
Abstract
The detection, distribution, and long-term behavior of 241Am in the terrestrial environment at the Waste Isolation Pilot Plant (WIPP) site were assessed using historical data from an independent monitoring program conducted by the Carlsbad Environmental Monitoring & Research Center (CEMRC), and its predecessor organization the Environmental Evaluation Group (EEG). An analysis of historical data indicates frequent detections of trace levels of 241Am in the WIPP environment. Positive detections and peaks in 241Am concentrations in ambient air samples generally occur during the March to June timeframe, which is when strong and gusty winds in the area frequently give rise to blowing dust. A study of long-term measurements of 241Am in the WIPP environment suggest that the resuspension of previously contaminated soils is likely the primary source of americium in the ambient air samples from WIPP and its vicinity. Furthermore, the 241Am/239 + 240Pu ratio in aerosols and soils was reasonably consistent from year to year and was in agreement with the global fallout ratios. Higher than normal activity concentrations of 241Am and 241Am/239 + 240Pu ratios were measured in aerosol samples during 2014 as a result of February 14, 2014 radiation release event from the WIPP underground. However, after a brief spike, the activity concentrations of 241Am have returned to the normal background levels. The long-term monitoring data suggest there is no persistent contamination and no lasting increase in radiological contaminants in the region that can be considered significant by any health-based standard.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Americium (Am) is an anthropogenic, α-emitting radionuclide produced in small quantities in nuclear reactors and by the decay of 241Pu. Its occurrence in the environment can be attributed to three sources: fallout from nuclear weapons testing, releases from nuclear reactors and reprocessing plants, and production and disposal of smoke detectors by producers and consumers. The americium isotopes range from 232Am to 247Am, of which 241Am is the most important isotope found in the environment. It is also the most prevalent isotope of americium in nuclear waste and because of the low penetration of α-radiation, 241Am only poses a health risk when ingested or inhaled (UNSCEAR, Report 1982). 241Am also emits 59.54 keV gamma radiation with an intensity of 36%. However, due to its low energy, the health risk from gamma is significantly lower than that from alpha emissions of 241Am. Not only is the half-life (t1/2) of 432.2 years much longer than that of its 241Pu parent (t1/2 = 14.3 years), but 241Am is much more radiotoxic and mobile. The 241Am subsequently decays to 237Np, also an α-emitter (t1/2 = 2.1 × 106 years). In the short-term assessment of dose, either due to atmospheric testing or severe reactor accidents, the contribution of 241Pu is important and often underestimated. However, in the long-term management of contamination due to fall out, nuclear accidents, and transuranic (TRU) waste disposal, 241Am and 237Np present much higher risks. In this context, understanding the spatial and temporal distribution of 241Am in the environment, at or near reprocessing and disposal facilities, is crucial for assessing the long-term radiological dose. This radionuclide is therefore of particular interest at the U.S. Department of Energy’s (USDOE) WIPP, where 239 + 240Pu and 241Am account for more than 99% of the total radioactivity slated for disposal (USDOE 2014).
The WIPP, located near Carlsbad in southeastern New Mexico, is the only operating deep geological repository for defense-related TRU and mixed transuranic waste (MTRU), i.e., waste containing both TRU and hazardous wastes, in the USA. The repository is mined into a thick Permian-age sequence of interbedded salt (halite) and anhydrite, known as the Salado Formation, ~ 655 m (2150 ft.) below ground surface that is essentially non-radiogenic. The facility is authorized by the WIPP Land Withdrawal Act (Public Law 102-579) n.d.) to dispose of 175,000 m3 of TRU waste. Since starting operations in March 1999, some 91,000 m3 of waste have been emplaced in the repository. The facility recently resumed waste disposal operations after a 3-year hiatus due to an accidental radiological release in February 2014. It is estimated that the WIPP repository will contain 1.20 × 104 kg of Pu isotopes and 203 kg of 241Am at closure (USDOE 2014). The transuranic actinides are the key constituents of nuclear waste for long-term considerations as many of them are α-emitters and have very long half-lives. Even though americium are less widely discussed compared to plutonium isotopes, the separation of americium from nuclear waste streams is a major goal of fuel reprocessing research. In this context, a clear understanding of the coordination and environmental behavior of the americium is essential for the safe management and disposal of radioactive waste (Welch et al. 2017; Dares et al. 2015). The stable oxidation state of Am in the environment is Am(III). However, formation of higher oxidation states of americium such as AmO2, Am(V)O2+, and Am(VI)O22+ have been reported in acidic media. Selective oxidation of trivalent americium has been used in the separation of americium from the lanthanides in nuclear waste streams (Dares et al. 2015).
Generally, as with the other transuranium elements, most of the americium present in the environment today is due to atmospheric nuclear tests that were conducted between 1945 and 1980. It is estimated that nuclear weapons testing dispersed some 13 PBq (1 PBq = 1015 Bq) of 239 + 240Pu and 3.1 PBq of 241Am into the atmosphere (UNSCEAR 1982). Nuclear reactor accidents, such as Chernobyl and Fukushima, and other releases from weapons production facilities have caused localized contamination in some areas. Since 241Am is the β-decay daughter of the short-lived 241Pu, its concentration continues to increase in the environment. Concentrations of 241Am are estimated to peak 70–80 years following an accidental release of 241Pu (US-EPA 1976). Generally, 241Am and plutonium isotopes have been measured as traces in environmental samples with a 241Am/239 + 240Pu activity ratio of ~ 0.37 and 238Pu/239 + 240Pu activity ratio of ~ 0.024 at latitudes of 40° to 50° N, indicating their global fallout origin (UNSCEAR 2000). However, the 241Am/239 + 240Pu ratio is expected to increase with time, reaching ~ 0.40 by 2035 (Leon Vintro et al. 1999).
The background radiation levels at the WIPP are influenced primarily by natural radioactivity and global fallout from nuclear weapons tests with a possible contributor being anthropogenic radioactivity remaining from Project Gnome. The Project Gnome was the first nuclear test conducted in the Plowshare Program that focused on developing peaceful uses for nuclear explosives (USAEC 1973). The December 1961 test detonated a 239Pu device, with a 3.0-kt equivalent TNT yield, ~ 366 m below ground surface in a thick salt deposit. The Gnome plume reportedly traveled mostly north-northwest not too far from the WIPP site. Although it had been planned as a contained explosion, radioactive materials were vented to the surface. The Gnome site is located only 8.8 km southwest of the WIPP site and falls within the pre-operational radiological surveillance area for the WIPP. The site was decontaminated several times following the test and surface contamination is now well below levels of public health and environmental concern. However, prior to the opening of the WIPP, low levels of 137Cs, 241Am, and 239 + 240Pu were still detectable in surface soils at the Gnome site (Faller 1994; Kenney et al. 1995). These contaminated soils remain likely sources of contamination in samples of environmental media collected to demonstrate regulatory compliance.
Another likely contributor to current background radiation levels at the WIPP is a recent accidental radiological release from the repository. On February 14, 2014, a waste container in the repository underwent a chemical reaction that caused it to overheat and rupture to release radionuclides, mostly 241Am, into the disposal room (USDOE 2015). A small amount of radioactivity escaped to the surface through the ventilation system and was detected approximately 1 km away from the facility. Source term estimation suggests a release of ~ 3.7 × 107 Bq of 241Am and 239 + 240Pu into the environment, which would have briefly increased the concentration of 241Am and 239 + 240Pu in surface air in the vicinity of the WIPP site (Thakur et al. 2016).
In this context, the variation in concentrations of 241Am and 239 + 240Pu in the WIPP environment is important, not only because they are the main component of the WIPP wastes, but also because of their global background activity. Since the atmospheric nuclear weapons tests of the 1950s and 1960s, plutonium, and to some extent 241Am, have become ubiquitous elements in the environment, including at the WIPP site. Several studies have assessed the distribution and long-term behavior of plutonium in the WIPP environment (Thakur et al. 2012; Hayes and Akbarzadeh 2014). However, 241Am has received much less attention than plutonium despite being the second most abundant actinide in TRU waste. In fact, there have been no studies on the temporal and spatial distribution of 241Am in the WIPP environment.
Radionuclides present in the environment, whether naturally occurring or anthropogenic, may result in radiation doses to humans. Therefore, environmental monitoring is required around nuclear facilities to characterize radiological baseline conditions, identify any releases, and determine the effects of releases should they occur. In this study, the variation of 241Am before and after the WIPP begin accepting TRU waste is assessed. Historical data, collected over more than a decade, are used to characterize the distribution and long-term behavior of 241Am in the WIPP environment. Isotopic ratios, derived from measurements in different environmental media, are analyzed to gain insight into likely sources contributing to the 241Am profile in the WIPP environment.
Experimental
Site description and sampling locations
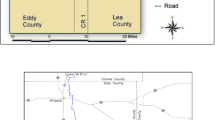
The WIPP facility, shown in Fig. 1, encompasses 41.4 km2 and consists of surface buildings, an array of vertical shafts, and a mined repository ~ 650 m below ground surface. This part of New Mexico is relatively flat, sparsely inhabited, with little surface water. The climate in this region is classified as cold, semi-arid. Winds are predominantly from the southeast and with speeds ranging from 3.7 to 6.3 ms−1. The annual average precipitation is 324 mm and the annual average temperature is 17 °C with average highs over 40 °C in the summer months and lows around 15 °C. In the winter months, daily temperatures are cooler with highs around 10 to 15 °C and lows reaching − 10 °C. The main population centers lie within 80 km of the site and are concentrated in and around the communities of Carlsbad, Hobbs, Eunice, Loving, Jal, Lovington, and Artesia, with an estimated population of 88,952.

Location of the Waste Isolation Pilot Plant (WIPP) in southeastern New Mexico, USA, including the location ambient air samplers and soil sampling areas
Lands in the immediate vicinity of the WIPP and Project Gnome sites are managed by the U.S. Department of the Interior Bureau of Land Management and are used for livestock grazing, potash mining, oil and gas exploration and production, and recreational activities. Environmental monitoring, to demonstrate compliance with the regulations, includes airborne particulates, soil, surface water, groundwater, sediments, and biota.
Airborne particulate sampling
Airborne particulate samples were collected in the vicinity of the WIPP site using high-volume air samplers at a volumetric flow rate of ~ 1.13 m3 min−1. These samplers are installed at monitoring stations around the site (Fig. 1) and include: (1) Onsite, located about 0.1 km northwest of the WIPP exhaust shaft; (2) Near Field, about 1 km northwest of the WIPP facility; and (3) Cactus Flats, about 19 km southeast of the WIPP facility. The sampling sites were selected based on their likelihood of intercepting accidentally released airborne radionuclides using an analysis of prevailing wind direction and speed scenarios. Particulate samples were collected on pre-weighed, 20 × 25 cm A/E™ glass fiber filters with a nominal 1.0 μm pore size (Pall German Laboratory, Ann Arbor, MI). Samplers are operated to maximize particulate loading without impacting air flow. However, filters were changed when the volumetric flow rate dropped below 0.99 m3 min−1, when there was a power outage, or when the sampler malfunctioned due to mechanical issues. Each filter was weighed after retrieval and the change in weight used to determine the weight of particulate material collected over the sampling interval. Actinide analyses were performed on individual filters. A typical sampling period lasts for about 3 to 4 weeks depending on the levels of particulate matter that accumulates on the filters. An average ~ 28,000 m3 air flow through these filters.
Soil sampling
Soil samples were collected from the two locations (Near Field and Cactus Flats) where the high-volume air samplers are stationed (Fig. 1). Bulk soil samples were collected with a trowel from a depth of 0–2 cm. At both locations, samples were taken at random orientations and distances (0–16 km) from the stations. Individual sampling locations were selected on the basis of having relatively flat topography, minimum surface erosion, and minimum surface disturbance by human or livestock activity.
Sample digestion
Filter particulate samples were prepared for radiochemical analyses by first ashing in a muffle furnace at 500 °C for 6 h. This was followed by wet digestion with a mixture of concentrated nitric (HNO3), hydrochloric (HCl), and hydrofluoric (HF) acids on a hot plate at 200 °C. Samples were then treated with concentrated perchloric acid (HClO4) and HNO3 to remove fluoride ions. The inside walls of the beaker were rinsed carefully with HNO3, to gather residual HF, and heated on a hot plate to ensure removal of residual HF from the matrix. The residues were then dissolved in 1.0 M HCl for subsequent actinide separation and analysis. Ashing at a higher temperature may result in loss of plutonium if only the HNO3 leaching method is employed. Wang et al. (2015) observed a loss of about 40% plutonium in soil samples at ashing temperature > 450 °C. More aggressive digestion such as combination of HNO3 and HF or alkali fusion method is necessary if samples are ashed at > 500 °C. Use of glass fiber filter in this case is also warranted HNO3 + HF digestion.
Soil samples were dried at 110 °C and blended to homogenize prior to digestion. A 5-g aliquot of each homogenized sample was dissolved by heating with a mixture of HNO3, HCl, and HF acids. The sample residues were then heated with HClO4 and boric acid (H3BO3) to remove HF. After cooling, samples were transferred to a 50-mL centrifuge tube and centrifuged at 3600 rpm for 10 min. The supernatant was filtered through a 0.45-μm filter and transferred to a 250-mL beaker. The actinides were subsequently separated as a group by co-precipitation on Fe(OH)3.
The resins used in this work were TEVA (Aliquot 336), TRU-resin (tri n-butyl phosphate, [TBP]), N-diisobutyl carbamoxyl methyl phosphine oxide (CMPO), and an anion exchange resin (Eichrom 1-×8, 100–200 mesh, chloride form) obtained from Eichrom Technologies, Inc. Chemical recoveries were determined using 242Pu and 243Am as yield tracers. Nitric, perchloric, and hydrochloric acids were prepared from reagent-grade acids (Fisher Scientific, Inc.). All other chemicals were ACS reagent grade and dilutions were made with de-ionized water.
Radiochemical separation
The actinides were concentrated in an iron hydroxide precipitate as Fe(OH)3. After decantation and centrifugation, the precipitate was dissolved in 10 mL of concentrated HNO3 and diluted to 20 mL to make the solution 8 M in HNO3. The oxidation state of plutonium as Pu(IV) was adjusted by adding 1 mL of 1 M NH4I with a 10-min wait step, after which 2 mL of 2 M NaNO2 was added. The sample solutions were then loaded onto anion exchange columns, pre-conditioned with 8 M HNO3. The columns were then washed with 3 × 10 mL 8 M HNO3. Finally, plutonium was eluted with 30 mL of 0.1 M NH4I + 10 M HCl. A two-step column separation process was used to ensure complete removal of any interference.
An Eichrom-TRU column (2 mL, 100–150 μm), pre-conditioned with 10 mL of 2 M HNO3, was used for the separation of americium. The fraction containing americium from the 8 M HNO3 eluate of the first anion exchange resin column was evaporated to dryness and re-dissolved in 10 mL of 2 M HNO3. A solution of NH4SCN was used to test for the presence of Fe3+. If Fe3+ was present, then 300 mg of ascorbic acid was added to reduce Fe3+ to Fe2+. The sample solutions were then loaded onto exchange columns and washed with 3 × 5 mL of 2 M HNO3. The columns were then washed with 10 mL of 2 M HNO3 + 0.05 M NaNO2. This step was used to oxidize any Pu3+, formed by the reduction of Pu4+ with ascorbic acid, back to Pu4+ to prevent any Pu3+ from co-eluting with the Am3+. Americium was then eluted with 20 mL of 4 M HCl.
Polonium is often present in significant amounts in particulate samples. Therefore, an additional step using an anion exchnage column was added to allow effective removal of polonium from the Am3+ fraction (Lemons et al. 2018). The americium fraction from the TRU column was evaporated to dryness with 1 mL of 50% H2SO4, to enhance destruction of any extractant in this solution, and then with 3 mL of HClO4. The samples were dissolved in 4 mL of 2 M HCl and loaded onto anion exchange columns (AG1-×8, 50–100 mesh, Cl− form, 10 mm dia. × 5 cm long) pre-conditioned with 10 mL of 1 M HCl. The columns were washed with 3 × 4 mL of 1 M HCl. The load and the washing solutions were collected in 50 mL polycarbonate centrifuge tubes.
A TEVA column was used to remove lanthanides, which are present in significant amount in soil samples, from the americium fraction. The americium fraction, stripped from the TRU columns, was re-dissolved in 10 mL of 3 M NH4SCN + 0.1 M HCOOH, warming gently as needed. The solution was loaded onto a TEVA cartridge (2 mL, 100–150 μm), previously conditioned with 10 mL of 3 M NH4SCN. The column was washed with 3 × 4 mL of 1.5 M NH4SCN + 0.1 M HCOOH to remove any lanthanides present as they interfere with alpha spectrometry peak resolution. Americium was then eluted with 15 mL of 2 M HCl and any residual NH4SCN destroyed by heating with 8–10 mL of HNO3:HCl (1:3). The solution was then evaporated to dryness with 3 mL of HClO4. The residue was dissolved in 4 mL of 2 M HCl and transferred to a 50-mL polycarbonate centrifuge tube with DI water. The individual actinides were then micro-co-precipitated with an Nd-carrier and HF (Hindman 1986) and counted using alpha spectrometry (Alpha Apex, Canberra).
Results and discussion
Temporal variations of 241Am in the atmosphere
Americium (241Am) is not produced directly by nuclear weapons detonation, but indirectly by the decay of 241 Pu as it disperses in the atmosphere or after terrestrial deposition. Because of high affinity of 241Am for particulates, the major mode of transport is atmospheric dispersion of particulate matter. Atmospheric concentrations of 241Am from nuclear weapons testing depend primarily on the amount of un-fissioned 241Am and 241Pu. There are also contributions from lower isotopes of plutonium that are neutron activated to 241Pu during weapons detonation. Spatially variable concentrations arise from transport and differences in residence time, of both 241Am and its precursor 241Pu, in the various atmospheric compartments (e.g., stratosphere and troposphere). In the test of a particular weapon, this depends on the amount of plutonium in the weapon, the explosive yield, the detonation height, and meteorological conditions at the time of detonation. High-yield tests by the USA were characterized by very high ratios of 241Pu to 239 + 240Pu, which produced relatively large quantities of 241Am (Roos et al. 1994). The atmospheric residence time can span several years, long enough to allow mixing with hemispheric air to be distributed worldwide (Perkins and Thomas 1980). A typical residence time of particulate debris in the troposphere is reported to be around 30–70 days whereas it is about 12–24 months in the stratosphere (Reiter 1975). The transfer of fallout from the stratosphere to the troposphere is seasonally modulated and for the northern hemisphere, it occurs mostly in the late winter and spring with little transfer occurring during summer and autumn.
During the large-scale testing of nuclear weapons, most of the fallout debris was injected into the stratosphere. Thus, the stratosphere serves as the main reservoir for bomb-derived radionuclides in the environment. However, after the signing of the partial atmospheric test-ban treaty in 1963, the concentrations of bomb-derived radionuclides in the stratosphere decreased significantly. While the French and Chinese continued atmospheric nuclear tests until the 1980s, most of these tests were of small yields that contributed only slightly to the atmospheric radionuclide concentrations. It is generally accepted that the current levels of plutonium and americium in the stratosphere are negligible and that most of americium in the air today is associated with resuspended soil and sediment, which were contaminated by weapons fallout.
It has been estimated that the globally deposited 241Pu from the nuclear weapons testing of 1950s and 1960s will ultimately produce about 5.5 × 1015 Bq (5.5 PBq) of 241Am (UNSCEAR Report 1982, Annex E). Additionally, ~ 1.5 × 1014 Bq (0.15 PBq, decay-corrected to 2017) of 241Am has been in-grown from the decay of ~ 6 × 1015 Bq (~ 6 PBq) of 241Pu released from the Chernobyl NPP accident in 1986 (UNSCEAR 2000, Annex J). The 241Am is therefore the only radionuclide whose concentration continues to increase, over time, in the environment. The activity concentrations of 239 + 240Pu in the stratosphere and troposphere (10.1–14.2 km altitude) over Switzerland, from 1973 to 1986 and from 2004 to 2011, were reported by the Alvarado et al. (2014). These authors also measured activity concentrations of 241Pu in high-altitude aerosol samples for the periods 1973–1977, 2007–2008, and 2010. The activity concentrations of 241Am during the same period in the atmosphere over Switzerland were estimated from the measured concentrations of 239 + 240Pu and the reported 241Am/239 + 240Pu ratios. There were several peaks in the stratospheric concentrations of 239 + 240Pu, 241Pu, and 241Am in 1970s due to the Chinese nuclear tests conducted between 1970 and 1976. After 1976, the stratospheric concentrations of these radionuclides decreased. There are no stratospheric data available for 239 + 240Pu during the period 1987 to 2003, nor for 241Pu between 1978 and 2006. These authors added very few new data on stratospheric plutonium isotopes (239 + 240Pu, 241Pu, and 238Pu) and 137Cs were added for the period between 2007 and 2010–2011. However, they found that the activity concentrations of these radionuclides in the stratosphere are about two to four orders of magnitude higher than previously assumed and suggested that the stratospheric mean residence time of these particles is 2.5–5 years.
Temporal variations of 241Am in surface air
There are very limited data on the surface air activity concentrations of 241Am in surface air. The concentrations of americium in surface air were not systematically monitored during the period 1959–1964, the time of the heaviest contributions from global fallout. The annual average concentrations of 241Am in surface air, prior to 1965, in the mid-latitudes of the northern hemisphere resulting from nuclear weapons testing between the 1950s and the 1980s were estimated using an atmospheric transport model and the amount of 241Am that would have been produced as a result of the testing (Bennett 1979). As shown in Fig. 2, surface air concentrations of 241Am, attributed to global fallout, increased rapidly and reached a maximum of 0.85 μBq/m3 in 1964. Since 1973, levels have been declining. It is this global fallout that created a legacy of 241Am on the terrestrial surface.

Predicated surface air concentrations of 241Am during 1951–1980 (From ref. Bennett 1979)
Temporal variations of 241Am in surface air at the WIPP
The background concentrations of 241Am in the vicinity of the WIPP site were measured by Lee et al. (1998). The aerosol samples were collected between February 23 and May 6, 1996 using high-volume samplers. Samplers concurrently obtained total suspended solids (TSP), particles > 10 μm (PM10) at a 5-m elevation, and PM10 at a 2-m elevation. Activity concentrations of 241Am in the samples were 0.008 ± 0.002 μBq/m3 in the PM10 at 2 m, 0.006 ± 0.002 μBq/m3 in the PM10 at 5 m, and 0.011 ± 0.001 μBq/m3 in the TSP at 5 m (Lee et al. 1998). The background concentrations of 239 + 240Pu, 241Am, and other radionuclides within and adjacent to the WIPP site were also measured by the Environmental Evaluation Group (EEG) for the period between 1985 and 2000 (Gray et al. 2000). Average pre-operational baseline concentrations reported in their study were 0.027 ± 0.11 μBq/m3 (n = 79) for 241Am, 2.3 ± 0.056 μBq/m3 (n = 88) for 239 + 240Pu, and 0.006 ± 0.062 μBq/m3 (n = 90) for 238Pu. None of the composite samples were statistically different from the lower limits of detection for 239 + 240Pu or 241Am.
A time series of 241Am airborne activity concentration, measured in aerosol filter samples collected from the Onsite, Near Field, and Cactus Flats stations from 2001 through 2013, is shown in Fig. 3a. The data show frequent detection of trace levels of 241Am and 239 + 240Pu. The activity concentrations of 241Am measured in the ambient surface air, in the WIPP vicinity, range from 0.00036–0.126 μBq/m3 at Onsite station, to 0.00012–0.05 μBq/m3 at Near Field station, and 0.00017–0.113 μBq/m3 at Cactus Flats station (CEMRC 2006). In general, 241Am activities peak in the March to June timeframe, which coincides with the period of strong and gusty winds in the area that frequently give rise to blowing dust. The activity concentrations of 239 + 240Pu, measured in aerosol samples from the three study sites, are shown in Fig. 3b. The detection of 241Am in surface air was not as frequent as that of 239 + 240Pu because of the normally low levels of 241Am in the environment. Current low levels are due to 241Am resulting primarily from the decay of 241Pu, rather than fallout. However, when detectable, the activity concentrations of 241Am in the high-volume samples closely tracked those of 239 + 240Pu. Most notably, strong springtime peaks in 241Am activity concentrations were evident in the samples from 2001 through 2002, and from 2011 through 2013. The amount of particulate deposits (mass loadings) on filters also tends to increase during the windy period at all three stations. Figure 3b also shows that the aerosol mass loadings, i.e., the mass of aerosols collected per unit volume of air, followed a seasonal pattern similar to that of 239 + 240Pu and 241Am activity concentrations. Although seasonality in 241Am data is not as pronounced as in the case of 239 + 240Pu (Fig. 3c), the data do show a typical seasonal variation with highs in the spring and lows in the summer. The observed seasonality in 241Am activity concentrations in the WIPP environment can therefore be attributed to the resuspension of contaminated soil and sediment.

Temporal patterns in atmospheric activity concentrations of 241Am (a), 239 + 240Pu (b), and aerosols mass loading on the filters (c) at three stations in the vicinity of the WIPP site
Temporal variations in surface air concentrations of 239 + 240Pu and 241Am activity concentrations have been observed since the early days of atmospheric monitoring. This cycle was confirmed by numerous data collected in the northern hemisphere. For example, Arnold and Wershofen (2000) showed seasonality in 239 + 240Pu concentrations in ground-level air from Germany. Salminen and Paatero (2009) observed a seasonal cycle in 239 + 240Pu, 241Pu, 241Am, and 238Pu concentrations in surface air of Sodankyla, Finland in 1963. A springtime enhanced activity of 239 + 240Pu was reported in the dry and wet deposition samples collected from Tsukuba, Japan, and Daejeon, South Korea (Hirose et al. 2003; Hirose et al. 2004). While the current americium/plutonium aerosol data (post-1984) and those collected during the era of atmospheric nuclear weapons testing (pre-1984) both show springtime peaks, the causes for the cycles are likely quite different. It has been suggested that the seasonal cycle in aerosol samples collected prior to the cessation of nuclear weapons testing is associated with a stratospheric-tropospheric exchange phenomenon, while the resuspension is believed to be the main source of 241Am in aerosol samples collected after the cessation of nuclear weapons testing.
In general, the 241Am activity concentrations in the ambient surface air after WIPP became operational are not statistically different from those measured prior to waste disposal operations. The exception is 2014 when higher-than-normal background levels of 241Am and 239 + 240Pu were detected in February and March, 2014. The increased activity concentrations are attributed to an accidental release from the repository due to a drum breach on February 14, 2014 (USDOE 2015). This event released moderate levels of radioactivity, mostly 241Am and 239 + 240Pu, into the underground air. A small amount of radioactivity escaped to the atmosphere through the ventilation system and was detected at the surface. The highest activity concentrations detected were 115.2 μBq/m3 for 241Am, and 10.2 μBq/m3 for 239 + 240Pu at Onsite station, which is only 0.1 km northeast of the exhaust shaft. At Near Field station, which is 1 km northwest of the exhaust shaft (i.e., the predominant wind flow direction), the activity concentrations were 81.4 μBq/m3 for 241Am and 5.78 μBq/m3 for 239 + 240Pu. A third station at Cactus Flats, located 19 km southeast (upwind) of the shaft, showed no increase in 241Am or 239 + 240Pu (CEMRC 2014). While levels of 241Am and 239 + 240Pu were above the pre-release background levels, it is important to note that these concentrations were below any level of public health or environmental concern. It should also be noted that the sampling rate for high-volume samplers, 1.13 m3 (40 cfm), is significantly greater than the 0.03–0.14 m3 (1–5 cfm) respiration rate for humans so any attempt to estimate internal dose from exposure to the reported levels would need to account for the volume differences. The regulatory limit for exposure to a standard adult member of the public from activities on the WIPP site is 1 mSv/year based on 10 CFR 835. According to DOE, the maximally exposed general member of the public would have received a dose of less than 0.34 mSv/year (34 mrem/year) in 2014 from the WIPP operations (USDOE 2014). The average person living in the USA receives an annual dose of about 6.2 mSv (620 mrem) from exposure to naturally occurring and medical sources of radiation (Thurston 2010).
Americium data from the WIPP environment were also compared with other national and international monitoring data to assess the worldwide concentrations of 241Am in surface air. At the Hanford Site in eastern Washington, USA, background concentrations of 241Am measured ranged from 0.015 to 0.033 μBq/m3 during the 1993–1994 period. These are significantly lower than the range of 0.20–1.1 μBq/m3 in the 100-Hanford’s K Area, and 0.12–0.93 μBq/m3 in the 200-East Area of the Hanford site measured during the 1999–2001 period (Poston et al. 2002). At the Los Alamos National Laboratory site in northern New Mexico, USA, the highest concentration measured was 0.25 μBq/m3 (quarterly composite) over the period 1986–1997 (Eberhart 1998). In Vilnius, Lithuania, measured 241Am concentrations ranged from 0.0003 to 0.5 μBq/m3 during the 1995–2003 periods compared to a range of 0.0005 to 0.025 μBq/m3 during the 2005–2006 periods (Lujaniené et al. 2012a). Surface air samples collected in 1963 from Sodankylä, Finland and measured in 2007 for the in-growth of 241Am from the 241Pu initially collected on these filters had 241Am concentrations ranging from < 0.5 to 50 μBq/m3 (Salminen and Paatero 2009). Lehto et al. (2006) reported 241Am and 239 + 240Pu concentrations of 1400 μBq/m3 and 0.014–2.5 μBq/m3, respectively, in surface air at Kurchatov, Kazakhstan (near ground zero) during the 2000–2001 timeframe.
A brief increase in surface air concentrations of 241Am and 239 + 240Pu was reported by many monitoring stations around Europe during 1986–1987 due to the Chernobyl accident. For example, average surface air concentrations of 241Am in Roskilde, Denmark were in the range 5.2–11.0 μBq/m3 during April–May, 1986 (Aarkrog 1988a). Levels measured in Austria were in the range 7.4–10.4 μBq/m3 (Irlweck and Wicke 1998). In Bragin, located about 55 km north of Chernobyl, the mean activity concentration of 241Am during this period was 40 μBq/m3 (Knatko et al. 1993). Similarly, elevated levels of 239 + 240Pu in the range 10–28 μBq/m3 were measured in Prague (Holgye and Filgas 1987); 1.2–89 μBq/m3 in Vienna (Holgye and Filgas 1987); 32 μBq/m3 in Nurmijärvi, Finland (Paatero et al. 2010); and 0.4–10.6 μBq/m3 in Belgrade, Serbia (Manić-Kudra et al. 1995). However, following a peak in 1986, the concentrations of 241Am and 239 + 240Pu diminished. The activity concentrations of 241Am and 239 + 240Pu measured in Vilnius, Lithuania in March–April, 2011 were 0.016 μBq/m3 and 0.044 μBq/m3, respectively (Lujaniené et al. 2012b).
Temporal variability of 241Am activity due to resuspension in the vicinity of WIPP
Given the length of time since the cessation of nuclear weapons testing, most of the 241Pu deposited on the land surface near the WIPP is expected to be fully decayed into 241Am and incorporated into the soil. This is the basis for the hypothesis that americium detected in aerosol samples is associated with resuspended soil particles. Resuspension is recognized as the predominant mechanism for maintaining residual plutonium and americium in surface air. Most radioactive material transported by wind is a result of saltation, which occurs at a typical threshold wind speed ranging from 6 to 13 ms−1 at 0.3 m (Stout and Arimoto 2010; Arimoto et al. 2005). Under these conditions, particles less than 50 μm may be suspended for extended periods of time. The effectiveness of wind transport in spreading radionuclides across the landscape also varies with particle size. For example, at the Hanford site, 241Am was reported to have been transported in the air on different particle sizes and reached maximum concentrations at different heights than those of 239 + 240Pu (Sehmel 1987). Over time, radionuclide concentrations decrease rapidly, especially at high wind speeds (Hollander 1994). Arimoto et al. (2005) have shown that under semi-arid conditions like those at the WIPP, plutonium-bearing aerosols tend to increase in ambient air samples as wind speeds approach 4 ms−1, reaching a maximum at 7 ms−1, and staying constant at wind speeds above 7 ms−1. These aerosols are readily trapped on the filter in an air monitoring station, leading to their frequent radionuclide detection during high-wind events.
Positive detections of 239 + 240Pu and 241Am in aerosol samples, from the three study sites, during the March to June timeframe when wind gusts are more common, are attributed to resuspension. As shown in Fig. 4, about 60–70% of positive 239 + 240Pu or 241Am detections are sampled during the gusty period. The apparent seasonality in plutonium activity concentrations is therefore due to the resuspension of contaminated soil particles. A linear relationship between the mass of dust retained on the filters and the 239 + 240Pu (r2 = 0.68) and 241Am (r2 = 0.69) concentrations is also evident at the three monitoring stations (Fig. 5).

Concentrations of 239 + 240Pu and 241Am in aerosol samples from ambient air collected during windy and non-windy seasons in the vicinity of the WIPP

A cross plot of 241Am concentrations and mass loading on the filters
These findings suggest that the detection of 239 + 240Pu and 241Am in the aerosol samples collected from ambient air around the WIPP facility is primarily due to the resuspension of contaminated soils. In the WIPP environment, the concentrations of 241Am and 239 + 240Pu were similar at all stations and a good correlation exists between 241Am and 239 + 240Pu concentrations (r2 = 0.70) even though neither 239Pu nor 240Pu are the immediate progeny of 241Am (Fig. 6).

A cross plot of 241Am and 239 + 240Pu in aerosol samples collected in the vicinity of the WIPP site
Near-surface distribution of 241Am in the WIPP soils
Surface soil concentrations of radionuclides are the primary source of nearby and downwind aerosol concentrations. Therefore, it is important to understand near-surface soil concentrations of 241Am at and around the WIPP site. Based on data collected after the WIPP become operational, 241Am concentrations in the Near Field soil ranged from 0.002 to 0.14 Bq/kg, with a mean value of 0.044 Bq/kg. The same dataset showed 241Am concentrations in a range from 0.007 to 0.26 Bq/kg, with a mean value of 0.068 Bq/kg in the Cactus Flats soil. The corresponding concentrations of 239 + 240Pu at these two locations ranged from 0.001 to 0.40 Bq/kg, with a mean of 0.11 Bq/kg in the Near Field soil, and 0.013 to 0.51 Bq/kg, with a mean of 0.2 Bq/kg in the Cactus Flats soil. Background concentrations have been established based on soil surveys conducted at these two locations during the 1996–1997 timeframe, prior to arrival of TRU wastes at the WIPP. Background concentrations of 241Am are in the range 0.075–0.11 Bq/kg (n = 9), whereas background 239 + 240Pu concentrations are in the range 0.037–0.30 Bq/kg (n = 16). A good correlation (r2 = 0.64) between 241Am and 239 + 240Pu concentrations at the two locations suggests an identical origin (Fig. 7).

A cross plot of 241Am and 239 + 240Pu in soil samples collected in the vicinity of the WIPP site
Although the concentration of 241Am in the surface soil at Cactus Flats is slightly higher than that in the surface soil at Near Field, there is no statistical difference between the 241Am concentrations at these two locations. Furthermore, there is no difference between the concentrations in soil samples collected before and after WIPP started receiving TRU waste. The deposition of radioactive fallout from weapons testing is also known to vary with latitude, being highest in middle latitudes of the northern hemisphere (UNSCEAR 1982). The range of 241Am concentrations in the Near Field soil is lower than background concentrations (0.6–1.7 Bq/kg) found in the Colorado Front Range, between Fort Collins and Colorado Springs (Hodge et al. 1996). The 241Am concentrations in the WIPP vicinity are also lower than the background range of 0.037–1.14 Bq/kg in soils at Rocky Flats, Colorado (Hulse et al. 1999) and the 0.74–107.3 Bq/kg range in soils around the Los Alamos National Laboratory in northern New Mexico. The 241Am and 239 + 240Pu levels in soil at various locations are summarized in Table 1.
Several studies have been conducted to assess the residual contamination levels at the Project Gnome site. The EEG conducted a soil survey of the area around the Gnome site over a 7-month period in 1994–1995 (Kenney et al. 1995). In 2006, CEMRC conducted a separate study in an attempt to determine concentrations and precise isotopic ratios of Gnome contamination. Both the EEG soil survey conducted 1994–1995 (Kenney et al. 1995) and the later study by CEMRC (CEMRC 2006) reported heterogeneous distributions of 241Am and plutonium isotopes in the near-surface soil. Concentrations of 241Am and 239 + 240Pu reported by EEG ranged from 0.59 to 7600 Bq/kg and 4.4 to 48,000 Bq/kg, respectively. The survey by CEMRC, some 6 years later, showed concentrations ranging from 0.043 to 346 Bq/kg for 241Am and 0.073 to 1550 Bq/kg for 239 + 240Pu. The large variation in radionuclides concentration is attributed to a spotty and inconsistent clean-up effort at the site (Faller 1994).
Depth distribution of 241Am in WIPP soils
Radionuclides deposited at the soil surface in the beginning are eventually transported downward into the soil profile. The depth distribution of 241Am and 239 + 240Pu was determined, using soil samples collected at and near the WIPP site, to evaluate vertical pattern of these radionuclides in a semi-arid environment. The depth distribution of 137Cs was also investigated because concentrations of 137Cs in soils, from atmospheric nuclear weapons testing, are considerably greater than 241Am and 239 + 240Pu concentrations. In addition, 137Cs being a gamma-emitter requires less effort for analysis. Although chemically quite different, 137Cs is expected to behave similarly to 239 + 240Pu in soils because it adheres tightly to cation-exchange surfaces and is relatively inert chemically (Coppinger et al. 1991) making it a good surrogate when the radionuclides of interest are also present.
As shown in Fig. 8, the highest concentrations of 241Am and 239 + 240Pu in soil profiles occurred at the 2 and 3 cm depths, respectively. The concentrations of these radionuclides drop off sharply within 10 cm of the soil surface and are at or below minimum detectable concentrations (MDC) by 20 cm. There is no indication that radionuclides have been transported or redistributed to any substantial degree within these profiles. These show ~ 86% of 137Cs, 90% of 239 + 240Pu, and 78% of 241Am are retained in the top 5 cm of soil around the WIPP site (CEMRC 2006). These results, coupled with soil column experiments, suggest that leaching and colloidal transport are not major transport mechanisms in the redistribution of surface-deposited radionuclides in WIPP soils. There has been no analysis of the depth distributions of 241Am and 239 + 240Pu in soils at the Project Gnome site.

Depth profiles of 241Am, 239 + 240Pu, and 137Cs in WIPP soils
There have been several studies of the vertical distribution of actinides in soil profiles contaminated as a result of nuclear fallout and/or accidental releases. Generally, these studies show higher concentrations in surface soils than in subsurface soils. Several authors have also reported exponentially decreasing relationships between plutonium and americium concentrations and soil depth (Turner et al. 2003; Łokas et al. 2013). Evidence of strong retardation can be seen with 239 + 240Pu and 137Cs in Rocky Flats soils and in soil profiles collected from an undisturbed area near the Nevada Test Site (Turner et al. 2003; Hulse et al. 1999). Retardation in the near-surface is also evident in upwind and downwind soils in the Hanford Site study of Price (1991). The author reported that 98–100% of the 137Cs, and 95–97% of the 239 + 240Pu remained in the top 5 cm of the soil profiles. The depth profile study of 239 + 240Pu and 241Am in sandy loam soil from the northeastern USA, conducted in 1972, also shows decreasing activity concentrations of these radionuclides with increasing soil depth (Bennett 1979). Although most of the 239 + 240Pu and 241Am was found in the top 5 cm, measureable amounts had reached the 20-cm depth with trace amounts observed in the 20- to 30-cm depth (Bennett 1979). Subsequent measurements at the same location in 1976 show a redistribution of activity to greater depths below which it levelled off. Soil profile studies of 241Am, 238Pu, and 239 + 240Pu at the Mururoa and Fagataufa Atolls, where French nuclear weapons tests were conducted over the period 1977 to 1974, show that more than 95% of the plutonium and americium remained in the top 5-cm layer (Irlweck and Hrnecek 1999). The profile of 241Am at Taranaki, a site at Maralinga in Southern Australia used for UK atomic weapons tests, also showed the majority of americium (77–96%) remained in the top 0–1 cm of soil in undisturbed areas (Burns et al. 1995; Cooper et al. 1994). In the few samples in which this is not the case, the top 1–2 cm is found to contain bulk of the activity.
High organic matter content in the near-surface is known to retain 241Am in the top soil layers. Boulyga et al. (2003) found that 80–95% of the plutonium and americium remained in the top 5-cm layer in turf podzol soil collected from Belarus (8–45 km to the north and northwest of Chernobyl NPP). In forest soils, in which organic matter content is typically high, ~ 76% of 241Am was found in the 0–5 cm layer (Mietelski et al. 2007). The retention of 241Am in the surface layer of organic-rich soil is also evident in data from the French Mercantour wetlands (Solovitch-Vella et al. 2007). Significant levels of 239 + 240Pu and 241Am (5–10 kBq/kg) have also been found in surface gley soils of West Cumbria in Northwest England, with the actinides enriched in the organic fraction (Livens and Singleton 1991).
Some studies have reported differences in the mobilities of 241Am and of 239 + 240Pu in soil. For example, in an acidic, sandy soil with low clay content from the Vosges Mountains in Eastern France, 241Am migrated at a rate one order of magnitude higher than that of 239 + 240Pu (Solovitch-Vella, 2007). Analysis of the vertical migration of 241Am and plutonium in Kapachi soils, near Chernobyl and in Eastern Europe, has shown that more than 80% of Chernobyl-derived 241Am and plutonium remains in the top 2 cm. 241Am appears to migrate faster than plutonium isotopes (Mboulou et al. 1998). These data suggest that while radionuclides migrate vertically through soils, transport generally does not extend much deeper than 20 cm unless the soils are disturbed (Anspaugh et al. 1975). Depending on soil properties, recharge rates, and pore water chemistry, surface-deposited radionuclides can take years to migrate just a few centimeters.
Radionuclide activity and atom ratios in the WIPP vicinity
The 241Am/239 + 240Pu activity ratios measured on samples collected at the three aerosol stations, for more than a 10-year time span, are shown in Fig. 9. As of December 2013, the mean activity ratios of 241Am/239 + 240Pu at the three aerosol stations were 0.38 ± 0.05 (range 0.30–0.42) at Onsite station, 0.37 ± 0.03 (range 0.33–0.44) at Near Field station, and 0.39 ± 0.03 (range 0.35–0.44) at the Cactus Flats station. In soil samples, the mean activity ratio of 241Am/239 + 240Pu was quite comparable to those measured in aerosols. In the Near Field soil, the mean 241Am/239 + 240Pu activity ratio was 0.42 ± 0.05 (range 0.34–0.49) whereas it was 0.39 ± 0.07 (range 0.29–0.51) in the Cactus Flats soil. The slightly higher range and mean in the aerosol samples are not surprising given the higher concentration of high-surface area materials that would be transported by wind compared to near-surface soil samples. Even with the differences in particle distributions of the soils and aerosols, the similarity in 241Am/239 + 240Pu activity ratios in the two studies is indicative of a connection between contaminated soil and aerosol. At the Gnome site, 8.8 km southwest of the WIPP, soil samples showed a 241Am/239 + 240Pu ratio of 0.26 ± 0.04 (n = 3; range 0.23–0.30), as reported by CEMRC (2006), and 241Am/239 + 240Pu ratio of 0.22 ± 0.12 (n = 9; range 0.12–0.50), as reported by Keeny et al. (1995).

Temporal variability in the 241Am/239 + 240Pu ratio measured in aerosol samples collected in the vicinity of the WIPP site
The ratios in the WIPP samples are in agreement with the mean activity ratio of these radionuclides in soils and sediments reported in different studies, indicating their global fallout origin (Table 2). For example, Jia et al. (1999) measured mean ratios of 0.35 ± 0.05 (range 0.23–0.43) in cultivated soil and 0.32 ± 0.06 (range 0.21–0.42) in uncultivated soil collected in 1997 from Urbino in the Marche Region of Central Italy. Bunzl and Kracke (1988) measured a ratio of 0.30 in soil from Southern Germany. Data from Prague, Czech Republic, showed average ratios of 0.52 for a forest soil, 0.47 for an uncultivated soil, and 0.48 for cultivated soil near the nuclear research center at Řež (Holgye et al. 2004). Breban et al. (2003) reported ratios of 0.43–0.87 from data collected at the Carpathian Mountains, Romania, in 1996. Measurements in South Korean soils during 1992–1994 showed a ratio of 0.43 (Sha et al. 1991). Leon Vintro et al. (1999) reported that in mid-latitude soils, the 241Am/239 + 240Pu ratio was 0.37 in the late 1980s and estimated that it would reach ~ 0.40 in 2000s, which is consistent with observations in the WIPP aerosol studies. Taking into account the contribution of 241Am from 241Pu decay, the observed 241Am/239 + 240Pu activity ratio confirms a global fallout origin.
Perturbations in the 241Am/239 + 240Pu ratios were observed in the aerosol samples of ambient air collected following the 2014 accidental radiation release at the WIPP. Both the Onsite and Near Field aerosol sampling stations detected elevated levels of 241Am and 239 + 240Pu as well as elevated 241Am/239 + 240Pu activity ratios. Early post-event analysis of WIPP environmental samples showed that the 241Am/239 + 240Pu ratio varied between 2.95 and 20.67 as the event progressed, but the average ratios remained fairly constant at about 10. Over the time period 2014–2016, the 241Am/239 + 240Pu ratio at Onsite station ranged from 3.06 to 11.31, whereas it ranged from 1.39 to 15.44 at Near Field station. These high activity ratios indicate the presence of 241Am and 239 + 240Pu particles associated with the breached waste drum that caused the release, and possibly the associated clean-up activities that followed.
The 238Pu/239 + 240Pu ratios measured in aerosols, at and near the WIPP site, varied from 0.069 to 0.18 with a mean value of 0.12 ± 0.04. The ratio in WIPP soil ranged from 0.06 to 0.19 with a mean value of 0.14 ± 0.05. Mean ratios of 238Pu/239 + 240Pu measured on soil samples from the Project Gnome site varied from 0.14 ± 0.025 (n = 8; range 0.06–0.28), as reported by CEMRC (2006) and 0.16 ± 0.02 (n = 9, range 0.14–0.18), as reported by Keeny et al. (1995). These values are clearly higher than the global fallout ratio of ~ 0.024 (Table 2). There is no logical explanation for these high 238Pu/239 + 240Pu ratios. However, they could be attributed to the sporadic detection of 238Pu because of the much lower levels of 238Pu normally found in the environment compared to 241Am and 239 + 240Pu. Only around 35% of the 238Pu present in the environment originated from weapons testing fallout with the remaining 65% coming from that released by the burn-up of the nuclear-powered satellite SNAP-9A in April 1964 over the South Pacific (Hardy et al. 1973). Studies have shown that the SNAP-derived 238Pu activity in surface air peaked 2–3 years later, depending upon the latitude, and was largely depleted from the atmosphere by 1971 (Perkins and Thomas 1980).
The 240Pu/239Pu isotopic ratio of Project Gnome soils is distinctly different from that of WIPP soils. Gnome-contaminated soils had a 240Pu/239Pu ratio of 0.114 ± 0.013 (range 0.07–0.168) (CEMRC 2006). Soils sampled from around the WIPP site showed a mean ratio of 0.175 ± 0.005 (range 0.193–0.146), whereas the global fallout ratio, based on a worldwide survey of terrestrial soils in 1970–1971, between 30 and 60° N, ranges from 0.12 to 0.21 with a mean of 0.176 ± 0.014 (Krey et al. 1976). The one exception in the northern hemisphere includes soils in the southwestern USA where fallout from the Nevada Test Site was also deposited. The 240Pu/239Pu atom ratio produced in a nuclear test is a function of the design and yield of the device being tested. The fallout produced by high-yield tests tend to have higher 240Pu/239Pu atom ratios than the fallout produced by low-yield tests. For example, 240Pu/239Pu ratios ranging from 0.21 to 0.36 have been measured in soils from Bikini Atoll (Muramatsu et al. 2001). However, fallout from Nagasaki, the Nevada Test Site, and the Semipalatinsk Test Site in the former Soviet Union, is characterized by lower 240Pu/239Pu ratios ranging from 0.03 to 0.08, with average values of 0.042, 0.035, and 0.036, respectively (Yamamoto et al. 1996). Therefore, WIPP soils do not appear to have been measurably affected by resuspension and transport of Gnome site soil, WIPP waste disposal operations, or the February 2014 accidental release, but reflect normal global fallout.
Global redistribution of radionuclides in the atmosphere
Several studies have shown that resuspension is a dominant mechanism in maintaining residual concentrations of 241Am and 239 + 240Pu in the post-fallout air. For example, concentrations in aerosols sampled near contaminated sites such as Rocky Flats, the Nevada Test site, the Marshall Islands (Shinn et al. 1997; Hulse et al. 1999), and in areas near the Semipalatinsk Test Site (Lehto et al. 2006) were elevated. Resuspension studies performed with 241Am indicate that the activity to mass ratio is greatest for particles < 45 μm in diameter (Burns et al. 1995). This is because fine soils, which have a higher surface area and capacity to sorb these radionuclides, have higher concentrations of 241Am than the remaining surface soil. These fine soils are also easily resuspended and transported by air, giving rise to a so-called enhancement factor. The enhancement factor for resuspended soil, in the inhalable fraction (< 7 μm), ranged from 3.7 to 32.5 at six sites at Taranaki in Southern Australia (Burns et al. 1995).
Shinonaga et al. (2014) reported long-distance transport of Fukushima-derived plutonium and uranium particles by wind. Environmental processes such as global dust storms, biomass burning, and/or wildfire have also been shown to play an important role in redistributing post-fallout radionuclides, including 241Am, in the atmosphere. Recent wildfire events of April and August 2015, in the Chernobyl exclusive zone, are estimated to have released about 12.5 TBq (1012 Bq) of radioactivity, mostly 137Cs, 90Sr, 239 + 240Pu, 238Pu, and 241Am, into the atmosphere (Evangeliou et al. 2016). These authors also concluded that during the fire events of August 2015, ~ 75% of (137Cs, 90Sr) and 59% of (239 + 240Pu, 238Pu, and 241Am) were exported from the Chernobyl exclusive zone and deposited mostly in Belarus, while during the spring fire event of 2016, ~ 93% of the 137Cs and 90Sr, and 97% of the 239 + 240Pu, 238Pu, and 241Am, were transported to Eastern European countries. Wotawa et al. (2006) reported that radionuclides resuspended by wildfires can be transported over intercontinental distances. Lujaniene et al. (2006) attributed detection of an elevated level of 137Cs (253 μBq/m3) in air over Vilnius, Lithuania, to the transport of Chernobyl-derived cesium from the Chernobyl exclusion zone in Ukraine following the wildfire event of 1992.
Soil dust is a major component of aerosols in the global atmosphere and dust transport events that move African Sahara dust and Asian Kosa dust are known to play a major role in redistribution of radionuclides in the atmosphere. A significant increase in anthropogenic radionuclide concentrations in air has been observed during Saharan dust events in Europe, in the USA, and Asia (Menut et al. 2009). The 239 + 240Pu activity concentrations in Saharan dust samples vary between 0.68 and 0.98 Bq/kg (Pham et al. 2017), which is significantly higher than the 0.096 Bq/kg usually found in Saharan soil. Sequential extraction studies were performed on aerosols collected in Lithuania after dust storms in September 1992 carried radioactive aerosols to the region from contaminated areas of the Ukraine and Belarus. The fraction distribution of 241Am in the aerosol samples was ~ 18% organically bound, ~ 10% oxide-bound, ~ 36% acid-soluble, and ~ 32% residual (Lujaniene et al. 1999). A very small amount of americium was found in the more readily extractable “exchangeable and water soluble” and “specifically adsorbed” fractions.
The primary source of anthropogenic radionuclides in dust transported globally is surface soil contaminated by global fallout, and possibly by regional fallout from the accidents at Chernobyl and Fukushima. The activity and atom ratios of dust samples collected during several past dust events confirm that they originate from global fallout origin, and/or mixing with local resuspended soil particles during transport.
Conclusion
The 241Am present in the environment today originated directly from nuclear reactors or indirectly from weapons testing. At nuclear facilities like the WIPP repository, being able to determine the origin of transuranium contaminants found in the environment is important for improving radiological protection programs and demonstrating regulatory compliance. Pre-operations measurements at the WIPP site established a background 241Am concentrations of 0.027 ± 0.11 μBq/m3 in aerosols and 0.066 ± 0.001 Bq/kg in soil. The 241Am activities in aerosol and soil are determined to be not significantly different from those due to atmospheric global fallout. The Gnome site is a potential local source of contamination of WIPP environmental samples and has a characteristic isotopic signature. Long-term monitoring of 241Am concentrations show that, except for a brief detection of elevated americium and plutonium in nearby ambient air samplers during the 2014 accidental release from the repository, there is no evidence of contamination that can be attributed to operations at the WIPP. The source of 241Am at the site is mostly wind-borne redistribution of resuspended soils contaminated by weapons testing. During most years studied, the peaks in 241Am activities, like that of 239 + 240Pu, generally occurred in the March to June timeframe, which is when strong and gusty winds in the area frequently give rise to blowing dust. The isotopic signature of 241Am in the WIPP environment is mainly from three different sources with 241Am/239 + 240Pu ratios of ~ 0.37 from global fallout, ~ 0.25 as a result of Project Gnome fallout, and ~ 10 due to the accidental radiation release in February 2014. Soil monitoring in the vicinity of the WIPP site suggests that the WIPP soils have received their plutonium and americium from global fallout during nuclear weapons testing and have not been measurably affected by resuspension and transport of Gnome site soils or by WIPP waste disposal operations. There is no reason to believe that WIPP is a source of transuranium contaminants in the environment that can be considered significant by any health-based standard.
References
Aarkrog A (1988a) Studies of Chernobyl debris in Denmark. Environ Int 14:149–155
Aarkrog A (1988b) The radiological impact of the Chernobyl debris compared with that from nuclear weapons fallout. J Environ Radioact 6:151–162
Aarkrog A, Dahlgaard H, Nilsson K (1984) Further studies of plutonium and americium at Thule, Greenland. Health Phys 46:29–44
Alvarado JAC, Steinmann P, Estier S, Bochud F, Haldimann M, Froidevaux P (2014) Anthropogenic radionuclides in atmospheric air over Switzerland during the last few decades. Nature Com 5:3030. https://doi.org/10.1038/ncomms4030
Anspaugh LR, Shinn JH, Phelps PL, Kennedy NC (1975) Resuspension and redistribution of plutonium in soils. Health Phys 29:571–582
Eberhart CF (1998) Ambient air sampling for radioactive air contaminants at Los Alamos National Laboratory: A large research and development facility. LA-UR-98-897, Los Alamos National Laboratory, Los Alamos, NM
Arimoto R, Webb JL, Conley M (2005) Radioactive contamination of atmospheric dust over southeastern New Mexico. Atom Environ 39:4745–4754
Arnold D, Wershofen H (2000) Plutonium isotopes in ground-level air in Northern Germany since 1990. J Radioanal Nucl Chem 243:409–413
Baskaran M, Asbill S, Santschi P, Davis T, Brooks J, Champ M, Makeyev V, Khlebovich V (1995) Distribution of 239,240Pu and 238Pu concentrations in sediments from the Ob and Yenisey Rivers and the Kara Sea. Appl Radiat Isot 46(11):1109–1119
Beasley TM, Kelley JM, Orlandini KA, Bond LA, Aarkrog A, Trapeznikov AP, Pozolotina VN (1998) Isotopic Pu, U, and Np signatures in soils from Semipalatinsk-21, Kazakh Republic and the southern Urals, Russia. J Environ Radioact 39:215–230
Bennett B.G (1979). Environmental aspects of americium. Rep. EML-348, Environmental Measurements Laboratory, U.S. Department of Energy, New York, New York
Boulyga SF, Zoriy M, Michael E, Ketterer ME, Becker JS (2003) Depth profiling of Pu, 241Am and 137Cs in soils from southern Belarus measured by ICP-MS and α and γ spectrometry. J Environ Monit 5:661–666
Breban DC, Moreno J, Mocanu N (2003) Activities of Pu radionuclides and 241Am in soil samples from an alpine pasture in Romania. J Radioanal Nucl Chem 258:613–617
Bunzl K, Kracke W (1988) Cumulative deposition of 137Cs, 238Pu, 239+240Pu and 241Am from global fallout in soils from forest, grassland and arable land in Bavaria (FRG). J Environ Radioact 8:1–14
Burns PA, Cooper MB, Lokan KH, Wilks MJ, Williams GA (1995) Characteristics of plutonium and americium contamination at the former UK atomic weapons test ranges at Maralinga and Emu. Appl Radiat Isot 46:1099–1107
Carlsbad Environmental Monitoring and Research Center (CEMRC) (1998) 1997 Report Carlsbad Environmental Monitoring & Research Center Waste-management Education & Research Consortium (WERC), New Mexico State University, Carlsbad, NM.
Carlsbad Environmental Monitoring and Research Center (CEMRC) (2006) Annual report. New Mexico State University, Carlsbad, NM
Carlsbad Environmental Monitoring and Research Center (CEMRC) (2014) Annual report. New Mexico State University, Carlsbad, NM
Cooper MB, Burns PA, Tracy BL, Wilks MJ, Williams GA (1994) Characterization of plutonium contamination at the former nuclear weapons testing range, at Maralinga in South Australia. J Radioanal Nucl Chem 177:161–184
Dares CJ, Lapides AM, Mincher BJ, Meyer TJ (2015) Electrochemical oxidation of 243Am(III) in nitric acid by a terpyridyl-derivatized electrode. Science 350:652–655
EPA (1976) Americium - Its behavior in soil and plant systems. Las Vegas, NV: Office of Research and Development, U.S. Environmental Protection Agency. EPA600/3-76-005. PB250797
Eriksson M, Lindahl P, Roos P, Dahlgaard, H, Holm, E (2008) U, Pu, and Am nuclear signatures of the Thule hydrogen bomb debris. Environ Sci Technol 42:4717–4722
Evangeliou N, Zibtsev S, Myroniuk V, Zhurba M, Hamburger T, Stohl A, Balkanski Y, Paugam R, Mousseau TA, Møller AP, Kireev SI (2016) Resuspension and atmospheric transport of radionuclides due to wildfires near the Chernobyl Nuclear Power Plant in 2015: an impact assessment. Sci Rep 6:26062. https://doi.org/10.1038/srep26062.
Faller F (1994) Residual soil radioactivity at the gnome test site in Eddy County, New Mexico, report no. EPA 600/R-94/117, July 1994. Washington, DC, Environmental Protection Agency
Gray DH, Kenney JW, Ballard SC (2000) Operational Radiation Surveillance of the WIPP Project by EEG During 1999. EEG-79, Environmental Evaluation Group, Albuquerque
Guogang J, Testa C, Desideri D, Roselli C (1998) Sequential separation and determination of plutonium, americium-241 and strontium-90 in soils and sediments. J Radioanal Nucl Chem 230:21–27
Hardy EP, Krey PW, Volchok HL (1973) Global inventory and distribution of fallout plutonium. Nature 241:444–445
Hindman FD (1986) Actinide separations for alpha spectrometry using neodymium fluoride coprecipitation. Anal Chem 56:1238–1241
Hirose K, Igarashi Y, Aoyama M, Kim CK, Kim CS, Chang BW (2003) Recent trends of plutonium fallout observed in Japan: plutonium as a proxy for desertification. J Environ Monit 5:302–307
Hirose K, Kim CK, Kim CS, Chang BW, Igarashi Y, Aoyama M (2004) Wet and dry deposition patterns of plutonium in Daejeon, Korea. Sci Total Environ 332:243–252
Hodge V, Smith C, Whiting J (1996) Radiocesium and plutonium: still together in “background” soils after more than thirty years. Chemosphere 32:2067–2075
Holgye Z, Schlesingerova E, Tecl J, Filgas R (2004) 238Pu and 239+240Pu, 241Am, 90Sr and 137Cs in the soils around nuclear research center REZ, near Prague. J Environ Radioact 71:115–125
Holgye Z, Filgas R (1987) Determination of 239+240Pu in surface air in several localities in Czechoslovakia in 1986 in connection with the Chernobyl radiation accident. J Radioanal Nucl Chem 119:21–28
Hollander W (1994) Resuspension factors of 137Cs in Hannover after the Chernobyl accident. J Aerosol Sci 25:789–792
Hulse SE, Ibrahim SA, Whicker FW, Chapman PL (1999) Comparison of 241Am, 239,240Pu and 137Cs concentrations in soil around rocky flats. Health Phys 76:275–287
Hayes RB, Akbarzadeh M (2014) Using isotopic ratios for discrimination of environmental anthropogenic radioactivity. Health Phys 107:277–291
Irlweck K, Hrnecek E (1999) 241Am concentration and 241Pu/239(240) Pu ratios in soils contaminated by weapons-grade plutonium. J Radioanal Nucl Chem 242:595–599
Irlweck K, Wicke J (1998) Isotopic composition of plutonium immissions in Austria after the Chernobyl accident. J Radioanal Nucl Chem 227:133–136
Jia C, Testa C, Desideri D, Guerra F, Roselli MA, Belli ME (1999) Soil concentration, vertical distribution and inventory of plutonium, 241Am, 90Sr and 137Cs in the Merche region of Central Italy. Health Phys 77:52–61
Kenney JW, Downes PS, Gray DH, Ballard SC (1995) Radionuclide baseline in soil near project gnome and the waste isolation pilot plant. EEG-58, Environmental Evaluation Group, Albuquerque, NM
Knatko VA, Mayall A, Drugachenok MA, Matveenko II, Mironov VP (1993) Radiation doses in southern Byelorussia from the inhalation of specific radionuclides following the Chernobyl accident. Radiat Prot Dosim 48:179–183
Krey PW, Hardy EP, Pachucki C, Rourke F, Coluzza J, Benson WK (1976) Mass isotopic composition of global fallout plutonium in soil. Transuranic nuclides in the environment. IAEA-SM-199/39. Vienna: IAEA, p. 671–678.
Lee MH, Clark S (2005). Activities of Pu and Am isotopes and isotopic ratios in a soil contaminated by weapons-grade plutonium, 39: 5512–5516
Lee SC, Orlandini KA, Webb J, Schoep D, Kirchner T, Fingleton DJ (1998) Measurement of baseline atmospheric plutonium-239, 240 and americium-241 in the vicinity of the waste isolation pilot plant. J Radioanal Nucl Chem 234:267–272
Lehto J, Salminen S, Jaakkola T, Outola I, Pulli S, Paatero J, Tarvainen M, Ristonmaa S, Zilliacus R, Ossintsev A, Larin V (2006) Plutonium in the air in Kurchatov, Kazakhstan. Sci Total Environ 366:206–217
Lemons B, Khaing H, Ward A, Thakur P (2018) A rapid method for the sequential separation of polonium, plutonium, americium and uranium in drinking water. Appl Radiat Isot 136:10–17.
Leon Vintro L, Mitchell PI, Condren OM, Downes AB, Papucci C, Delfanti R (1999) Vertical and horizontal fluxes of plutonium and americium in the western Mediterranean and the Strait of Gibraltar. Sci Total Environ 237/238:77–91
Litaor MI (1995) Spatial analysis of plutonium-239+ 240 and americium-241 in soils around Rocky Flats, Colorado. J Environ Qual 24:506–516
Litaor M, Allen LA (1996) Comprehensive appraisal of 241Am in soils around Rocky Flats, Colorado. Health Phys 71:347–357
Livens FR, Singleton DL (1991) Plutonium and americium in soil organic matter. J Environ Radioact 13:323–339
Łokas EJW, Mietelski JW, Ketterer ME, Kleszcz K, Wachniew P, Michalska S, Miecznik M (2013) Sources and vertical distribution of 137Cs, 238Pu, 239+240Pu and 241Am in peat profiles from southwest Spitsbergen. Appl Geochem 28:100–108
Lujanienė G, Aninkevičius V, Lujanas V (2009) Artificial radionuclides in the atmosphere over Lithuania. J Environ Radioact 100:108–119
Lujaniené G, Valiulis D, Bycenkiene S, Sakalys J, Povinec PP (2012a) Plutonium isotopes and 241Am in the atmosphere of Lithuania: a comparison of different source terms. Atmos Environ 61:419–427
Lujaniené G, Bycenkiene S, Povinec PP, Gera M (2012b) Radionuclides from the Fukushima accident in the air over Lithuania e measurement and modeling approaches. J Environ Radioact 114:71–80
Lujaniene G, Sapolaite J, Remeikis V, Lujanas V, Jermolajev A (2006) Cesium, americium and plutonium isotopes in ground level air of Vilnius. Czech J Phys Suppl D 56:D55–D66
Lujaniene G, Lujanas V, Jankunaite D, Ogorodnikov BI, Mastauskas A, Ladygiene R (1999) Speciation of radionuclides of the Chernobyl origin in aerosol and soil samples. J Environ Radioact 49:107–114
Manić-Kudra S, Paligorić D, Novković D, Smiljanić R, Milošević Z, Subotić K (1995) Plutonium isotopes in the surface air at Vinča-Belgrade site in May 1986. J Radioanal Nucl Chem 199:27–34
Mboulou MO, Hurtgen C, Hofkens K, Vandecasteele C (1998) Vertical distributions in the Kapachi soil of the plutonium isotopes (238Pu, 239,240Pu, 241Pu), of 241Am, and of 243,244Cm, eight years after the Chernobyl accident. J Environ Radioact 39:231–237
Menut L, Masson O, Bessagnet B (2009) Contribution of Saharan dust on radionuclide aerosol activity levels in Europe? The 21-22 February 2004 case study. J Geophys Res 114:D16202
Mietelski JW, Kubica B, Gaca P, Tomankiewicz E, Blazej S, Tuteja-Krysa M, Stobiski M (2007) 238Pu, 239+240Pu, 241Am, 90Sr and 137Cs in mountain soil samples from the Tatra National Park (Poland). J Radioanal Nucl Chem 275:523–533
Muramatsu Y, Hamilton T, Uchida S, Tagami K, Yoshida S, Robison W (2001) Measurement of 240Pu/239Pu isotopic ratios in soils from the Marshall Islands using ICP-MS. Sci Total Environ 278:151–159
Paatero J, Hameri K, Jaakkola T, Jantunen M, Koivukoski J, Saxen R (2010) Airborne and deposited radioactivity from the Chernobyl accident—a review of investigations in Finland Boreal. Environ Res 15:19–33
Perkins RW, Thomas CW (1980) Worldwide fallout. In: Hanson WC (ed) Transuranic elements in the environment. Technical Information Center, US Dept of Energy, Springfield, pp 53–82.
Pham MK, Chamizo E, Balbuena JLM, Juan-Carlos Miquel JC, Martín J, Osvath I, Pavel P, Povinec PP (2017) Impact of Saharan dust events on radionuclide levels in Monaco air and in the water column of the northwest Mediterranean Sea. J Environ Radioact 166:2–19
Poet SE, Martell EA (1972) Plutonium-239 and americium-241contamination in the Denver area. Health Phys 23:537–548
Popov L, Mihailova G, Naidenov I (2010) Determination of activity ratios of 238, 239+240, 241Pu, 241Am 134, 137Cs and 90Sr in Bulgarian soils. J Radioanal Nucl Chem 285:223–237
Poston TM, Hanf RW, Dirkes RL and Morasch LF (2002) Hanford site environmental report for calendar year 2000. PNNL-13910, Pacific Northwest National Laboratory, Richland
Price RR (1991) The depth distribution of 90Sr, 137Cs, and 239,240Pu in soil profile samples. Radiochim Acta 54:145–147
Reiter ER (1975) Stratospheric-tropospheric exchange processes. Rev Geophys Space Phys 4:459–474
Roos P, Holm E, Persson RBR, Aarkrog A, Nielsen SP (1994) Deposition of 210Pb, 137Cs, 239+240Pu, 238Pu, and 241Am in the Antarctic Peninsula area. J Environ Radioact 24:235–251
Salminen S, Paatero J (2009) Concentrations of 238Pu, 239+240Pu and 241Pu in the surface air in Finnish Lapland in 1963. Boreal Environ Res 14:827–836
Sehmel GA (1987) Transuranic resuspension. In: Pinter JE III, Alberts JJ, McLeod KW, Schreckhise RG (eds) Environmental research on actinide elements. Office of Science and Technical Information; CONF-841142, Washington, DC, pp 157–192
Sha L, Yamamoto M, Kumura K, Ueno K (1991) 239+240Pu, 241Am and 137Cs in soils from several areas in China. J Radioanal Nucl Chem Lett 155:45–53
Shinn JH, Homan DN, Robison WL (1997) Resuspension studies in the Marshall Islands. Health Phys 73:248–257
Shinonaga T, Steier P, Lagos M, Ohkura T (2014) Airborne plutonium and non-natural uranium from the Fukushima DNPP found at 120 km distance a few days after reactor hydrogen explosions. Environ Sci Technol 48:3808–3814
Solovitch-Vella N, Pourcelot L, Chen VT, Froidevaux P, Gauthier-Lafaye F, Stille P, Aubert D (2007) Comparative migration behavior of Sr-90, Pu-239+240 and Am-241 in mineral and organic soils of France. Appl Geochem 22:2526–2535
Srncik M, Wallner G, Hrnecek E, Steier P, Wallner A, Bossew P (2008) Vertical distribution of 238Pu, 239(40)Pu, 241Am, 90Sr and 137Cs in Austrian soil profiles. Radiochim Acta 96:733–738
Stout JE, Arimoto R (2010) Threshold wind velocities for sand movement in the Mescalero Sands of southeastern New Mexico. J Arid Environ 74:1456–1460
Thakur P, Ballard S, Nelson R (2012) Plutonium in the WIPP environment: its detection, distribution and behavior. J Environ Monit 14:1604–1615
Thakur P, Lemons BG, White CR (2016) The magnitude and relevance of the February 2014 radiation release from the waste isolation pilot plant repository in New Mexico, USA. Sci Total Environ 565:1124–1137
Thurston J (2010) NCRP Report No. 160: ionizing radiation exposure of the population of the United States, National Council on Radiation Protection and Measurements, Bethesda
Turner M, Rudin M, Cizdziel J, Hodge V (2003) Excess plutonium in soil near the Nevada Test Site, USA. Environ Pollut 125:193–203
United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) 2000, Sources and effects of ionizing radiation, Report to the General Assembly, with Scientific Annexes Vol. I United Nations, New York
United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) 1982, Sources and effects of ionizing radiation, Report to the General Assembly, Annexe E. United Nations, New York.
USAEC (1973) Gnome/Coach site disposal options. U.S. Atomic Energy Commission NVO-131, Las Vegas, NV
USDOE (2014) Title 40 CFR Part 191 Subparts B and C Compliance Recertification Application 2014.DOE/WIPP-14-3503. SOTERM-III. Appendix SOTERM-2014.
USDOE (2015). U.S. Department of Energy Accident Investigation Report, Phase-II. Radiological Release Event at the Waste Isolation Pilot Plant on February 14, 2014. Washington, DC: U.S. Department of Energy. Accessible at: http://www.wipp.energy.gov/Special/AIB_WIPP%20Rad_Event%20Report_Phase%20II.pdf
USDOE (1995) Geochemical characterization of background surface soils: background soils characterization program. Rocky Flats Environmental Technology Site, Golden CO
USDOE (2014) Waste Isolation Pilot Plant Annual Site Environmental Report for 2014. DOE/WIPP-15-8866, Carlsbad Field Office, Carlsbad
Wang Z, Yang G, Zheng J, Cao L, Yu H, Zhu Y, Tagami K, Uchida S (2015) Effect of ashing temperature on accurate determination of plutonium in soil samples. Anal Chem 87:5511–5515
Welch JM, Mgller D, Knoll C, Wilkovitsch M, Giester G, Ofner J, Lendl B, Weinberger P, Georg Steinhauser G (2017) Picomolar traces of americium(III) introduce drastic changes in the structural chemistry of terbium(III): a break in the “gadolinium break”. Angew Chem Int Ed 56:13264–13269
WIPP Land Withdrawal Act (Public Law 102-579). The Waste Isolation Pilot Plant Land Withdrawal Act as amended by public law 104-201 (H.R. 3230, 104th congress).
Wotawa G, De Geer L-E, Becker A, D’Amours R, Jean M, Servranckx R, Ungar K (2006) Inter- and intra-continental transport of radioactive cesium released by boreal forest fires. Geophys Res Lett 33:L12806
Yamamoto M, Komura K, Sakanoue M (1983) 241Am and plutonium in Japanese rice-field surface soils. J Radiat Res 24:237–249
Yamamoto M, Tsukatani T, Katayama Y (1996) Residual radioactivity in the soil of the Semipalatinsk-21 Nuclear Test Site in the former USSR. Health Phys 71:142–148
Yammoto M, Tsumura A, Katayama Y, Tsukatani T (1996) Plutonium isotopic composition in soil from the former Semipalatinsk Nuclear Test Site. Radiochim Acta 72:209–215
Acknowledgements
This research is supported by grant from US Department of Energy, Carlsbad Field Office of DOE through Grant No. DE-EM 0002423. Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the sponsors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Georg Steinhauser
Rights and permissions
About this article

Cite this article
Thakur, P., Ward, A.L. Sources and distribution of 241Am in the vicinity of a deep geologic repository. Environ Sci Pollut Res 26, 2328–2344 (2019). https://doi.org/10.1007/s11356-018-3712-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-3712-5