
Abstract
In this work, activated carbon-supported copper(II) oxide (CuO/AC) was prepared and used to degrade heavy oil refinery wastewater (HORW) by catalytic ozonation with the aim to develop low-cost and high-efficient supported-catalysts for degrading real recalcitrant industrial wastewater. Supported-catalyst CuO/AC was characterized by X-ray diffraction (XRD), N2-physisorption, scanning electronic microscope (SEM), transmission electron microscope (TEM), and X-ray fluorescence (XRF). The degradation was mainly evaluated by chemical oxygen demand (COD), total organic carbon (TOC), 5-day biochemical oxygen demand (BOD5), biodegradability and toxicity. Compared with unsupported-catalyst CuO or the mixed system of activated-AC and unsupported-catalyst CuO, supported-catalyst CuO/AC with reduced cost exhibited significantly enhanced activity for degrading HORW (5.0 g CuO-5%/AC, 90 mg/L O3, and 7.3 pH). TEM analysis showed that the high activity of supported-catalyst CuO-5%/AC might be ascribed to the fact that CuO particles were small and highly dispersed on AC. Mass spectrum spectrometry (MS) analysis revealed that the organic components in HORW were first degraded to small molecule oxidation products, which were then oxidized or mineralized further. The influence of CuO loading, CuO/AC dose, ozone dose and initial pH on the degradation efficiency was also investigated. The results of the present work showed that CuO/AC could be a promising supported-catalyst for catalyzing ozonation degradation of HORW.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy oil refinery wastewater (HORW) is generated during the processing of heavy crude oil (HCO) in petrochemical industry. HORW contains high concentration of recalcitrant organic compounds such as aliphatic hydrocarbons, aromatic hydrocarbons, naphthenic acid, aliphatic acid, and phenols. Those recalcitrant compounds not only contribute to high level of COD but also have detrimental and harmful effects on plant and aquatic life as well as surface and ground water sources (Cai 2011; El-Naas et al. 2009). Generally, light oil refinery wastewater (LORW) is treated by a series of physico-chemical operations such as oil/water separation, gas flotation and coagulation, followed by biological treatment aiming at degrading the residual dissolved organic compounds. HORW is usually treated by the same operations as that used for treating LORW, but biological treatment process is inefficient in degrading residual organic compounds in HORW due to their complicated molecular structures, acute biotoxicity, and poor biodegradability (Dong et al. 2019; El-Naas et al. 2014). In recent years, the volume of HORW produced has shown an increasing trend since more and more HCO is processed in refineries around the world. On the other hand, more stringent environmental legislation has been implemented according to effluents standards. Therefore, the pressure is mounting on refineries and researchers to develop new methods or technologies for treating HORW.
In recent decades, catalytic ozonation has been intensively studied in the treatment of high-concentrated organic wastewater (Hassani El et al. 2019; Kermani et al. 2018). Especially, heterogeneous catalytic ozonation process (HCOP) is considered as the most potential technology for treating real industrial wastewater containing high concentration of recalcitrant organic pollutants due to its high efficiency, easy operability, and no secondary pollution (Sania et al. 2019; Liu et al. 2019a, b; Polat et al. 2015). The high efficiency of HCOP is ascribed to heterogeneous catalysts, which play a role in transforming ozone into more reactive species over the active functional groups of the catalyst surface and/or adsorbing the targeted pollutants to react with the dissolved ozone (Chen et al. 2015). Notably, Munir et al. (2019) reported recently that UV-light irradiation could also enhance the performance of O3 process for treating paper and pulp industry wastewater near wastewater pH. Additionally, Wang et al. (2019) also studied metal-free catalytic ozonation on surface-engineered graphene. They prepared reduced graphene oxide (rGO) and N-doped rGO via microwave reduction. rGO synthesized by microwave reduction demonstrated higher activity than rGO prepared by thermal anneal in argon, and N-doping could improve the catalytic activity further. Despite those above, many research works have been done to treat wastewater by HCOP with unsupported-catalysts such as Fe2O3, MnO2, Al2O3, CoO, MoO3, MgO, CuO, NiO, TiO2, SnO2, and so on (Nawaz et al. 2017; Lv et al. 2010; Vittenet et al. 2015; Manivel et al. 2015; Liu et al. 2019a, b; Zhang et al. 2009; Yang et al. 2014; Zeng et al. 2009). Moreover, most of those works have focused on degrading model organic compounds or simulated wastewater with single component. However, the components of real industrial wastewater such as HORW are very complex. Besides, what should be specially mentioned is that most preparation processes of unsupported-catalysts are usually complex and uncontrollable, which cause high preparation cost and unstable catalytic activity. This partly hampers HCOP from going into industrial application for treating real wastewater in large scale.
A major trend that addresses this issue is to develop low-cost and high-efficient catalysts such as heterogeneous supported-catalysts (Chen et al. 2017). Heterogeneous supported-catalysts can be prepared by impregnation method in which catalysts are prepared just by impregnating supports with metal-salt solutions (Jiang 2006). The impregnation method is very simple and controllable, which will reduce the preparation cost and improve the performance stability of catalysts. Additionally, in supported-catalyst systems, catalyst particles are well dispersed on supports, which will enhance the activities of catalysts and reduce the dosage of catalysts in treatment. This will further reduce the treatment cost. For instance, Ikhlaq et al. (2019) prepared iron-loaded rice husk ash by impregnation method and which was used as catalyst to decolorize methylene blue solution by catalytic ozonation process. The results indicated that catalytic ozonation with iron-loaded rice husk ash was more effective than Fenton-like process near wastewater pH values. Further, HCOP undergoes an absorption-catalytic oxidation degradation mechanism; supports with large specific areas in supported-catalysts favor absorbing the targeted pollutants, leading to the enhancement of degradation efficiency.
AC is a porous material with a large specific surface area, which is also low-cost, abundant, chemically stable, and environmentally friendly (Hossain et al. 2018), showing that it can be a good support material for preparing heterogeneous supported-catalysts for HCOP (Chen et al. 2014a, b). In this work, CuO/AC was prepared simply by impregnating AC with Cu (NO3)2 solution and was used to catalyze ozonation degradation of HORW with the aim to develop low-cost and high-efficient heterogeneous supported catalysts for degrading real recalcitrant industrial wastewater by HCOP. In experiments, CuO/AC was characterized by XRD, N2-physisorption, SEM, TEM, and XRF. The influences of factors, including CuO loading, catalyst dose, O3 dose, and pH, on the COD removal were investigated. Further, the degradation mechanism of CuO/AC catalyzed ozonation process was preliminary explored.
Experimental
HORW sample characteristics
The HORW samples were obtained from Tahe Petrochemical Co., Ltd., Sinopec, which is a main heavy oil refinery with a capacity of 5 Mt/a. The streams of HORW mainly came from the dehydration of heavy oil tanks and electric desalting tanks, as well as the condensate from units of atmospheric vacuum distillation and delayed coking. The characteristics of HORW samples are listed in Table 1; it was noted that COD in HORW was high, whereas BOD5 was low, and the ratio of BOD5/COD was only about 0.09, indicating the poor biodegradability of HORW. A considerable amounts of total suspended solid (TSS), volatile suspended solid (VSS), TDS (H2S, HS−, and S2−), and SO42− were also detected out in HORW.
Materials and methods
Chemicals including NaOH (CAS No. 1310-73-2), H2SO4 (CAS No. 7664-93-9), Cu (NO3)2·3H2O (CAS No.10031-41-3), Na2S2O3·5H2O (CAS No.10102-17-7), and KI (CAS No. 7681-11-0) used in the present study were of analytical grade purity, and all of them were purchased from Shanghai Chemical Reagent Co., Ltd. (China). Oxygen with a purity of more than 99%, bought from Sichuan, Xindu Industrial Gas Co., Ltd. (China), was used to preparation of O3. Coconut shell–based granular AC purchased from Sichuan Changzheng Chemical Reagent Co., Ltd. (China) was crushed and sieved to 40~60-mesh size for use. The concentration of O3 in the gas mixture of O2 and O3 was determined by iodometric (KI) method via introducing the mixture of O2 and O3 into KI solution through the gas sampling port to form iodin (I2), which was then titrated with standard Na2S2O3 solution. The volume of the mixture of O2 and O3 was monitored with a gas flowmeter.
Preparation and characterization of supported-catalyst CuO/AC
AC of 40~60-mesh size was first washed sequentially with 10 m% NaOH solution, 1:1(v/v) H2SO4 solution for 10 h, respectively, and distilled water until pH about 7.0 to remove impurities and dried in air at room temperature for 2 days. Afterwards, the washed AC was dried in an air oven at 110 °C for 24 h and the resulted AC was designated as pretreated-AC.
Supported-catalysts CuO/AC were prepared using impregnation method, taking the preparation of CuO/AC with CuO loading of 1 wt% as an example. Twenty grams of pretreated-AC was dipped in 36 mL Cu (NO3)2 (0.074 M) for 8 h, then the sample was dried in air at 105 °C for 10 h, and subsequently calcined at 350 °C in N2 for 3 h; the obtained sample was desiganated as CuO-1%/AC. Further, CuO-3%/AC, CuO-5%/AC, and CuO-7%/AC samples were also prepared using the same procedure by varying the concentration of Cu (NO3)2 solution.
Activated-AC was prepared by calcinating the pretreated-AC under the same conditions as that for the preparation of supported-catalyst CuO/AC. Unsupported-catalyst CuO was also prepared by calcinating solid chemical agent Cu(NO3)2·3H2O under the same conditions as that for the preparation of supported-catalyst CuO/AC.
XRD pattern of samples was recorded on an X-ray diffractometer (D/Max-IIIA) with Cu-Kα (λ = 1.54 Å) radiation.
Specific surface area, pore volume, and average pore size of CuO/AC were determined by N2-physisorption using Micromeritics ASAP 2020 instrument at − 196 °C. Before analysis was performed, all samples were degassed at 150 °C to vacuum 10−3 Torr for 5 h. Specific surface area and pore volume were calculated using Brunauer-Emmet-Teller (BET) and t-plot methods, respectively. Detailed information is shown in Electronic Supplementary Material (ESI).
The morphology of CuO/AC was determined by SEM (Hitachi S-4800). CuO content in CuO/AC was determined by XRF (Panalytical Axios).
Degradation of HORW
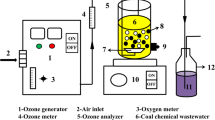
The catalytic ozonation degradation experiments were performed in a system shown in Fig. S2, which was composed of an oxygen (O2) steel cylinder (40 L), an O3 generator (< 10 g/h), a flowmeter (20~40 L/h), a column steel reactor (60 mm in inner diameter and 500 mm in height), and an exhaust gas adsorption bottle. The catalyst was sandwiched between porous supporting plates and porous splint in the reactor in order to avoiding catalysts effluence before 1 L targeted HORW was added in the reactor. O2 passed through the O3 generator to produce O3, feeding into the reactor. The O3 concentration in the experiments was controlled by adjusting the flow of O2 and the voltage of the O3 generator. Excess O3 in the exhaust was trapped with an exhaust gas absorption bottle that was a cylindrical glass bottle with volume of 3 L, containing 2 L Na2S2O2 solution (2 M). The temperature of the reactor was maintained at 60 °C (original temperature of HORW) by recirculating warm water from a thermostatic water bath in hollow shell around the cylindrical body of the reactor. During each 90 min degradation experiment, water samples of 5 mL each were taken with a glass suction pipette with volume of 5 mL at 15-min interval for immediately analyzing COD, BOD5, and TU respectively without storage. After degradation, water samples of 50 mL each were taken with a glass suction pipette with volume of 50 mL for immediate analyzations of TSS, VSS, TDS, and electrospray ionization-Fourier transform-ion cyclotron resonance mass spectrometry (ESI-FT-ICR MS) respectively. Two milliliters of Na2S2O3 solution (0.1 M) was added to each sample to cease the reaction after sampling at various intervals.
Anaerobic-aerobic bio-treatment is a process typically used to degrade LORW. Can the process also be applied to degrade HORW effectively? Therefore, as for comparison, the biodegradation experiments of HORW using a sequential anaerobic-aerobic process were carried out in experimental setup consisting of an up-flow anaerobic sludge blanket (UASB) reactor followed by an aerobic packed bed biofilm reactor (PBBR). Detailed descriptions of experimental procedure, schematic diagram and the corresponding degradation results are shown in ESI.
Before formal degradation experiments, some pilot experiments were carried out with aim to determine experimental conditions. Each data point of results for each experiment was measured three times in parallel. In order to avoid the randomness in the process, each experiment was repeated, and similar results were obtained.
Analyzation of HORW samples
The pH was measured by a PHS-3C meter (LeiCi, China). The determination of BOD5 was performed in an OxiTop system (CHALLENGE, USA). COD was analyzed by dichromate method according to GB11914-89 of PR China. TOC was analyzed by a TOC-VCPH analyzer (Shimadzu, Japan). The toxicity test of HORW was analyzed by the Vibrio fischeri method (Farré et al. 2001). Chemical composition characterization in HORW was carried out on an Apex-ultra FT-ICR MS (Bruker Daltonics, USA) in the negative-ion mode with a 9.4-T actively shielded magnet. The concentrations of copper ion (Cu2+) in treated HORW samples were determined by inductively coupled plasma atomic emission (ICP-AES) technique. The TDS content was determined by iodometry method (Luo 2011); SO42− content was determined by EDTA complexometric titration method (Zhang et al. 2007).
Results and discussion
Characterization of the catalysts
Figure 1 depicts the XRD patterns of the pretreated-AC and CuO/AC samples. As shown in the XRD pattern of the pretreated-AC, there were two broad peaks centering at about 24° and 44° indexed to the (002) and (100) plane diffractions of amorphous carbon, respectively (Islam et al. 2016; Raj and Joy 2015). The pattern of CuO-1%/AC was similar to that of AC, but extremely vague diffraction signals between 34 and 40° were observed, indicating that new species formed on the surface of AC. The two small and clear peaks centering at 35.31° and 38.71° in the pattern of the CuO-3%/AC sample were indexed to characteristic peaks of CuO (PDF0045-0973), suggesting that copper was loaded on AC in forms of CuO. As CuO loading increased, the characteristic peaks of CuO in the patterns of CuO-5%/AC and CuO-7%/AC samples became stronger. No other diffraction peaks such as Cu2O and Cu was found in XRD patterns; this might be attributed to the fact that catalyst precursor was calcined at low temperature (350 °C), and no reduction reaction between CuO and AC took place.

XRD patterns of pretreated-AC and CuO/AC catalyst
Figure 2 shows N2 adsorption-desorption isotherms of pretreated-AC, CuO-1%/AC, CuO-3%/AC, CuO-5%/AC, and CuO-7%/AC. All the isotherms are type I according to the IUPAC; the major consumption of N2 in the adsorption-desorption isotherms occurred at relatively low pressure (< 0.2) and reached a plateau at high relative pressure. These results showed the micropore structure characteristics of pretreated-AC and CuO/AC catalysts. However, as it is shown in Fig. 2, loading CuO had obvious influence on the adsorption kinetics of AC. The adsorption plateaus for CuO-1%/AC and CuO-3%/AC were notably higher than that of pretreated-AC, which revealed that the adsorption capacities of CuO-1%/AC and CuO-3%/AC were higher than that of the pretreated-AC, and the micropore volumes of CuO-1%/AC and CuO-3%/AC are larger than that of AC. Those were confirmed by the texture properties shown in Table 2, and the specific surface areas of CuO-1%/AC and CuO-3%/AC were larger than that of pretreated-AC. This is because that the H2O (steam) which was adsorbed on AC worked as an activation agent during calcination (Rodríguez-Reinoso et al. 1995), and it reacted with C atoms to form new pore structures with similar size to that in the pretreated-AC, leading to the increase in specific area. Notably, as Table 2 shows, the specific surface area of the activated-AC was much larger than that of the pretreated-AC, which experimentally indicated that calcination treatment had induced the steam activation of AC. Meanwhile, as Fig. 3 shows, steam activation also corroded the pore structures of AC. Figure 3a and b present that there were obviously more pores in fresh CuO-1%/AC than that in the pretreated-AC. However, the specific surface area decreased when CuO loading increased to 5 wt% and 7 wt%, which might be ascribed to the reason that AC activation intensified as CuO loading increased because Cu2+ ions catalyzed the steam activation to form larger pores, and the intensified activation also corroded pore structures of AC to bare small pores (Fig. 3c–e). Besides, the CuO particles grew larger as CuO loading increased and part of pores in AC were blocked by CuO particles (Fig. 3d, e), which might be another reason for the decrease in specific area. Figure 3f shows that after 4 cycles of degradation treatment, the pore structure of CuO-5%/AC collapsed partly.

N2 adsorption-desorption isotherms of pretreated-AC, CuO-1%/AC, CuO-3%/AC, CuO-5%/AC, and CuO-7%/AC

SEM images of pretreated-AC (a), fresh CuO-1%/AC (b), fresh CuO-3%/AC (c), fresh CuO-5%/AC (d), fresh CuO-7%/AC (e), and CuO-5%/AC used for 4 cycles (f)
The unsupported-catalyst CuO particles and the supported-catalyst CuO-5%/AC partices were characterized by TEM technology. Figure 4a shows that the sizes of unsupported-catalyst CuO particles were large and were mainly distributed in the range of 250~350 nm. It was also observed in Fig. 4a that CuO particles aggregated with each other slightly. By contrast, Fig. 4b shows that CuO particles in supported-catalyst CuO-5%/AC were much smaller and were mainly distributed in the range of 40~70 nm. CuO particles in supported-catalyst CuO-5%/AC were highly dispersed on AC, and no obvious agglomeration was observed.

TEM images of unsupported-catalyst CuO particles (a) and supported-catalyst CuO-5%/AC (b). The insets in the TEM images are size distribution histograms of CuO particles
Catalyzed ozonation degradation of HORW
COD removal results of HORW by HCOP (5.0 g CuO-5%/AC, 7.3 pH and 90 mg/L O3) with supported-catalyst CuO-5%/AC are shown in Fig. 5. For comparison, degradation experiments of HORW by pretreated-AC, activated-AC, O3, activated-AC + O3, CuO + O3, and activated-AC + CuO + O3 systems were also carried out and the corresponding results are presented in Fig. 5 too. The pretreated-AC (5.0 g pretreated-AC and 7.3 pH) and activated-AC (5.0 g activated-AC and 7.3 pH) have very limited capacities to remove COD, and COD removal by the latter was slightly higher than that by the former. This was because COD removal capacity of AC derived from its adsorption of organic pollutants, and the specific surface area of activated-AC was larger than that of the pretreated-AC (shown in Table 2). COD removal by single O3 (90 mg/L O3 and 7.3 pH) increased as treatment time prolonged and COD removal of about 22.0% was obtained after treatment. COD removal efficiency of HORW by activated-AC + O3 (5.0 g activated-AC, 90 mg/L O3 and 7.3 pH) was greater than that by single O3, indicating that some function groups on the surface of AC catalyzed the decomposition of ozonation into •OH radicals to some extent (Tian et al. 2016). Notably, the COD removal efficiency of HORW by system CuO + O3 (0.25 g CuO, 90 mg/L O3 and 7.3 pH) was obviously enhanced, and after treatment, COD removal of about 52.0% was obtained, which indicated that CuO was a good catalyst for HCOP. The efficiency of system activated-AC + CuO + O3 (4.75 g activated-AC, 0.25 g CuO, 90 mg/L O3, and 7.3 pH) in COD removal of HORW was greater than that of system CuO + O3, which was attributed to the additive effect in catalytic activities of activated-AC and CuO. Interestingly, COD removal efficiency of HORW by system CuO-5%/AC + O3 (5.0 g CuO-5%/AC, 90 mg/L O3 and 7.3 pH) was remarkably greater than that by system CuO + O3 and system activated-AC + CuO + O3. In such case, COD removal efficiency of about 72.0% was obtained after treatment, indicating that the catalytic activity of supported-catalyst CuO-5%/AC was much higher than that of CuO and the mixture of activated-AC and CuO, although the amounts of CuO in systems CuO-5%/AC + O3, CuO + O3, and activated-AC + CuO + O3 were equal. As TEM images (Fig. 4) showed, this might be ascribed to the fact that CuO particles in supported-catalyst CuO/AC were much smaller and highly dispersed, leading to the high catalytic activity of CuO-5%/AC. Despite the high activity of CuO-5%/AC, COD did not reduce in a linear manner. Instead, it reduced quickly in first 45 min and then slowly as treatment time prolonged, which might be attributed to the generation of intermediates oxygen-containing compounds in later stage such as aldehydes and carboxylic acids. Those compounds are active-low in degradation reactions. Besides, the inherent recombination of •OH radicals might be another reason for it.

COD removal of HORW by different systems
Figure 6 shows the COD and BOD5 reduction trend, and biodegradability (BOD5/COD ratio curve) of HORW samples treated by HCOP and single ozonation. As shown in Fig. 6a, COD concentration in HORW treated by HCOP with CuO-5%/AC (5.0 g CuO-5%/AC, 90 mg/L O3, and 7.3 pH) was on a clear decline along with the extended treatment time. As for BOD5, it first decreased, then increased, and last decreased slowly again with treatment time prolonged. Notably, BOD5/COD ratio increased from 0.076 to 0.33 at 45 min, indicating that anti-biodegradable organic components became biodegradable after treatment. The slight decrease of BOD5/COD ratio to 0.31 at 90 min was mainly due to mineralization of biodegradable substances. By contrast, COD removal by single ozonation was low and the biodegradability of HORW did not improved obviously (Fig. 6b).

COD, BOD5, and BOD5/COD ratio changing trend of HORW samples treated by CuO-5%/AC catalyzed ozonation (a) and single ozonation (b)
The toxicity of HORW was analyzed by Vibrio fischeri method, basing on a mechanism that the luminescence of bacteria Vibrio fischeri is directly associated with respiratory activity, and it therefore provides a good indicator of metabolic activity and the general cytotoxicity of a compound or a mixture of compounds (Rodriguez-Ruiz et al. 2015). As Fig. 7 shown, the toxicity of HORW treated by HCOP with CuO-5%/AC slightly increased in first 15 min treatment, then decreased quickly in an approximate linear manner until at 75 min and afterward slowly down, revealing that intermediates with higher toxicity formed in initial 15 min and then were further converted to substances with low toxicity. Toxicity of HORW decreased by about 67.1% after treatment by HCOP. However, toxicity decreased by only about 21.0% in the case of single ozonation. Toxicity analysis results combining with COD results (Fig. 6a, b) confirmed further that the toxicity of HORW dominantly derived from organic petroleum compounds. Additionally, as Table 1 shows, after treatment by HCOP, TSS and VSS were reduced remarkably and no TDS was detected out within analyzation sensitivity. While SO42− content increased obviously, this was ascribed to the oxidation conversion of TDS.

Toxicity changing trend of HORW samples treated by CuO-5%/AC catalyzed ozonation and single ozonation
As for comparison, HORW was also treated by sequential anaerobic-aerobic process for a period of 11 days and the corresponding degradation results were presented in Fig. S3, showing that after treatment COD was reduced by only 21.8% averagely and BOD5/COD ratio was enhanced slightly. Biological treatment results further proved the anti-biodegradable characteristic of HORW. As Table 3 shows, the results obtained in the present study were also compared with some other studies on degrading petroleum refinery wastewater by HCOP.
The experimental results (not shown) showed the dosage of unsupported-CuO 4 times to CuO in supported-CuO-5%/AC if the same degradation results were obtained. Based on the treatment of HORW in lab-scale in this work, the catalyst cost of unsupported-CuO was 0.62 dollar per cubic meter of HORW, while the catalyst cost for supported-CuO-5%/AC was 0.23 dollar per cubic meter of HORW.
Degradation mechanism discussion
Active species for high efficiency of CuO/AC catalyzed ozonation
HCOP is a kind of advanced oxidization technology, and it is commonly accepted that the high efficiency of advanced oxidization is ascribed to the generation of more •OH radicals. To verify it also happened in CuO/AC catalyzed ozonation, tert-butyl alcohol (TBA) (5 mL, 0.5 mol/L), typical •OH radical inhibitor, was added in systems to scavenge •OH radicals produced during treatment (Feng et al. 2016). Influences of TBA concentrations on the inhibition of COD removal were investigated with the aim to determine the optimal adding amount of TBA, the results (seeing in Fig. S4) showed that the optimal concentration of TBA was 0.5 mol/L. Figure 8 presents the influence of TBA introduction on COD of HORW samples treated by HCOP (5.0 g CuO-5%/AC, 7.6 pH and 90 mg/L O3). After treatment, the COD of HORW treated without adding TBA decreased by about 72.0%, while in the case of adding TBA initially (at 0 min), COD reduced by only about 17.5%, revealing the strong inhibition effect of TBA toward COD removal, which was ascribed to the fact that the formed •OH radical was scavenged by TBA.

Influence of adding TAB on COD removal
To prove further that the high degradation efficiency of CuO/AC catalyzed ozonation is due to the generation of •OH radicals, TBA was also added in system at different intervals during degradation. It is found in Fig. 8 that COD was reduced sharply before adding TBA, while COD reduction trend slowed down immediately upon adding TBA at 15 and 30 min during treatment. This was ascribed to the fact that •OH radicals generated during the treatment were scavenged by TBA, and only O3 left in the systems. The oxidation-reduction potential of O3 is much less than that of •OH radical. Besides, O3 is unstable in water, and its lifetime is short and its solubility is low. All those factors lead to COD reduction trend became slow after adding TBA. Therefore, it was confirmed that organic components in HORW were dominantly degraded via oxidization with active species •OH radicals.
Chemical composition analysis of HORW
ESI-FT-ICR MS is a sensitive analyzation technology, which was employed in the present study to determine organic components in HORW during treatment with aim to reveal decomposition mechanism of organic compounds. Figure S5 presents ESI-FT-ICR MS spectra of HORW samples treated by HCOP (5.0 g CuO-5%/AC, 90 mg/L O3 and 7.6 pH) and single O3 (90 mg/L O3 and 7.6 pH). Figure S5A demonstrates that there were about 6538 organic compounds (signal peaks) in original HORW with an S/N above 4 detected in the range of 175 < m/z < 415 at an average mass resolving power of more than 100,000, and among which 634 compounds were indexed to oxygen (O)-containing compounds. As shown in Fig. S5B, after 90 min treatment by single O3, peak intensities and molecular weight distribution did not change obviously. However, after 45 min treatment by HCOP, the molecular weight distribution of polar organic compounds changed significantly (Fig. S5C), many new small molecular products emerged in the range of 175 < m/z < 250, and O-compounds increased to 821, and the relative peak intensity of compounds, particularly in molecular weight fraction with 250 < m/z < 415, were markedly decreased. This indicated a pronounced degradation of organic compounds, and that O-compounds are intermediates. With treatment time extended to 90 min, the relative peak intensity of compounds were decreased further, and total signal peaks and O-compound peaks decreased to 4821 and 386 respectively (Fig. S5D). All this showed a pronounced degradation of organic components, in which initial large organic molecules were decomposed step by step. Therefore, it was concluded that organic components in HORW were dominantly decomposed via oxidization with •OH radicals in a way that original big organic molecules were first degraded to small molecule oxidation products, which were then oxidized or mineralized further.
Influences on degradation
Influence of CuO loading in CuO/AC
Figure 9 shows COD removal of HORW samples treated by CuO-x%/AC catalyzed ozonation with O3 dose of 90 mg/L, 7.6 pH and CuO-x%/AC dose of 5.0 g. After treatment, COD removal increased from 27.7 to 47.1% and to 72.5% as CuO loading increased from 1 to 3 wt%, and to 5 wt%, respectively; then, however, it decreased to 60.9% when CuO loading increased to 7 wt% further. As aforementioned, heterogeneous catalytic ozonation undergoes absorption-catalytic oxidation mechanism, and the activity of supported-catalyst relates not only to catalyst loading but also to adsorption property such as specific surface area. As shown in Fig. 9, COD removal efficiency was significantly enhanced as CuO loading increased from 1 to 5 wt%, indicating that at this stage the degradation activity was dominantly determined by CuO loading because the number of catalytic active sites increased as CuO loading increased. The degradation activity, however, decreased remarkably when CuO loading increased to 7 wt%, which was probably due to that CuO particles grew into larger size (revealed by the broaden diffraction peaks of CuO in Fig. 1) and adsorption properties of CuO/AC deteriorated (Table 2; Fig. 3). Here, 5 wt% of CuO seemed to be the optimal catalyst loading.

Influence of CuO loading on COD removal
Influence of CuO/AC dose
Figure S6 shows the COD removal of HORW over various CuO-5%/AC catalysts does of 0.5 g, 1.5 g, 3.5 g, and 5.0 g with 90 mg/L O3 and 7.2 pH. It was clear that adding catalyst dose promoted COD removal, which was probably due to more surface active sites for more catalyst dose (Chen et al. 2014a). After treatment, COD removal efficiency of 58.87%, 62.3%, and 81.2% was obtained over CuO-5%/AC catalyst dose of 0.5 g, 1.5 g, and 3.5 g, respectively. However, when catalyst dose increased to 5.0 g, the COD removal was close to the case of 3.5 g catalyst dose. Therefore, 3.5 g of CuO-5%/AC seemed to be the optimal catalyst dose considering cost-cutting.
Influence of O3 concentration
Figure S7 shows COD removal of HORW samples over 3.5 g CuO-5%/AC catalyst with 7.5 pH and different O3 concentrations. The COD removal efficiency of 62.2%, 69.3, 74.4%, and 75.3% was obtained at O3 concentrations of 30, 60, 90, and 120 mg/L, respectively. COD removal increased gradually as O3 concentration increased, which was due to that increasing O3 concentration favored generating more •OH radicals. Besides, what was more important was that increasing O3 concentration (means increasing gas mixture flow) could change the dispersion state of catalysts in system, which therefore benefited for the enhancement of HORW degradation efficiency (Chen et al. 2014a, b). During experiments with 30 mg/L O3, the channeling phenomenon was existed in the reaction system, suggesting insufficiently fluidized state of CuO-5%/AC catalyst. For O3 concentration of 60 mg/L in the system, the channeling phenomenon weakened to some extent. Fully fluidized state of catalysts was realized, when the O3 concentration was increased to 90 mg/L. According to the mass transfer theory (Benitez 2002), fluidization state made the interface between gas–liquid–solid contact each other fully, which can promote heterogeneous catalytic reaction. Additionally, the driving force for O3 mass transfer increased as O3 concentration increased. Above factors resulted in the enhancement of COD removal. On other hand, O3 accumulated in water as the O3 mass transfer rate increased, and the concentration of O3 in HORW increased to a saturation value. Thus, the COD reduction removal did not increase further when ozone concentration increased to 120 mg/L. Therefore, the O3 concentration of 90 mg/L was reasonable considering the O3 utilization and COD removal efficiency.
Influence of initial pH
The influence of various initial pHs (adjusted by HCl and NaOH solutions) on COD and TOC removal of HORW is illustrated in Fig. S8a with 30 mg/L O3 and 3.5 g CuO-5%/AC. COD and TOC removal showed a “volcano-type” changing trend with initial pH increased from 1.02 to 11.11. High COD and TOC removal efficiency was obtained at weak acid and neutral condition, while COD and TOC removal was low in strong acid or alkaline condition. In experiments, CuO/AC showed a good acid-base stability at pH 5–8, and CuO was found to easily leach out from the surface of catalyst at pH less than 4 (Fig. S8b). Therefore, the low COD and TOC removal efficiency in strong acidic condition might be ascribed to the leach out of CuO from catalyst in the manner of Cu2+. As to the low COD and TOC removal, efficiency in strong alkaline condition might be due to the generation of carbonate and bicarbonate in mineralization of organic pollutants, which are the typical •OH radical scavengers (Xiong et al. 2003). Additionally, the surface hydroxyl groups in neutral state on the surface of heterogeneous catalyst favor the decomposition of O3 to attain relative high catalytic performance, and the surface hydroxyl groups are dominant only when the pH of the solution is at about the point of zero charge pH (pHPZC) (Zhang et al. 2008). Herein, the pHPZC of CuO-5%/AC catalyst was 6.28 and that was why COD and TOC removal was the highest at pH 6.03. The performance stability of CuO-5%/AC in the degradation of HORW by HCOP (3.5 g CuO-5%/AC, 90 mg/L O3 and 7.3 pH) was also investigated and the results (shown in Fig. S9) showed that the performance of CuO-5%/AC decreased obviously after 4 cycles.
Conclusion
The degradation of HORW by HCOP with supported-catalyst CuO/AC was investigated. The results showed that compared with unsupported-CuO or the mixed system of activated-AC and unsupported-CuO, supported-catalyst CuO/AC with reduced cost demonstrated significantly enhanced activity for the degradation of HORW by HCOP. The high activity of supported-catalyst CuO/AC might be ascribed to the fact that CuO particles were much smaller and highly dispersed on AC. The organic components in HORW were predominantly degraded via oxidization with •OH radicals in a way that original big organic molecules were first degraded to small molecule oxidation products with low toxicity and improved biodegradability, which were then further oxidized or mineralized. CuO loading, catalyst dose, O3 dose, and initial pH had influences on the degradation. CuO/AC could be a promising heterogeneous supported-catalyst for the degradation of HORW by HCOP.
References
Benitez J (2002) Principles and modern applications of mass transfer operations. Wiley, New York, pp 125–134
Cai Y (2011) Project design for treatment of super heavy oil refinery wastewater by biological process. Modern Chem Ind 31(1- supplement):366–369
Chen C, Chen H, Guo X et al (2014a) Advanced ozone treatment of heavy oil refining wastewater by activated carbon supported iron oxide. J Ind Eng Chem 20:2782–2791
Chen C, Li Y, Ma W et al (2017) Mn-Fe-Mg-Ce loaded Al2O3 catalyzed ozonation for mineralization of refractory organic chemicals in petroleum refinery wastewater. Sep Purif Technol 183:1–10
Chen C, Wei L, Guo X et al (2014b) Investigation of heavy oil refinery wastewater treatment by integrated ozone and activated carbon-supported manganese oxides. Fuel Process Technol 124:165–173
Chen C, Yu J, Yoza BA et al (2015) Role and potential of spent fluid catalytic cracking catalyst assisted ozonation of petrochemical wastewater. J Environ Manag 152:58–65
Deng F, Qiu S, Chen C et al (2015) Heterogeneous Catalytic Ozonation of Refinery Wastewater over Alumina-Supported Mn and Cu Oxides Catalyst. Ozone-Sci Eng 37:546–555
Dong HH, Jiang X, Sun S et al (2019) A cascade of a denitrification bioreactor and an aerobic biofilm reactor for heavy oil refinery wastewater treatment. RSC Adv 9:7495–7504
El-Naas MH, Al-Zuhair S, Al-Lobaney A et al (2009) Assessment of electrocoagulation for the treatment of petroleum refinery wastewater. J Environ Manag 91:180–185
El-Naas MH, Alhaija MA, Al-Zuhair S (2014) Evaluation of a three-step process for the treatment of petroleum refinery wastewater. J Environ Chem Eng 2:56–62
Farré M, Ferrer I, Ginebreda A et al (2001) Determination of drugs in surface water and wastewater samples by liquid chromatography–mass spectrometry: methods and preliminary results including toxicity studies with Vibrio fischeri. J Chromatogr A 938:187–197
Feng M, Yan L, Zhang X et al (2016) Fast removal of the antibiotic flumequine from aqueous solution by ozonation: influence of factors, reaction pathways, and toxicity evaluation. Sci Total Environ 541:167–175
Hassani El K, Kalnina D, Maris T et al (2019) Enhanced degradation of an azo dye by catalytic ozonation over Ni-containing layered double hydroxide nanocatalysts. Sep Purif Technol 210:764–774
Hossain MZ, Wu W, Xu WZ et al (2018) High-surface-area mesoporous activated carbon from hemp bast fiber using hydrothermal processing. C 4(3):38–43
Islam MS, Ang BC, Gharehkani S et al (2016) Adsorption capacity of activated carbon synthesized from coconut shell. Carbon Lett 20:1–9
Ikhlaq A, Munir HMS, Khan A et al (2019) Comparative study of catalytic ozonation and Fenton-like processes using iron-loaded rice husk ash as catalyst for the removal of methylene blue in wastewater. Ozone-Sci Eng 41(3):250–260
Jiang SP (2006) A review of wet impregnation—an alternative method for the fabrication of high performance and nano-structured electrodes of solid oxide fuel cells. Mater Sci Eng A 418:199–210
Kermani M, Kakavandi B, Farzadkia M et al (2018) Catalytic ozonation of high concentrations of catechol over TiO2@Fe3O4 magnetic core-shell nanocatalyst: optimization, toxicity and degradation pathway studies. J Clean Prod 192:597–607
Luo Z (2011) The research of determination of sulfides by iodometric method. Guangzhou Chem Ind 12:113–115 (in Chinese)
Liu H, Chen L, Ji L (2019a) Ozonation of ammonia at low temperature in the absence and presence of MgO. J Hazard Mater 376:125–132
Lv A, Hu C, Nie Y et al (2010) Catalytic ozonation of toxic pollutants over magnetic cobalt and manganese co-doped γ-Fe2O3. Appl Catal B-Environ 100:62–67
Liu D, Wang C, Song Y, Wei Y, He L, Lan B, He X, Wang J (2019b) Effective mineralization of quinoline and bio-treated coking wastewater by catalytic ozonation using CuFe2O4/Sepiolite catalyst: efficiency and mechanism. Chemosphere 227:647–656
Munir HMS, Ferozea N, Ikhlaq A et al (2019) Removal of colour and COD from paper and pulp industry wastewater by ozone and combined ozone/UV process. Desalin Water Treat 137:154–161
Manivel A, Lee GJ, Chen CY et al (2015) Synthesis of MoO3 nanoparticles for azo dye degradation by catalytic ozonation. Mater Res Bull 62:184–191
Nawaz F, Cao H, Xie Y, Xiao J, Chen Y, Ghazi ZA (2017) Selection of active phase of MnO2 for catalytic ozonation of 4-nitrophenol. Chemosphere 168:1457–1466
Polat D, Balcı İ, Özbelge TA (2015) Catalytic ozonation of an industrial textile wastewater in a heterogeneous continuous reactor. J Environ Chem Eng 3:1860–1871
Rodriguez-Ruiz A, Etxebarria J, Boatti L, Marigomez I (2015) Scenariotargeted toxicity assessment through multiple endpoint bioassays in a soil posing unacceptable environmental risk according to regulatory screening values. Environ Sci Pollut Res 22:13344–13361
Raj KG, Joy PA (2015) Coconut shell based activated carbon-iron oxide magnetic nanocomposite for fast and efficient removal of oil gas. J Environ Chem Eng 3:2068–2075
Rodríguez-Reinoso F, Molina-Sabio M, Gonzalez MT (1995) The use of steam and CO as activating agents in the preparation of activated carbons. Carbon 33:15–23
Sania ON, Navaei fezabadya AA, Yazdanib M et al (2019) Catalytic ozonation of ciprofloxacin using γ-Al2O3 nanoparticles in synthetic and real wastewaters. J Water Process Eng 32:100894
Tian G, Wu Q, Li A, Wang W et al (2016) Promoted ozonation for the decomposition of 1, 4-dioxane by activated carbon. Water Sci Tech-Water Sup 17:613–620
Vittenet J, Aboussaoud W, Mendret J et al (2015) Catalytic ozonation with γ-Al2O3 to enhance the degradation of refractory organics in water. Appl Catal A-Gen 504:519–532
Wang Y, Cao H, Chen C et al (2019) Metal-free catalytic ozonation on surface-engineered graphene: microwave reduction and heteroatom doping. Chem Eng J 355:118–129
Xiong Y, He C, Karlsson HT, Zhu X (2003) Performance of three-phase three-dimensional electrode reactor for the reduction of COD in simulated wastewater-containing phenol. Chemosphere 50:131–136
Yang Y, Cao H, Peng P, Bo H (2014) Degradation and transformation of atrazine under catalyzed ozonation process with TiO2 as catalyst. J Hazard Mater 279:444–451
Zeng YF, Liu ZL, Qin ZZ (2009) Decolorization of molasses fermentation wastewater by SnO2-catalyzed ozonation. J Hazard Mater 162:682–687
Zhang Y, Cui F, Mu Y et al (2007) An improvement in quantitative determination of SO42− in oilfield produced water by EDTA complexometry. Oilfield Chem 4:369–371 (in Chinese)
Zhang T, Li CJ, Ma J et al (2008) Surface hydroxyl groups of synthetic a-FeOOH in promoting •OH generation from aqueous ozone: property and activity relationship. Appl Catal B-Environ 82:131–137
Zhang X, Li X, Qin W (2009) Investigation of the catalytic activity for ozonation on the surface of NiO nanoparticles. Chem Phys Lett 479:310–315
Funding
This work was supported by the open found of State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation (grant no. PL1128) and the fund of the Back-bone of Young and Middle-age Personal Training Plan of Southwest Petroleum University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Vítor Pais Vilar
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 7018 kb)
Rights and permissions
About this article

Cite this article
Wang, W., Yao, H. & Yue, L. Supported-catalyst CuO/AC with reduced cost and enhanced activity for the degradation of heavy oil refinery wastewater by catalytic ozonation process. Environ Sci Pollut Res 27, 7199–7210 (2020). https://doi.org/10.1007/s11356-019-07410-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-07410-1