103:2810–2815. https://doi.org/10.1161/01.CIR.103.23.2810
Pope CA, Bhatnagar A, McCracken JP et al (2016) Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circ Res 119:1204–1214. https://doi.org/10.1161/CIRCRESAHA.116.309279
Power MC, Kioumourtzoglou M-A, Hart JE, Okereke OI, Laden F, Weisskopf MG (2015) The relation between past exposure to fine particulate air pollution and prevalent anxiety: observational cohort study. BMJ 350:h1111. https://doi.org/10.1136/bmj.h1111
Provoost S, Maes T, Pauwels NS, vanden Berghe T, Vandenabeele P, Lambrecht BN, Joos GF, Tournoy KG (2011) NLRP3/caspase-1-independent IL-1 production mediates diesel exhaust particle-induced pulmonary inflammation. J Immunol 187:3331–3337. https://doi.org/10.4049/jimmunol.1004062
Pun VC, Manjourides J, Suh H (2017) Association of ambient air pollution with depressive and anxiety symptoms in older adults: results from the NSHAP study. Environ Health Perspect 125:342–348. https://doi.org/10.1289/EHP494
Rebuli ME, Speen AM, Martin EM et al (2019) Wood smoke exposure alters human inflammatory responses to viral infection in a sex-specific manner: a randomized, placebo-controlled study. Am J Respir Crit Care Med:rccm.201807-1287OC. https://doi.org/10.1164/rccm.201807-1287OC
Risom L, Møller P, Loft S (2005) Oxidative stress-induced DNA damage by particulate air pollution. Mutat Res Mol Mech Mutagen 592:119–137. https://doi.org/10.1016/j.mrfmmm.2005.06.012
Rivas-Santiago CE, Sarkar S, Cantarella P et al (2015) Air pollution particulate matter alters antimycobacterial respiratory epithelium innate immunity. Infect Immun 83:2507–2517. https://doi.org/10.1128/IAI.03018-14
Roqué PJ, Dao K, Costa LG (2016) Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. NeuroToxicology 56:204–214. https://doi.org/10.1016/j.neuro.2016.08.006
Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P (2019) Environmental exposures and asthma development: autophagy, mitophagy, and cellular senescence. Front Immunol 10
Sayan M, Mossman BT (2015) The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol 13:51. https://doi.org/10.1186/s12989-016-0162-4
Ségala C, Poizeau D, Mesbah M, Willems S, Maidenberg M (2008) Winter air pollution and infant bronchiolitis in Paris. Environ Res 106:96–100. https://doi.org/10.1016/J.ENVRES.2007.05.003
Siddique S, Banerjee M, Ray MR, Lahiri T (2011) Attention-deficit hyperactivity disorder in children chronically exposed to high level of vehicular pollution. Eur J Pediatr 170:923–929. https://doi.org/10.1007/s00431-010-1379-0
Sigaud S, Goldsmith CAW, Zhou H, Yang Z, Fedulov A, Imrich A, Kobzik L (2007) Air pollution particles diminish bacterial clearance in the primed lungs of mice. Toxicol Appl Pharmacol 223:1–9. https://doi.org/10.1016/j.taap.2007.04.014
Sun B, Shi Y, Yang X, Zhao T, Duan J, Sun Z (2018a) DNA methylation: a critical epigenetic mechanism underlying the detrimental effects of airborne particulate matter. Ecotoxicol Environ Saf 161:173–183. https://doi.org/10.1016/j.ecoenv.2018.05.083
Sun Y, Wang Y, Yuan S et al (2018b) Exposure to PM2.5 via vascular endothelial growth factor relationship: meta-analysis. PLoS ONE 13. https://doi.org/10.1371/journal.pone.0198813
Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF (2002) Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol 39:935–942. https://doi.org/10.1016/S0735-1097(02)01715-1
Tarantini L, Bonzini M, Apostoli P, Pegoraro V, Bollati V, Marinelli B, Cantone L, Rizzo G, Hou L, Schwartz J, Bertazzi PA, Baccarelli A (2009) Effects of particulate matter on genomic DNA methylation content and iNOS promoter methylation. Environ Health Perspect 117:217–222. https://doi.org/10.1289/ehp.11898
Thompson JE (2018) Airborne particulate matter: human exposure and health effects. J Occup Environ Med 60:392–423. https://doi.org/10.1097/JOM.0000000000001277
Tsai S-S, Chang C-C, Yang C-Y (2013) Fine particulate air pollution and hospital admissions for chronic obstructive pulmonary disease: a case-crossover study in Taipei. Int J Environ Res Public Health 10:6015–6026. https://doi.org/10.3390/ijerph10116015
Turner MC, Krewski D, Pope CA et al (2011) Long-term ambient fine particulate matter air pollution and lung cancer in a large cohort of never-smokers. Am J Respir Crit Care Med 184:1374–1381. https://doi.org/10.1164/rccm.201106-1011OC
Uh S-T, Koo SM, Kim Y, Kim K, Park S, Jang AS, Kim D, Kim YH, Park CS (2017) The activation of NLRP3-inflammsome by stimulation of diesel exhaust particles in lung tissues from emphysema model and RAW 264.7 cell line. Korean J Intern Med 32:865–874. https://doi.org/10.3904/kjim.2016.033
Vandini S, Bottau P, Faldella G, Lanari M (2015) Immunological, viral, environmental, and individual factors modulating lung immune response to respiratory syncytial virus. Biomed Res Int 2015:1–7. https://doi.org/10.1155/2015/875723
Vert C, Sánchez-Benavides G, Martínez D, Gotsens X, Gramunt N, Cirach M, Molinuevo JL, Sunyer J, Nieuwenhuijsen MJ, Crous-Bou M, Gascon M (2017) Effect of long-term exposure to air pollution on anxiety and depression in adults: a cross-sectional study. Int J Hyg Environ Health 220:1074–1080. https://doi.org/10.1016/j.ijheh.2017.06.009
Wang H, Song L, Ju W et al (2017) The acute airway inflammation induced by PM2.5 exposure and the treatment of essential oils in Balb/c mice. Sci Rep 7:44256. https://doi.org/10.1038/srep44256
Woo SH, Lee SM, Park KC, Park GN, Cho B, Kim I, Kim J, Hong S (2018) Effects of fine particulate matter on Pseudomonas aeruginosa adhesion and biofilm formation in vitro. Biomed Res Int 2018:1–10. https://doi.org/10.1155/2018/6287932
World Health Organization (2006) Air quality guidelines global update 2005. World Health Organization, Copenhagen
World Health Organization (2013) HealtH effects of particulate matter, WHO. World Health Organization, Copenhagen
World Health Organization (2014) 7 million premature deaths annually linked to air pollution. In: World Health Organ. http://www.who.int/mediacentre/news/releases/2014/air-pollution/en/
World Health Organization (2016) Ambient (outdoor) air quality and health. In: World Health Organ. http://www.who.int/en/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health
Xing Y-F, Xu Y-H, Shi M-H, Lian Y-X (2016) The impact of PM2.5 on the human respiratory system. J Thorac Dis 8:E69–E74. https://doi.org/10.3978/j.issn.2072-1439.2016.01.19
Xu Q, Wang S, Guo Y, Wang C, Huang F, Li X, Gao Q, Wu L, Tao L, Guo J, Wang W, Guo X (2017) Acute exposure to fine particulate matter and cardiovascular hospital emergency room visits in Beijing, China. Environ Pollut 220:317–327. https://doi.org/10.1016/j.envpol.2016.09.065
Yang B, Chen D, Zhao H, Xiao C (2016) The effects for PM2.5 exposure on non-small-cell lung cancer induced motility and proliferation. SpringerPlus 5:2059. https://doi.org/10.1186/s40064-016-3734-8
Yang S, Lee S-P, Park J-B, Lee H, Kang S-H, Lee S-E, Kim JB, Choi S-Y, Kim Y-J, Chang H-J (2019) PM2.5 concentration in the ambient air is a risk factor for the development of high-risk coronary plaques. Eur Heart J 20:1355–1364
Yang L, Li C, Tang X (2020) The impact of PM2.5 on the host defense of respiratory system. Front Cell Dev Biol 8
Ying Z, Yue P, Xu X, Zhong M, Sun Q, Mikolaj M, Wang A, Brook RD, Chen LC, Rajagopalan S (2009) Air pollution and cardiac remodeling: a role for RhoA/Rho-kinase. Am J Physiol-Heart Circ Physiol 296:H1540–H1550. https://doi.org/10.1152/ajpheart.01270.2008
Zanobetti A, Dominici F, Wang Y, Schwartz JD (2014) A national case-crossover analysis of the short-term effect of PM2.5 on hospitalizations and mortality in subjects with diabetes and neurological disorders. Environ Health 13:38. https://doi.org/10.1186/1476-069X-13-38
Zhang Z, Hong Y, Liu N (2017) Association of ambient particulate matter 2.5 with intensive care unit admission due to pneumonia: a distributed lag non-linear model. Sci Rep 7:8679. https://doi.org/10.1038/s41598-017-08984-x
Zhang R, Liu G, Jiang Y, Li G, Pan Y, Wang Y, Wei Z, Wang J, Wang Y (2018) Acute effects of particulate air pollution on ischemic stroke and hemorrhagic stroke mortality. Front Neurol 9:827. https://doi.org/10.3389/fneur.2018.00827
Zhang Z, Dong B, Li S, Chen G, Yang Z, Dong Y, Wang Z, Ma J, Guo Y (2019) Exposure to ambient particulate matter air pollution, blood pressure and hypertension in children and adolescents: a national cross-sectional study in China. Environ Int 128:103–108. https://doi.org/10.1016/j.envint.2019.04.036
Zhao H, Li W, Gao Y, Li J, Wang H (2014) Exposure to particular matter increases susceptibility to respiratory Staphylococcus aureus infection in rats via reducing pulmonary natural killer cells. Toxicology 325:180–188. https://doi.org/10.1016/j.tox.2014.09.006
Zheng D, Fei-yan D, Ya-dong Y et al (2013) Effects of PM2.5 exposure on Klebsiella Pneumoniae clearance in the lungs of rats. Chin J Tuberc Respir Dis 36:836–840
Zhong J, Colicino E, Lin X et al (2015a) Cardiac autonomic dysfunction: particulate air pollution effects are modulated by epigenetic immunoregulation of toll-like receptor 2 and dietary flavonoid intake. J Am Heart Assoc 4. https://doi.org/10.1161/JAHA.114.001423
Zhong J, Urch B, Speck M, Coull BA, Koutrakis P, Thorne PS, Scott J, Liu L, Brook RD, Behbod B, Gibson H, Silverman F, Mittleman MA, Baccarelli AA, Gold DR (2015b) Endotoxin and β-1,3-d-glucan in concentrated ambient particles induce rapid increase in blood pressure in controlled human exposures. Hypertension 66:509–516. https://doi.org/10.1161/HYPERTENSIONAHA.115.05342
Zhou H, Kobzik L (2007) Effect of concentrated ambient particles on macrophage phagocytosis and killing of Streptococcus pneumoniae. Am J Respir Cell Mol Biol 36:460–465. https://doi.org/10.1165/rcmb.2006-0293OC
Acknowledgments
The authors thank the help of Universidad de Antioquia UdeA, Corporacion Universitaria Remington and Universidad Cooperativa de Colombia, for their support in the project.
Availability of data and materials
Not applicable.
Funding
This study was financed by Colciencias (141580763047), Universidad Cooperativa de Colombia, and Corporación Universitaria Remington.
Author information
Authors and Affiliations
Contributions
Funding acquisition and project administration: JCH. Writing-original draft: RDAP, NAT, DMG, JCH. Writing-review and editing: RDAP, NAT, DMG, JPL, JFN, JCH. Approval of manuscript for publication: RDAP, NAT, DMG, JPL, JFN, JCH.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they do not have a conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Disclaimer
The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Additional information
Responsible editor: Lotfi Aleya
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Arias-Pérez, R.D., Taborda, N.A., Gómez, D.M. et al. Inflammatory effects of particulate matter air pollution. Environ Sci Pollut Res 27, 42390–42404 (2020). https://doi.org/10.1007/s11356-020-10574-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-10574-w