
Abstract
To understand the atmospheric chemistry of hydrofluoroethers, we have studied the oxidation of a highly fluorinated compound n-C2F5CF(OCH3)CF(CF3)2 (HFE-7300) by OH/Cl oxidants. Here, we have employed M06-2X functional along with a 6-31 + G(d,p) basis set to obtain the optimized structures, various forms of energies, and different modes of frequencies for all species. We have characterized energies of all species on the potential energy surface, and it indicates that H-abstraction from n-C2F5CF(OCH3)CF(CF3)2 by Cl atom is kinetically more dominant than the H-abstraction reaction initiated by OH radical. In contrast, the calculated energy change (ΔrH°298 and ΔrG°298) results govern that OH-initiated H-abstraction reaction is highly exothermic and spontaneous compared to the Cl-initiated H-abstraction reaction. Rate constants are estimated using transition state theory as well as canonical variation transition state theory at the temperature range 200–1000 K and 1 atm pressure. The calculated rate constants of the H-abstraction channels are found to be in good agreement with the reported experimental rate constant at 298 K. Moreover, we have estimated the atmospheric lifetimes of HFE-7300 for the reaction with OH radical and Cl atom and are found to be 1.75 and 153.93 years, respectively. Additionally, the global warming potentials for HFE-7300 molecule are also estimated for 20-, 100-, and 500-year time horizons. Further, subsequent aerial oxidation of product radical (n-C2F5CF(OCH2)CF(CF3)2) in the presence of NO radical is performed, and it produced alkoxy radical via formation of peroxy radical. This alkoxy radical undergoes unimolecular decompositions via two different ways and formed n-C2F5CF(OCHO)CF(CF3)2 and n-C2F5CF(OH) CF(CF3)2 products.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrofluoroethers (HFEs) have emerged as the third-generation replacement of chlorofluorocarbons (CFCs), hydrofluorocarbons (HFCs), and hydrochlorofluorocarbons (HCFCs). HFEs have almost negligible ozone depletion potential (ODP) due to the absence of Cl atom. However, HFEs show moderately low global warming potential (GWPs) because of the absorption of IR radiation by C–F bonds (Devotta et al. 1994; Seikya and Misaki 1996; Bivens and Minor 1998; Sekiya and Misaki 2000; Blowers et al. 2008; Bravo et al. 2011a, 2011b. Highly fluorinated HFEs such as HFE-7500 (n-C3F7CF(OC2H5)CF(CF3)2), HFE-7300 (n-C2F5CF(OCH3)CF(CF3)2), HFE-7200 (n-C4F9OC2H5), HFE-7100 (n-C4F9OCH3), HFE-7000 (n-C3F7OCH3), etc. have various applications in the field of refrigerants and blowing and cleaning agents and as fuel additives, solvents, and medical products (Tsai 2005). Due to the wide range of applications, it is essential to explore the atmospheric impact of HFEs through the reaction with atmospheric oxidants and find its end degradation products.
In various degradation studies of HFEs (Papadimitriou et al. 2004; Díaz-de-Mera et al. 2009; Oyaro et al. 2004; Mishra et al. 2016; Tokuhashi et al. 2000), we observed that H atom can easily be abstracted from HFEs by oxidants such as OH radical and Cl atom. Note that the atmospheric lifetimes of HFEs or any volatile organic compounds (VOCs) are generally governed by the photolytic degradation (Kurylo and Orkin 2003). However, reaction with Cl atom is found to be faster than with the OH radical in the troposphere, although the global atmospheric concentration for OH radicals is known to be approximately two orders greater in magnitude than that of Cl atom concentration (Finlayson and Pitts Jr 2000). So, it is difficult to neglect the contribution of Cl atom toward the degradation of HFEs or VOCs compared to the OH radicals; especially in the coastal areas, where the concentration of Cl atom is higher than the OH radical (Spicer et al. 1998).
Oxidation of HFEs occurring at the tropospheric region is responsible for the production of corresponding fluorinated esters (FESs) (Bravo et al. 2011a, 2011b; Oyaro et al. 2004; Wallington et al. 1997; Ninomiya et al. 2000; Christensen et al. 1998; Wallington et al. 2002). For example, during the degradation of HFE-7100 (C4F9OCH3) and HFE7000 (C3F7OCH3), the fluoroakylformates C4F9OC(O)H and n-C3F7OC(O)H are produced as the major degradation products, respectively (Wallington et al. 1997; Ninomiya et al. 2000). On the other hand, degradation product fluoroalkylacetate C4F9OC(O)CH3 is formed from the HFE-7200 (C4F9OCH2CH3) (Christensen et al. 1998). The produced FESs have also the same number of C–F bonds in their molecular structures and actively contribute to global warming. Consequently, these HFEs lead to the formation of trifluoroacetic acid (TFA), which is classified as harmful to the agricultural and aquatic ecosystem (Jordan and Frank 1999).
Among HFEs, HFE-7300 (1,1,1,2,2,3,4,5,5,5-decafluoro-3-methoxy-4-trifluoromethyl-pentane) is also nonflammable, thermally stable, and non-ozone-depleting HFEs and used in heat transfer, lubricant deposition, electronic testing, and cleaning applications (M™ Novec™ 7300 Engineered Fluid n.d.). Though this compound is widely applicable, the Environmental Protection Agency (EPA) reported that it causes eye irritation, skin irritation, and respiratory tract irritation upon exposure as well as may be harmful to the ingestion of HFE-7300. Moreover, it is also recomm
ended an acceptable exposure limit (AEL) of 100 ppm on an 8-hour time-weighted average (TWA) (Federal Register 2017).
Therefore, to understand the atmospheric chemistry leading to its impact on global warming and climate change, the detailed kinetics and mechanistic degradation pathway studies of the HFE-7300 (n-C2F5CF(OCH3)CF(CF3)2) with OH radical and Cl atom are an urgent issue, which is discussed in this article. In this regard, Ana Rodríguez et al. (2014) first reported the rate constant values of 1.11 × 10−14 and 1.50 × 10−14 cm3 molecule−1 s−1 at 298 K for the reaction of n-C2F5CF(OCH3)CF(CF3)2 with OH radical and Cl atom, respectively, using GC/FID and GC/MS techniques. In this work, they have also reported the atmospheric lifetimes of HFE-7300 of 2.96 and 211.40 years for the reaction with OH radical and Cl atom, respectively. They also detected the degradation product n-CF3CF2CF(OCHO)CF(CF3)2 in the oxidation of HFE-7300 with the oxidants. These reported experimental measurements only provide the overall rate constant, and hence it is not possible to envisage the detailed mechanism and thermochemistry of reaction channels. Thus, to find the full insight of mechanistic pathways, kinetics, and thermochemistry of the titled reaction, quantum chemical calculations are carried out.
In this present investigation, we have performed the theoretical investigation using the density functional theory (DFT) method to explore the H-abstraction reaction pathways, thermochemistry, and kinetics of HFE-7300 reaction with OH radical and Cl atom. We have also determined the atmospheric lifetimes and global warming potentials (GWPs) of HFE-7300 to understand its impact on the atmosphere. In addition to this, we have further considered the aerial degradation pathways and thermochemistry of the product radical in the presence of NO radical and find the consequential end products.
Computational details
The meta-hybrid exchange and correlation density functional M06-2X (Zhao and Truhlar 2008) method along with standard basis set 6-31 + G(d,p) is employed for initial geometry optimization of all electronic structures, i.e., reactants (Rs), reaction complexes (RCs), transition states (TSs), product complexes (PCs), and product radicals and products (Ps). In this work, we have considered M06-2X functional (Zhao and Truhlar 2008) for the optimization and energetic calculations as it has been widely applicable with average mean absolute errors of about 1.3, 1.2, and 0.5 kcal mol−1, respectively, for thermochemical, barrier height, and non-covalent interaction calculations. In our titled reaction, only first- and second-row elements, i.e., C, H, F, O, and N atoms, are present, so we have chosen 6-31 + G(d,p) basis set along with M06-2X functional.
It is important to note that inthe case of product radical (n-C2F5CF(OCH2)CF(CF3)2) and alkoxy radical (n-C2F5CF(OC(O)H2)CF(CF3)2 ), the spin contaminations, i.e., < S2 >, are not significant because their value 0.7540 and 0.7543, respectively, before annihilation is slightly larger than the expected value of < S2 > = 0.7500. In addition to this, we have also performed the vibrational frequencies to verify the nature of all the stationary points and thermochemistry of the species. Further, intrinsic reaction coordinate (IRC) (Gonzalez and Schlegel 1989) calculation is carried out at the same level of theory to understand the progressive changes of molecular structures of Rs toward the TSs and then products in the minimum energy path (MEP). This Minnesota M06-2X functional is used widely for studying the chemical reactions involving radicals and found to be reliable for mechanism and rate constant calculation results (Rao et al. 2018; Ponnusamy et al. 2018; Wei et al. 2018; Gour et al. 2014; Hashemi et al. 2016; Paul et al. 2018); Paul et al. 2019; Here, all the calculations are performed using the Gaussian 09 program package (Frisch et al. 2009).
Results and discussion
To investigate the H-abstraction from –CH3 site of HFE-7300 molecule, we have first optimized and scanned the electronic structure of HFE-7300 along with the dihedral angle C − C − O − C at M06-2X/6-31 + G(d,p) level of theory. At the time of scanning, the rotation of the dihedral angle in the title molecule is considered from −180° to 180° in 10° increments. The scanned plot is shown in Fig. 1.

The electronic energies of the different conformers of HFE-7300 vs. the dihedral angle C − C − O − C (shown in shaded color) at M06-2X/6-31 + G(d,p) level of theory
Among the 36 optimized conformers of HFE-7300, we have obtained one most stable conformer having a total electronic energy (Eele) of −1641.2269751 Hartree (shown in Fig. 1). Therefore, H-abstraction reactions from HFE-7300 by OH radical and Cl atom are carried out from the most stable optimized conformer.
Reaction channels, electronic structures, and frequencies
Our optimized most stable conformer structure indicates that –OCH3 site of n-C2F5CF(OCH3)CF(CF3)2 has two equivalent hydrogen atoms and one nonequivalent hydrogen atom. However, the two equivalent hydrogen atoms are in the very close proximity of the nearest fluorine atoms. Due to this steric hindrance and weak interactions with fluorine atoms, H-abstractions from this site by OH radical and Cl atom are not easily feasible. Thus, here, one H-abstraction from n-C2F5CF(OCH3)CF(CF3)2 molecule is considered for each OH radical- and Cl atom-initiated reactions. The H-abstraction reaction channels (R1 and R2) are given below:
First, to know the nature and feasibility of the above primary reaction channels (R1 and R2), we have determined the enthalpy of reaction (ΔrH°298) and Gibb’s free energy (ΔrG°298) values at M06-2X/6-31 + G(d,p) level of theory, and values are reported in Table 1.
From Table 1, it is observed that both primary H-abstraction reaction channels (R1 and R2) are exothermic (ΔrH°298 < 0) and spontaneous (ΔrG°298 < 0) in nature. However, we found that OH-initiated H-abstraction reaction (i.e., R1) is more exothermic and thermodynamically more feasible than Cl atom-initiated H-abstraction reaction channel (R2).
For the reaction channels R1 and R2, the optimized geometries along with the bond lengths (in Å) of C–F, C–H, C–O, O–H, and H–Cl bonds of all stable species (Rs, RCs, PCs, and Ps) and transition states (TSs) are depicted in Fig. 2.

Optimized geometries of all species for primary H-abstraction reactions at M06-2X/6-31 + G(d,p) level of theory. Bond lengths are given in Å
We have observed that a reaction complex RC1 is formed in between reactants (n-C2F5CF(OCH3)CF(CF3)2 and OH) and TS1 for primary reaction channel R1. In RC1, two weak interactions (i.e., between the O atom of OH radical and H atom of −OCH3 site of distance 2.633 Å and H atom of OH radical and O atom of −OCH3 site of distance 2.153 Å) play a significant role in stabilizing the RC1. As the reaction progressed, in TS1, C–H bond has undergone a change to 1.182 Å from the C–H equilibrium bond length 1.091 Å in n-C2F5CF(OCH3)CF(CF3)2 molecule (i.e., 8.34% elongation). Additionally, a weak bond of distance 1.368 Å is formed between the O atom of OH radical and H atom of −OCH3 site of n-C2F5CF(OCH3)CF(CF3)2 molecule. The percentage of elongation of this bond from the equilibrium O–H bond length in the H2O molecule is approx. 41.37%. Thus, we observed a lower lengthening during the bond formation process than the bond breaking. It indicates that the transition state will be reactant-like and the reaction R1 will advance via “early transition state” as expected from Hammond’s postulate (Hammond 1955). Similarly, for the R2 channel, the RC2 is stabilized due to the weak interaction of the distance 2.995 Å between the Cl atom and H atom of the −OCH3 site of the n-C2F5CF(OCH3)CF(CF3)2 molecule (see Fig. 2). In TS2, we observed the lengthening of the C–H bond to 1.399 Å in correspondence to equilibrium distance of C–H bond (1.091 Å) in n-C2F5CF(OCH3)CF(CF3)2 (i.e., approx. 28.23% elongation), while the forming weak H–Cl bond distance is 1.471 Å. This weak H–Cl bond is elongated by 14.92% in corresponding to the H–Cl bond in the free HCl molecule. It explains that the barrier of the reaction R2 is close to the product like and the reaction passes through the late transition state which further follows the Hammond’s postulate (Hammond 1955). Further, we have obtained two product complexes (PC1 and PC2) with greater stability in comparison to the product radical n-C2F5CF(OCH2)CF(CF3)2 along with the elimination of H2O/HCl.
All the species involved in R1 and R2 channels are verified by real positive vibrational frequencies, while the TSs are confirmed by having only one imaginary frequency. The imaginary frequency for TS1 and TS2 are found to be 1152i cm−1 and 1002i cm−1, respectively. We have reported all possible modes of vibrational frequency values (cm−1) of all the species in supporting Table S1. Further, all the transitions states are verified by IRC calculations in which TS connects the reactant and product-like structure smoothly.
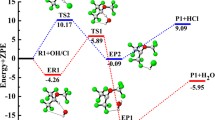
Zero-point corrected total energy of all species along with the relative energy w.r.t HFE-7300 + OH/Cl for the primary reaction channels (R1 and R2) is given in supporting Table S2 at M06-2X/6-31 + G(d,p) level of theory. Using the data given in Table S2, a schematic potential energy diagram of the oxidation of HFE-7300 + OH/Cl reaction is constructed and shown in Fig. 3.

Potential energy diagram for the HFE-7300 + OH/Cl reactions at M06-2X/6-31 + G(d,p) level of theory
We observed that the energy barriers of TS1 and TS2 are 2.90 kcal mol−1 and 0.84 kcal mol−1 with relative to the reactants, respectively. Since these energy barriers are small, the H-abstraction reactions are taking place via the formation of reaction complex (RC1 and RC2). The energy differences between the RC1 and TS1 and RC2 and TS2 are found to be 6.87 and 2.25 kcal mol−1. These energy differences suggest that the H-abstraction reaction channel R2 is kinetically more dominate than the reaction channel R1. Further, we have shown that product complexes PC1 and PC2 for reaction channels (R1 and R2) are formed with relative energies −19.59 and −2.82 kcal mol−1, respectively, before the formation of product radical (n-C2F5CF(OCH2)CF(CF3)2). We noticed that the PC1 and PC2 are energetically more stabilized than the product radical. The potential energy result indicates that the product (n-C2F5CF(OCH2)CF(CF3)2 + H2O) is energetically more stable than the product (n-C2F5CF(OCH2)CF(CF3)2 + HCl), which is in accordance with the thermochemistry results as discussed above.
Kinetic theories and rate constant calculations
To understand the reaction kinetics, the rate constants of the H-abstraction reaction channels are evaluated using the transition state theory (TST) (Laidler 2004) and canonical variation transition state theory (CVTST) (Garrett et al. 1986; Truhlar and Garrett 1984). The rate constant values are obtained within the temperature range 250–1000 K for these two kinetic theories with the help of KiSThelP program (Canneaux et al. 2014). In previous studies (Díaz-de-Mera et al. 2009; Rao et al. 2018), we observed that rate constants of H-abstraction from HFEs initiated by oxidants are independent of the total pressure. So, we have not determined the pressure dependence rate constants in this work.
First, the rate constant of the H-abstraction reaction channels are calculated from the TST theory by using the following Eq. 1
In Eq. 1, σr stands for the symmetry number which depends on the reaction path degeneracy, and Γ(T) is the tunneling correction factor at temperature T, which is calculated by Eckart’s unsymmetrical barrier method (Johnston and Heicklen 1962). ΔE# is the barrier height and other terms have the usual meaning. Here Q‡TS and QR are the total partition functions for TS and R, respectively, and these are obtained by using harmonic oscillator approximations. Due to the spin-orbit coupling, partition functions are also corrected for OH radical and Cl atom. It is important to note that a fast equilibrium supposition has been made between the reactants and pre-reactive complex for calculation of rate constant as mentioned by Singleton and Cvetanovic (Singleton and Cvetanovic 1976).
Next, the rate constants are calculated from the CVTST (Garrett et al. 1986; Truhlar and Garrett 1984) theory, which is based on the notion of varying the TS dividing surface along the reaction coordinate that minimizes the rate constant with the use of the following equations:
Here, kGT and kCVTST indicate the rate constants generalized from TST and CVTST. QGT and ΦR represent partition functions for TS and reactants, respectively, and VMEP(s) is the potential energy of the generalized TS at reaction coordinates. The rate constant values obtained from the TST and CVTST theories within the temperature range of 250–1000 K for reaction channels (R1 and R2) are given in Table 2.
The calculated rate constants (kOH) for HFE-7300 + OH reaction are found to be 1.81 × 10−14 cm3 molecule−1 s−1 and 8.62 × 10−16 cm3 molecule−1 s−1 using TST and CVTST, respectively. The calculated rate constant value (at 298 K) using TST is close to the experimental value 1.11 × 10−14 cm3 molecule−1 s−1 reported by Ana Rodríguez et al. (2014). Further, the calculated rate constants (kCl) for HFE-7300 + Cl reaction are found to be 3.46 × 10−12 cm3 molecule−1 s−1 and 2.06 × 10−13 cm3 molecule−1 s−1 using TST and CVTST, respectively. In this case, rate constant value reported by CVTST is found to be in good agreement with the experimental rate constant value 1.50 × 10−13 cm3 molecule−1 s−1 reported by Ana Rodríguez et al. (2014). The better value of kcl obtained at CVTST than the TST is because the energy barrier is very low for HFE-7300 + Cl reaction.
Further, in Fig. 4(a, b), we have shown the temperature dependence on the change in the value of kOH and kcl. It is observed that kOH and kcl are increasing with the increasing temperatures.

Variation of rate constants versus temperature for a HFE-7300 + •OH reaction and b HFE-7300 + Cl reaction along with experimental rate constant.
We have also fitted our calculated rate constant values of the titled reaction within the temperature range 250–450 K by using the modified three parameters Arrhenius expression (( i.e., k = ATn exp(−Ea/RT)), the results are shown below:
For TST theory
$$ {\mathrm{k}}_{\mathrm{OH}}=6.51\times {10}^{-23}\ {\mathrm{T}}^{3.47}\exp \left(-90.71/\mathrm{T}\right) $$(4)$$ {\mathrm{k}}_{\mathrm{Cl}}=1.43\times {10}^{-19}\ {\mathrm{T}}^{2.83}\ \exp \left(250.43/\mathrm{T}\right) $$(5)For CVTST theory
$$ {\mathrm{k}}_{\mathrm{OH}}=1.54\times {10}^{-15}\ {\mathrm{T}}^{0.20}\ \exp \left(-515.14/\mathrm{T}\right) $$(6)$$ {\mathrm{k}}_{\mathrm{Cl}}=6\times {10}^{-18}\ {\mathrm{T}}^{1.73}\exp \left(170.07/\mathrm{T}\right) $$(7)
Atmospheric lifetime and global warming potentials
Equation 8 serves the purpose of the evaluation of the OH radical-driven atmospheric lifetime of HFEs. For the evaluation of lifetime in Eq. 8, it takes into account the use of 272 K as scaled temperature and the atmospheric lifetime of methyl chloroform (MCF).
where \( {\uptau}_{OH}^{MCF} \)represents the lifetime of HFE-7300 and kHFE and kMCF represent the rate constants for the reactions of HFE-7300 and methyl chloroform (MCF) with OH radicals at 272 K and \( {\uptau}_{OH}^{MCF}\kern0.5em \)= 5.99 years (Spicer et al. 1998). Taking kMCF = 6.14 × 10−15 (Spivakovsky et al. 2000) and our calculated kHFE−7300 = 1.29 × 10−14 cm3 molecule−1 s−1 (TST) at 272 K, the estimated lifetime is found to be 2.91 years, which is reasonably in good agreement with the value of 5.24 years reported by Ana Rodríguez et al. (2014).
The atmospheric lifetime τOH and τCl of HFE-7300 molecule can be obtained by assuming that its elimination from troposphere occurs only by the reactions with OH radicals and Cl atom, respectively. The value of τOH and τCl can be obtained from the following Eq. 9 and Eq. 10.
Here, [OH] and [Cl] represent the global weighted average OH radical and Cl atom concentrations, and its values are 1.0 × 106 molecule cm−3 and 1.0 × 104 molecule cm−3, respectively (Hein et al. 1997; Wingenter et al. 1996). By taking the value of the calculated rate constant kOH = 1.81 × 10−14 cm3 molecule−1 s−1 and kCl = 2.06 × 10−13 cm3 molecule−1 s−1 at 298 K, the individual atmospheric lifetimes (τOH and τCl) for the titled molecule are calculated using Eqs. 9 and 10. The calculated τOH and τCl values are found to be 1.75 years and 153.93 years, respectively, which is reasonably in good agreement with the reported value of 2.96 years (OH-initiated) and 211.40 years (Cl initiated) at 298 K given by Ana Rodríguez et al. [22]. The atmospheric lifetimes of HFE-7300 are found in years, which suggest that it can contribute to the increase of GWPs. Thus, we have estimated the GWPs (Hammitt et al. 1996) of the title molecule.
GWP is an essential atmospheric parameter, and it provides the relative measure of how much heat a greenhouse gas traps in the atmospheric environment. Hodnebrog et al. (2013) gives an expression for the estimation of GWP of any VOCs or HFEs with respect to CO2, which is given as
where AGWPi and AGWPCO2 are the absolute GWP of gas i (i.e., HFE-7300) and reference gas CO2, respectively. AGWPi (Hodnebrog et al. 2013) depends on radiative forcing efficiency (Aior RE) and lifetime (τ) of HFE-7300 molecule, and it is expressed as:
Here, H is the time horizon and the value of Ai is given in the unit of W m−2 ppb−1. The infrared intensities and the wave numbers of harmonic vibrational modes are obtained at M06-2X level of theory as suggested by Bravo et al. (2010, 2011a, b), and then these wave numbers are scaled by \( {\overline{\nu}}_{scal}=0.977{\overline{\nu}}_{calc}+11.664 \)cm−1. After that, the radiative forcing efficiency (RE) of HFE-7300 molecule is calculated by the expression (13) given by Pinnock et al. (1995).
Here, Ak is the absorption cross section in cm2 molecule−1, and F\( \left({\overline{\nu}}_k\right) \) represents the instantaneous, cloudy sky, radiative forcing per unit cross section per wave number.
In Table 3, we have given the calculated RE and GWP values of HFE-7300 for the particular time interval 20, 100, or 500 years of time horizon. The estimated RE of the HFE-7300 is found to be 0.38 W m−2 ppb−1 which is slightly lower than the reported RE given by Ana Rodríguez et al. (2014). To calculate the GWPs, we have used the updated values of AGWP for CO2 of 2.495 × 10−14, 9.171 × 10−14, and 32.17 × 10−14 W m−2 yr (kgCO2)−1 for time horizons of 20, 100, and 500 years, respectively, as given by Hodnebrog et al. (2013). Our estimated GWP values of HFE-7300 at 272 K are 736, 196, and 56 for the 20, 100, and 500 years, respectively. These calculated GWPs at 272 K are found to be lower than the GWPs estimated by Rodríguez et al. (2014), which is because our calculated lifetime and RE values are slightly lower than their values. Further, we have also estimated the GWPs of HFE-7300 at 298 K for the reaction with OH radical and Cl atom and given in Table 3. The GWP values are found to be 445, 118, and 34 for the OH-initiated reaction and 4746, 4943, and 2848 for Cl-initiated reaction with HFE-7300 for the 20, 100, and 500 years, respectively. Our computed GWP values indicate that these values are significantly lower than the CFC-11 (Hodnebrog et al. 2013). Thus, it may contribute to increase the GWPs and make a significant contribution to the radiative forcing of climate change.
Aerial degradation of product radical [n-C2F5CF(OC•H2)CF(CF3)2]
As we have discussed in the above section, the product radical, i.e., n-C2F5CF(OC•H2)CF(CF3)2, is formed from the H-abstraction reaction of n-C2F5CF(OCH3)CF(CF3)2 by OH radical and Cl atom, which further oxidized with O2 molecule to produce peroxy radical n-C2F5CF(OC(OO•)H2)CF(CF3)2. This peroxy radical readily reacts with NO radical (which has significantly higher concentration in the troposphere than the OH radical) and ultimately formed n-C2F5CF(OC(O•)H2)CF(CF3)2 radical (known as alkoxy radical) via the formation of n-C2F5CF(OC(OONO2)H2)CF(CF3)2 intermediate. Since alkoxy radicals act as crucial intermediates in the atmospheric oxidation of halogenated hydrocarbons (Orlando et al. 2003; Vereecken and Francisco 2012; Gour et al. (2017, 2019), these radicals have become the important species for experimental and theoretical studies among the researchers. Due to the significant role played by alkoxy radicals formed in the destruction of a variety of volatile organic compounds, we have addressed the importance of studying the fate of n-C2F5CF(OC(O)H2)CF(CF3)2 radical from the viewpoint of understanding its role in the atmosphere. To the best of our knowledge, we have not observed any detailed theoretical elucidation of aerial degradation pathways of product radical. The degradation of n-C2F5CF(OCH2)CF(CF3)2 and unimolecular decomposition of n-C2F5CF(OC(O)H2)CF(CF3)2 in the atmosphere are envisaged to occur via the following reactions:
First, the thermochemistry of the above reactions (R3 to R6) is calculated at the same level of theory. The values of ΔH°r,298 and ΔG°r,298 for reaction channels R3 to R6 are also listed in Table 1. From the Table, we observed that the formation of n-C2F5CF(OC(OO)H2)CF(CF3)2 radical and subsequent formation of n-C2F5CF(OC(O•)H2)CF(CF3)2 radical (i.e., R3 and R4) are highly exothermic (i.e., ΔH°r,298 < 0) and spontaneous (ΔG°r,298 < 0) in nature. Further, it is found that the unimolecular decomposition reaction of R5 is thermodynamically not feasible (i.e., endothermic and non-spontaneous). On the other hand, the unimolecular decomposition reaction of R6 (i.e., n-C2F5CF(OC(O)H2)CF(CF3)2 → n-C2F5CF(OH)CF(CF3)2 + CHO) is thermodynamically feasible with the ΔH°r,298 and ΔG°r,298 values of−1.66 and−15.21 kcal mol−1 , respectively.
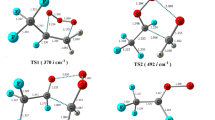
Further, geometries of the all species involved in reaction channels R3 to R6 along with TSs are optimized at the same level of theory as used earlier and are shown in Fig. 5. In this unimolecular decomposition reaction channels R5 and R6, transition states (TS3 and TS4) are characterized by only one imaginary frequency obtained at 979i cm−1 and 537i cm−1, respectively, and other species are characterized by positive frequencies which are also given in Table S1. In the optimized structure of TS3, the length of C–H bond of CH2O site elongated to 1.718 Å with respect to C–H bond of alkoxy radical of distance 1.103 Å. At the same time in TS3, C–O bond of –CH2O site decreased to 1.204 Å from 1.330 Å as well as C–O bond of ether linkage shrinkage to 1.395 Å from 1.439 Å. In the same way in TS4, we observed the increases of the length of C–O bond of ether linkage from 1.439 to 1.983 Å and decreases of the C–O bond of –CH2O site from 1.330 to 1.196 Å.

Optimized geometries of all the species in the aerial degradation of product radical in the presence of NO radical at M06-2X/6-31 + G(d,p) level of theory
Moreover, the zero-point corrected total energies of all the aerial degradation reactions (R3–R6) are given in the Tables S2. Following the observed changes in energies, we have constructed the PES diagram for the degradation pathway of product radical from the Table S2, and it is shown in Fig. 6. From the figure, we have found that the relative energy of the n-C2F5CF(OC(OO)H2)CF(CF3)2 w.r.t P1 radical and O2 is −30.96 kcal mol−1. It suggests that the peroxy radical formation from P1 radical is the kinetically dominant reaction, which is consistent with the thermodynamic results. Further, the relative energy of the n-C2F5CF(OC(O)H2)CF(CF3)2 radical formed from C2F5CF(OC(OO)H2)CF(CF3)2 radical in the presence of NO radical is −48.31 kcal mol−1. This n-C2F5CF(OC(O)H2)CF(CF3)2 radical undergoes two unimolecular decomposition reactions (R5 and R6) through the formation of transition states (TS3 and TS4). The relative energy of TS3 and TS4 are found to be −26.21 and −21.61 kcal mol−1. The relative energy of the products (n-C2F5CF(OCHO)CF(CF3)2 + H and n-C2F5CF(OH)CF(CF3)2 + CHO) are −33.93 and −49.97 kcal mol−1, respectively. These observations indicate that reaction channel R5 is a kinetically dominant reaction than the reaction channel R6. However, products formed from the R6 reaction channel is energetically more stable than the products formed from the R5 reaction channel. In addition to this, the R6 reaction channel is also thermodynamically more feasible than the R5 reaction channel. Moreover, Ana Rodríguez et al. (2014) proposed the decomposition of n-C2F5CF(OC(O)H2)CF(CF3)2 radical to n-CF3CF2CF(OCHO)CF(CF3)2 and n-CF3CF2CF(O)CF(CF3)2, but do not discussed about the feasibility of the decomposition reactions. In our case, we observed one similar product, i.e., CF3CF2CF(OCHO)CF(CF3)2, as proposed by Rodríguez et al. (2014). But we observed other decomposition product n-CF3CF2CF(OH)CF(CF3)2 with the elimination of CHO radical than their reported n-CF3CF2CF(O)CF(CF3)2 product with the elimination of CH2O molecule.

Schematic potential energy diagram for the aerial degradation of product radical in the presence of NO at M06-2X/6-31 + G(d,p) level of theory
Conclusions
The OH radical and Cl atom initiated H-abstraction reactions of n-C2F5CF(OCH3)CF(CF3)2, and subsequent degradation of its product radical has been studied in detailed at M06-2X/6-31 + G(d,p) level of theory. Energy values for all species involved in reaction channels along with intermediates and transition states have been represented by potential energy diagrams. Results show that Cl atom-initiated H atom abstraction reaction (R2) is kinetically more dominant than OH radical-initiated H atom abstraction reaction, while thermodynamic values of the abstraction reactions (R1 and R2) indicate that reaction channel R1 is thermodynamically more feasible. Further, a good agreement of our calculated rate constant has been found with the experimental rate constant obtained by using TST for the HFE-7300 reaction with OH radical. On the other hand, the rate constant reported for the HFE-7300 reaction with Cl atom is found to be close to the experimental rate constant obtained by using CVTST. The estimated atmospheric lifetimes for HFE-7300 by the reactions with OH radical and Cl atom are found to be 1.75 years and 154 years, respectively, which are reasonably in good agreement with reported lifetimes by Rodríguez et al. (2014). Due to the slightly higher atmospheric lifetime of HFE-7300, our GWP analyses suggest that it may make a moderate contribution to increasing global warming.
In addition to these primary reaction investigations, we have further carried out a study of the aerial degradation of product radical in the presence of NO radical via the formation of n-C2F5CFOCH2(O)CF(CF3)2 radical. The thermodynamic calculations of the unimolecular decomposition channels show that the reaction channel R6 is thermodynamically more feasible than R5 reaction, whereas potential energy calculations suggest that R5 is kinetically dominant. During the study of the decomposition reactions, we obtained n-C2F5CF(OCHO)CF(CF3)2 (1,1,1,2,2,3,4,5,5,5-decafluoro-4-(trifluoromethyl)pentan-3-yl formate) and n-C2F5CF(OH)CF(CF3)2 (1,1,1,2,2,3,4,5,5,5-decafluoro-4-(trifluoromethyl)pentan-3-ol) two end products which have similar no. of C–F bonds as that of HFE-7300 molecule and may be further contribute to the global warming potentials by absorbing the IR radiation. Our present study signifies that both primary H-abstraction reactions and subsequent aerial degradation reactions of product radical are important to understand the atmospheric chemistry of such types of compounds.
References
Bivens DB, Minor BH (1998) Fluoroethers and other next generation fluids. Int J Refrig 21:567–576
Blowers P, Moline DM, Tetrault KF, Wheeler RNR, Tuchawena SL (2008) Global warming potentials of hydrofluoroethers. Environ Sci Technol 42:1301–1307
Bravo I, Aranda A, Hurley MD, Marston G, Nutt DR, Shine KP, Smith K, Wallington TJ (2010) Infrared absorption spectra, radiative efficiencies, and global warming potentials of perfluorocarbons: Comparison between experiment and theory. J Geophys Res-Atmos 115:D24317 (1-12)
Bravo I, Díaz-de-Mera Y, Aranda A, Moreno E, Nutt DR, Marston G (2011a) Radiative efficiencies for fluorinated esters: indirect global warming potentials of hydrofluoroethers. Phys Chem Chem Phys 13:17185–17193
Bravo I, Marston G, Nutt DR, Shine KP (2011b) Radiative efficiencies and global warming potentials using theoretically determined absorption cross-sections for several hydrofluoroethers (HFEs) and hydrofluoropolyethers (HFPEs). J Quant Spectrosc Radiat Transf 112:1967–1977
Canneaux S, Bohr F, Henon E (2014) KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J Comput Chem 35:82–93
Christensen LK, Sehested J, Nielsen OJ, Bilde M, Wallington TJ, Guschin A, Molina LT, Molina MJ (1998) Atmospheric chemistry of HFE-7200 (C4F9OC2H5): reaction with OH radicals and fate of C4F9OCH2CH2O• and C4F9OCHO•CH3 radicals. J Phys Chem A 102:4839–4845
Devotta S, Gopichand S, Pendyala VR (1994) Comparative assessment of some HCFCs, HFCs and HFEs as alternatives to CFC11. Int J Refrig 17:32–39
Díaz-de-Mera Y, Aranda A, Bravo I, Moreno E, Martínez E, Rodríguez A (2009) Atmospheric HFEs degradation in the gas phase: reaction of HFE-7500 with Cl atoms at low temperatures. Chem Phys Lett 479:20–24
Federal Register, 2017, 82 (No.139), 33809 (https://www.govinfo.gov/content/pkg/FR-2017-07-21/pdf/2017-15379.pdf).
Finlayson BJ, Pitts JN Jr (2000) Chemistry of the upper and lower atmosphere: theory, experiments, and application. Academic Press, San Diego
Frisch MJ et al (2009) Gaussian 09. Revision D.01. Gaussian, Inc, Wallingford, CT.
Garrett BC, Truhlar DG, Schatz GC (1986) Test of variational transition state theory and multidimensional semiclassical transmission coefficients methods against accurate quantal rate constants for H+ H2/HD, D+ H2, and O+ H2/D2/HD, including intra-and intermolecular kinetic isotope effects. J Am Chem Soc 108:2876–2881
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J Chem Phys 90:2154–2161
Gour NK, Deka RC, Singh HJ, Mishra BK (2014) A computational perspective on mechanism and kinetics of the reactions of CF3C(O)OCH2CF3 with OH radicals and Cl atoms at 298 K. J Fluor Chem 160:64–71
Gour NK, Mishra BK, Sarma PJ, Begum P, Deka RC (2017) Tropospheric degradation of HFE-7500 [n-C3F7CF(OCH2CH3)CF(CF3)2] initiated by OH radicals and fate of alkoxy radical [n-C3F7CF (OCH (O)CH3)CF(CF3) 2]: a DFT investigation. J Fluor Chem 204:11–17
Gour NK, Borthakur K, Paul S, Deka RC (2019) Tropospheric degradation of 2-fluoropropene (CH3CF=CH2) initiated by hydroxyl radical: reaction mechanisms, kinetics and atmospheric implications from DFT study. Chemosphere 238:124556
Hammitt JK, Jain AK, Adams JL, Wuebbles DJ (1996) A welfare-based index for assessing environmental effects of greenhouse-gas emissions. Nature 381:301–303
Hammond GS (1955) A correlation of reaction rates. J Am Chem Soc 77:334–338
Hashemi SR, Saheb V, Hosseini SMA (2016) Theoretical studies on the mechanism and kinetics of the hydrogen abstraction reactions from C4F9OC2H5 (HFE-7200) by OH and Cl radicals. J Fluor Chem 187:9–14
Hein R, Crutzen PJ, Heimann M (1997) An inverse modeling approach to investigate the global atmospheric methane cycle. Glob Biogeochem Cycles 11:43–76
Hodnebrog Ø, Etminan M, Fuglestvedt JS, Marston G, Myhre G, Nielsen CJ, Shine KP, Wallington TJ (2013) Global warming potentials and radiative efficiencies of halocarbons and related compounds: a comprehensive review. Rev Geophys 51:300–378
Johnston HS, Heicklen J (1962) Tunnelling corrections for unsymmetrical Eckart potential energy barriers. J Phys Chem 66:532–533
Jordan A, Frank H (1999) Trifluoroacetate in the environment. Environ Sci Technol 33:522–527
Kurylo MJ, Orkin VL (2003) Determination of atmospheric lifetimes via the measurement of OH radical kinetics. Chem Rev 103:5049–5076
Laidler KJ (2004) Chemical kinetics, 3rd edn. Pearson Education, Delhi
Mishra BK, Gour NK, Bhattacharjee D, Deka RC (2016) Atmospheric chemistry of HFE-7000 (i-C3F7OCH3) and isofluoro-propyl formate (i-C3F7OC(O) H): reactions with OH radicals, atmospheric lifetime and fate of alkoxy radical (i-C3F7OCH2O•)–a DFT study. Mol Phys 114:618–626
M™ Novec™ 7300 Engineered Fluid (n.d.) Product Information (https://multimedia.3m.com/mws/media/338713O/3m-novec-7300-engineered-fluid.pdf)
Ninomiya Y, Kawasaki M, Guschin A, Molina LT, Molina MJ, Wallington TJ (2000) Atmospheric chemistry of n-C3F7OCH3: reaction with OH radicals and Cl atoms and atmospheric fate of n-C3F7OCH2O· radicals. Environ Sci Technol 34:2973–2978
Orlando JJ, Tyndall GS, Wallington TJ (2003) The atmospheric chemistry of alkoxy radicals. Chem Rev 103:4657–4690
Oyaro N, Sellevåg SR, Nielsen CJ (2004) Study of the OH and Cl-initiated oxidation, IR absorption cross-section, radiative forcing, and global warming potential of four C4-hydrofluoroethers. Environ Sci Technol 38:5567–5576
Papadimitriou VC, Kambanis KG, Lazarou YG, Papagiannakopoulos P (2004) Kinetic study for the reactions of several hydrofluoroethers with chlorine atoms. J Phys Chem A 108:2666–2674
Paul S, Deka RC, Gour NK (2018) Kinetics, mechanism, and global warming potentials of HFO-1234yf initiated by O3 molecules and NO3 radicals: insights from quantum study. Environ Sci Pollut Res 25:26144–26156
Paul S, Gour NK, Deka RC (2019) Mechanistic investigation of the atmospheric oxidation of bis (2-chloroethyl) ether (ClCH2CH2OCH2CH2Cl) by OH and NO3 radicals and Cl atoms: a DFT approach. J Mol Model 25:43 (1-9)
Pinnock S, Hurley MD, Shine KP, Wallington TJ, Smyth TJ (1995) Radiative forcing of climate by hydrochlorofluorocarbons and hydrofluorocarbons. J Geophys Res-Atmos 100:23227–23238
Ponnusamy S, Sandhiya L, Senthilkumar K (2018) Atmospheric oxidation mechanism and kinetics of hydrofluoroethers, CH3OCF3, CH3OCHF2, and CHF2OCH2CF3, by OH radical: a theoretical study. J Phys Chem A 122:4972–4982
Rao PK, Deka RC, Gour NK, Gejji SP (2018) Understanding the atmospheric oxidation of HFE-7500 (C3F7CF(OC2H5)CF(CF3)2) initiated by Cl Atom and NO3 radical from theory. J Phys Chem A 122:6799–6808
Rodríguez A, Rodríguez D, Moraleda A, Bravo I, Moreno E, Notario A (2014) Atmospheric chemistry of HFE-7300 and HFE-7500: temperature dependent kinetics, atmospheric lifetimes, infrared spectra and global warming potentials. Atmos Environ 96:145–153
Seikya A, Misaki S (1996) A continuing search for new refrigerants. Chemtech 26:44–48
Sekiya A, Misaki S (2000) The potential of hydrofluoroethers to replace CFCs, HCFCs and PFCs. J Fluor Chem 101:215–221
Singleton DL, Cvetanovic RJ (1976) Temperature dependence of the reaction of oxygen atoms with olefins. J Am Chem Soc 98(22):6812–6819
Spicer CW, Chapman EG, Finlayson-Pitts BJ, Plastridge RA, Hubbe JM, Fast JD, Berkowitz CM (1998) Unexpectedly high concentrations of molecular chlorine in coastal air. Nature 394:353–356
Spivakovsky CM, Logan JA, Montzka SA, Balkanski YJ, Foreman-Fowler M, Jones DBA, Horowitz LW, Fusco AC, Brenninkmeijer CAM, Prather MJ, Wofsy SC (2000) Three-dimensional climatological distribution of tropospheric OH: update and evaluation. J Geophys Res-Atmos 105:8931–8980
Tokuhashi K, Takahashi A, Kaise M, Kondo S, Sekiya A, Yamashita S, Ito H (2000) Rate constants for the reactions of OH radicals with CH3OCF2CHF2, CHF2OCH2CF2CHF2, CHF2OCH2CF2CF3, and CF3CH2OCF2CHF2 over the temperature range 250 − 430 K. J Phys Chem A 104:1165–1170
Truhlar DG, Garrett BC (1984) Variational transition state theory. Annu Rev Phys Chem 35:159–189
Tsai WT (2005) Environmental risk assessment of hydrofluoroethers (HFEs). J Hazard Mater 119:69–78
Vereecken L, Francisco JS (2012) Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem Soc Rev 41:6259–6293
Wallington TJ, Schneider WF, Sehested J, Bilde M, Platz J, Nielsen OJ, Christensen LK, Molina MJ, Molina LT, Wooldridge PW (1997) Atmospheric chemistry of HFE-7100 (C4F9OCH3): reaction with OH radicals, UV spectra and kinetic data for C4F9OCH2· and C4F9OCH2O2· radicals, and the atmospheric fate of C4F9OCH2O· radicals. J Phys Chem A 101:8264–8274
Wallington TJ, Hurley MD, Fedotov V, Morrell C, Hancock G (2002) Atmospheric chemistry of CF3CH2OCHF2 and CF3CHClOCHF2: kinetics and mechanisms of reaction with Cl atoms and OH radicals and atmospheric fate of CF3C(O•)HOCHF2 and CF3C(O•)ClOCHF2 radicals. J Phys Chem A 106:8391–8398
Wei B, Sun J, Mei Q, An Z, Wang X, He M (2018) Theoretical study on gas-phase reactions of nitrate radicals with methoxyphenols: mechanism, kinetic and toxicity assessment. Environ Pollut 243:1772–1780
Wingenter OW, Kubo MK, Blake NJ, Smith TW Jr, Blake DR, Rowland FS (1996) Hydrocarbon and halocarbon measurements as photochemical and dynamical indicators of atmospheric hydroxyl, atomic chlorine, and vertical mixing obtained during Lagrangian flights. J Geophys Res-Atmos 101:4331–4340
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215–241
Acknowledgments
Dr. SP is thankful to the University Grant Commission (UGC), New Delhi, for providing financial support from Dr. D. S. Kothari Post-Doctoral Fellowship (Award letter no: F.4-2/2006(BSR)/CH/16-17/0152). S. D. Baruah is thankful to the Department of Science and Technology (DST), New Delhi, for providing him INSPIRE fellowship (No.IF160658).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict(s) of interest.
Additional information
Responsible Editor: Philipp Gariguess
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM1
(DOCX 34 kb)
Rights and permissions
About this article

Cite this article
Paul, S., Mishra, B.K., Baruah, S.D. et al. Atmospheric oxidation of HFE-7300 [n-C2F5CF(OCH3)CF(CF3)2] initiated by •OH/Cl oxidants and subsequent degradation of its product radical: a DFT approach. Environ Sci Pollut Res 27, 907–920 (2020). https://doi.org/10.1007/s11356-019-06975-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-06975-1