
Abstract
Arsenate and phosphate have similar properties due to their electrochemical structures, but their environmental impacts are unique. The abundance and competition of arsenate and phosphate determine their bioavailability and leachability; thus, it is essential to understand their fate in the soil environment. In this study, the effects of pH and surface loading on the competitive adsorption of arsenate and phosphate on four iron (hydr)oxides were evaluated by employing the Langmuir isotherm, competitive sorption ratio (CSR), and competition effect (CE). The stability and transformation of the iron (hydr)oxides were also assessed. Various adsorption patterns were observed in the single and mixed treatments by controlling the addition of oxyanions, pH, surface loading, and type of iron (hydr)oxides. Arsenate was preferentially adsorbed at a low pH, whereas phosphate showed the opposite trend. The CEAs(V),P(V) was close to zero at low surface density (no competition) and sequentially changed to negative or positive values with increasing surface density, indirectly indicating the sequential development of promotive and competitive effects. Transformation in goethite was identified at a high pH with the presence of oxyanions, except that no transformation was observed upon the addition of oxyanions and with pH change. However, the stability of the iron (hydr)oxides decreased at a low pH and with the presence of phosphate, arsenate, or both. The hematite showed a significant promotive effect regardless of the pH. Our study revealed that the pH, surface loading, and type of iron (hydr)oxides are intercorrelated and simultaneously affect the adsorption characteristics of oxyanions and the stability of iron (hydr)oxides.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic and phosphorus are in group VII of the periodic table and have similar electrochemical properties; however, their environmental impacts are extremely different (Woolson et al. 1971; Luengo et al. 2007). Arsenic has been known as a poison throughout history (Vahidnia et al. 2007), and phosphorus had acted as a limiting nutrient for agricultural productivity worldwide (Elser et al. 2007). Recent anthropogenic activities in agriculture and mining have accelerated arsenic contamination in over 20 countries, and overfertilization with phosphorus has caused severe eutrophication of water systems worldwide (Barberis et al. 1995; Jain and Ali 2000; Leermakers et al. 2006; Elser et al. 2007). Arsenate and phosphate are the major species of arsenic and phosphorus in the oxic rhizosphere (Truog 1930; Masscheleyn et al. 1991; Lu and Zhu 2010). They are the most notorious elements controlling the fate of arsenic and phosphorus in the soil environment because the fluctuation of environmental conditions not only causes the transition of oxyanion species but also leads to the transformation of soil components and dramatically changes the sorption characteristics (Kwon et al. 2010; Michael Bolanz et al. 2013; Han and Ro 2018).
Various elements coexist in the soil environment, and their competition for adsorption onto the soil components determines their bioavailability and leachability (Zhang and Selim 2008; Scheckel et al. 2009; Liu et al. 2017). Thus, it is essential to understand their competitive adsorption under environmental changes (Violante 2013). Arsenate and phosphate have relatively high affinity for metal (hydr)oxides, but controversial results regarding their competition and structural configuration were found in previous studies. For example, the As(V)/P(V) ratio calculated in the previous literature showed significantly different competitive sorption patterns (Fig. S1) (Jain and Loeppert 2000; Hongshao and Stanforth 2001; Zhang and Selim 2008; Frau et al. 2010; Goh and Lim 2010; Carabante and Grahn 2010). In addition, numerous studies confirmed a bidentate complex as a major structural configuration of arsenate and phosphate on iron (hydr)oxides (Tejedor-Tejedor and Anderson 1990; Farquhar et al. 2002; Sherman and Randall 2003; Antelo et al. 2005; Brechbühl and Christl 2012; Neupane et al. 2014; Liu et al. 2015), but a few studies have identified the presence of a monodentate complex (Waychunas et al. 1993; Fendorf et al. 1997; Loring et al. 2009) or the transition of a monodentate to a tridentate complex upon changing the environmental conditions (Han and Ro 2018).
Previous studies showed diverse results in the adsorption of arsenate and phosphate on iron (hydr)oxides; the results were controversial, and a comparison of published data is challenging due to diverse interpretations and experimental setup conditions (for example, pH, ionic strength, solution/solid ratio, and surface loading) (Puls et al. 1991; Kim et al. 2012; Abdala et al. 2015; Li et al. 2015; Han and Ro 2018). We found that there is no simple equation to quantify the degree of competition to compare and interpret numerous studies. Recent studies by Bolanz et al. (2013) showed that the presence of oxyanions, their concentration, and the pH control the iron (hydr)oxide transformation via various processes, but previous adsorption experiments focused on the stability of iron (hydr)oxides. Most studies synthesized the iron (hydr)oxides in their laboratories, allowing for a scientific result with proper sorbent characterization but possibly generating impurities in the sorbent. Dai et al. (2018) studied the effect of impurities on iron (hydr)oxide formation and showed that the surface of the nanoparticle was enriched with impure ions, altering the adsorption characteristics. Therefore, four commercial and nanosized iron (hydr)oxides, goethite, hematite, maghemite, and magnetite, were selected to compare the adsorption characteristics and obtain results that allow for comparison with other studies.
In this study, single and mixed adsorption isotherms of arsenate and phosphate were investigated. The commercial and nanosized goethite, hematite, maghemite, and magnetite were employed to evaluate the competitive adsorption characteristics. The adsorption data were compared using two simple equations to interpret the sorption mechanism during the competitive sorption. In addition, the structural stability of the iron (hydr)oxides was evaluated under environmental changes (competing ion, pH, and surface loading) by measuring the iron dissolution rate and morphological transformation via transmission electron microscopy analysis.
Materials and methods
Physiochemical characterization
The nanosized iron (hydr)oxides were obtained from Sigma-Aldrich (USA) and US Research Nanomaterials (USA). We measured the pH, electrical conductivity (EC), point of net zero charge (PNZC), surface area (SA), crystal structure, and elemental composition of each of the iron (hydr)oxides. The pH and EC were measured using an Orion 5 Star (Thermo, USA) in a 1:200 (W/V) dilution. The PNZC was measured using the pH drift method (Tewari and Campbell 1976). The SA was obtained using an ASAP 2010 (Micromeritics, USA) with the BET isotherm (N2 and 77 K). To identify the crystal structure, X-ray diffraction (XRD), and high-resolution transmission electron microscopy (HRTEM) were employed. A D8 Advance (Bruker, Germany) with CuKα radiation from 5 to 90° was employed for XRD, and a JEM-3010 (Jeol, Japan) was used for HRTEM measurement. The diffractogram was compared with the reference from the American Mineralogist Crystal Structure Database. A Gatan digital camera and an energy dispersive spectroscopy (Gatan, USA) were employed in the HRTEM to characterize the morphology and elemental concentration. For sample preparation for the HRTEM, one drop of the sample was placed onto a carbon film on a copper grid (C300-25, Ted Pella Inc., USA) and dried overnight in a clean chamber. The sample grid was placed in a 60-mm Petri dish (SPL, Korea) and sealed with parafilm for further analysis. Digital Micrograph software (Gatan, USA) was used to analyze the HRTEM image. The characteristics of the iron (hydr)oxides are summarized in Table S1, and the XRD diffractograms of the iron (hydr)oxides, including the reference, are illustrated in Fig. S2. The pH, EC, PNZC, and SA measurements were conducted at least in triplicate.
Batch experiment
For the sorption isotherm, a single-batch experiment of arsenate or phosphate and a dual-batch experiment of arsenate and phosphate were conducted by varying the pH (4, 7, and 10) and surface loading (0, 0.1, 1, 5, and 10 mM) in triplicate. Briefly, 50 mL of oxyanion solution at different concentrations was mixed with 0.25 g of dried iron (hydr)oxide in a 50-mL conical tube. The initial pH of solution was adjusted to the target pH by adding 1 M HCl or NaOH, and the pH of the mixture was controlled every 12 h. All solutions contained 0.1 M NaCl to maintain the ionic strength under the soil environment, and a vertical shaker (Daehan, Korea) was used at 200 rpm at room temperature (25 °C ± 2 °C) for 48 h of incubation. After the incubation, the sample was centrifuged at 4200 RCF for 1 h, and 20 mL of supernatant was filtered with a 0.2-μm PTFE syringe filter (Advantec, Japan). The supernatant was acidified with a drop of 10 M HCl solution, and inductively coupled plasma-optical emission spectrometry (Icap-7200, Thermo, USA) was employed to measure the oxyanion and iron concentration in the solution. The adsorbed concentration was calculated by subtracting the initial concentration from the aqueous concentration. The iron dissolution rate was calculated by dividing the surface area and reaction time by the total amount of iron in the solution determined from the ICP-OES measurement. All reagents were obtained from Sigma-Aldrich (USA) and were at least 98% pure.
Data analysis and fitting
The Langmuir isotherm was used to evaluate the sorption characteristics of the iron (hydr)oxides. The Langmuir model is expressed as follows:
where Γeq and Ceq are the equilibrium concentrations of oxyanions in the adsorbed surface (molecule nm−2) and solution (mM), respectively, and Γmax and KL are the maximum surface density (molecule nm−2) and Langmuir constant (L mmol−1), respectively. The isotherm fitting was conducted with the dynamic fit wizard function in Sigmaplot 10 (Systat, USA), and we constrained 0 < Γmax < 100 and 0 < KL < 1000 and employed 1000 fits with 2000 iterations.
Two equations were employed, the competitive sorption ratio (CSR) and the competitive effect (CE), to better understand the competition between arsenate and phosphate (Violante and Pigna 2002; Zhu et al. 2011). The CSR was obtained from the study of Violante and Pigna (2002), and we developed the CE to compare the adsorption difference between single and mixed treatments. The equations are expressed as follows:
where ΓAs(V),d and ΓP(V),d are the adsorbed arsenate and phosphate in the dual-batch experiment, respectively, and ΓAs(V),s and ΓP(V),s are the adsorbed arsenate and phosphate in the single-batch experiment, respectively. CAs(V),d and CP(V),d are the initial concentrations in the dual-batch experiment, and CAs(V),s and CP(V),s are the initial concentrations in the single-batch experiment. The CSR indicates the preference between the oxyanions on the sorbent during the competitive adsorption; a CSRAs(V)/P(V) greater than 1 indicates that the adsorption of arsenate is more predominant than that of phosphate, and vice versa. If the CSRAs(V)/P(V) is close to 1, no preferential sorption was favored. We developed the CE to quantitatively demonstrate the change in adsorption via promotion or competition; a CE close to zero implies that no competitive effect occurs; positive or negative values indicate competitive or promotive effects, respectively. All calculations were conducted in Excel software (Microsoft, USA).
Results and discussion
Physiochemical characteristics
The physiochemical characteristics of goethite, hematite, maghemite, and magnetite are summarized in Table S1. The pH values of goethite, hematite, maghemite, and magnetite were 5.72, 9.02, 5.11, and 5.34, and the PNZC values were 5.66, 9.41, 4.66, and 4.63, respectively. The EC was 0.08–0.14 μS cm−1, and the SAs from the BET isotherm were 84, 38, 36, and 9.3 m2 g−1 for goethite, hematite, maghemite, and magnetite, respectively. The cell volume and SA were theoretically calculated from the TEM-measured size of each crystal structure, 0.5825, 0.3027, 0.5906, and 0.1386 nm3 for the cell volume and 103.4, 38.1, 24.7, and 15.4 m2 g−1 for the SA of goethite, hematite, maghemite, and magnetite, respectively. There was a minor difference in the proposed size of the nanosized iron (hydr)oxide and the TEM measurement result, with a rod shape found for the goethite and a spherical shape for the hematite, maghemite, and magnetite; the sizes were 50.3 × 10.8, 58.1, 53.7, and 109.4 nm, respectively (Fig. S3).
XRD measurement was conducted to confirm the crystal structure of the iron (hydr)oxides, and the results are illustrated in Fig. S2. The measured XRD pattern was compared with the previously reported pattern from the American mineralogist crystal structure database, showing a significant match with each crystal structure from the database. The XRD patterns between the maghemite and magnetite were significantly similar, which has been reported in previous studies (Dar and Shivashankar 2014; Ruíz-Baltazar et al. 2015). Small peaks at 21.9 and 25.4° were found, showing the difference between the two crystal structures. Based on our observations, nanosized goethite, hematite, maghemite, and magnetite without unknown sample contamination were confirmed.
Adsorption results and competitive sorption ratio
The batch experiment results at the maximum concentration (10 mM) are illustrated for four iron (hydr)oxides and three pH conditions (Fig. 1). The adsorbed oxyanion concentrations per gram of iron (hydr)oxide (qs) for arsenate, phosphate, and the mixture on the goethite were 0.34, 0.37, and 0.31 mmol g−1, respectively, at pH 4; 0.35, 0.32, and 0.26 mmol g−1, respectively, at pH 7; and 0.29, 0.23, and 0.27 mmol g−1, respectively, at pH 10. The qs values for arsenate, phosphate, and the mixture on the hematite were 0.29, 0.23, and 0.17 mmol g−1, respectively, at pH 4; 0.20, 0.20, and 0.21 mmol g−1, respectively, at pH 7; and 0.06, 0.13, and 0.13 mmol g−1, respectively, at pH 10. For the maghemite, the qs values for arsenate, phosphate, and the mixture were 0.17, 0.28, and 0.28 mmol g−1, respectively, at pH 4; 0.18, 0.19, and 0.15 mmol g−1, respectively, at pH 7; and 0.07, 0.14, and 0.07, respectively, at pH 10. For the magnetite, they were 0.08, 0.14, and 0.09 mmol g−1, respectively, at pH 4; 0.11, 0.16, and 0.11 mmol g−1, respectively, at pH 7; and 0.01, 0.15, and 0.06 mmol g−1, respectively, at pH 10. The maximum qs was observed in the goethite treatments at pH values of 4 and 7, and the minimum qs was observed in the maghemite treatment at pH 10. General patterns in qs were observed: a gradual decrease with increasing pH and a significant correlation between the qs value and the surface area of the adsorbent; the general patterns were described in previous studies (Dixit and Hering 2003; Roberts et al. 2004; Mahmood et al. 2014).

Stacked bar graph of the adsorbed oxyanion per unit mass in the arsenate, phosphate and mixed treatments under 10 mM of total oxyanion input on the goethite, hematite, maghemite, and magnetite at pH 4, 7, and 10. The error bar indicates the standard error from the Langmuir fitting result. An asterisk (*) indicates that the average value of the adsorbed oxyanions from two single treatments is significantly higher than the sum of the adsorbed oxyanions from the mixed treatment, and two asterisks (**) indicate the opposite
A distinctive pattern was observed in which the goethite and magnetite showed higher average values in the single treatments than in the mixed treatments at all pH conditions, but hematite showed lower average values in the single treatments than in the mixed treatments at all pH conditions. The maghemite showed similar results to those of hematite at pH 4 but showed the opposite results at pH 7 and 10. In addition, the qs of phosphate did not gradually decrease with increasing pH in the magnetite treatment, and the single arsenate and mixed treatments showed maximum qs at pH 7, whereas there was no significant difference for phosphate. The qs of phosphate in the hematite, maghemite, and magnetite was higher than the qs of arsenate, except for the hematite at pH 4, and the qs of arsenate was higher than that of phosphate in the goethite at pH 7 and 10, whereas the opposite pattern was observed at pH 4. The results show that the arsenate, phosphate, and their mixture showed different adsorption characteristics on the iron hydroxides and that the pH controlled the adsorption characteristics. To better explain the adsorption isotherm, the qs was converted to surface density (Γs) to address the effects of SA and surface loading. The terms surface loading and surface density indicate the total amount of oxyanion per SA of sorbent and the adsorbed amounts of oxyanion per SA of sorbent, respectively.
The Langmuir adsorption parameters (Γmax and KL) were fitted at pH 4, 7, and 10 in the arsenate, phosphate, and mixed treatments (Table 1). The Γmax values of the arsenate treatment at pH 7 were 2.45, 3.46, 3.50, and 100 molecule nm−2 for goethite, hematite, maghemite, and magnetite, respectively, and the Γmax values of the phosphate treatment at pH 7 were 2.24, 3.38, 3.54, and 11.4 molecule nm−2, respectively. In the mixed treatment at pH 7, the Γmax values of arsenate were 0.846, 2.03, 1.23, and 3.93 molecule nm−2 for goethite, hematite, maghemite, and magnetite, respectively, and the Γmax values of phosphate were 0.846, 2.03, 1.23, and 3.93 molecule nm−2, respectively. The adjusted coefficient of determination was 0.820 to 0.999. An unexpected error was found in the Γmax of the arsenate treatment at pH 7 on the magnetite because the maximum constraint for the fitting was reached. An unsaturated fitting curve was observed, and precipitation was assumed based on the unsaturated curves; thus, HRTEM and XRD analysis were conducted to identify the precipitates. However, the size measurement and XRD showed no significant difference (data not shown). Based on the results and previous studies, it is presumed that the unsaturated curve was caused by surface precipitation or reduction of arsenate to arsenite with the oxidation of magnetite. Numerous studies reported precipitation of arsenate on the surface of iron (hydr)oxides, but no study has confirmed the coprecipitation at circumneutral pH. Precipitation was also not detectable in our HRTEM and XRD analysis. Numerous studies reported that reduction of arsenate to arsenite could increase the adsorption (Jain and Loeppert 2000; Liu et al. 2015), but there was insufficient information to measure the reduction (for example, X-ray absorption near-edge spectroscopy). In addition, the magnetite easily oxidizes under ambient conditions, and the transformation is not easily detected if maghemite is formed or if only a thin surface is transformed (Schwaminger et al. 2017).
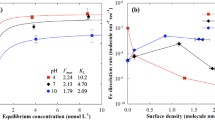
With increasing pH from 4 to 10, the Γmax decreased or was similar in most treatments, but the Γmax in all magnetite treatments and arsenate treatments on maghemite increased or increased then decreased. In contrast, the KL decreased or decreased then increased with increasing pH from 4 to 10 in most treatments, except for arsenate and the mixed treatment on goethite and phosphate and the mixed treatment on maghemite. However, the trends in competitive adsorption in response to a change in pH are complicated; thus, Γmax was applied in Eq. (2) to interpret the results more easily, as illustrated in Fig. 2a. As described above, the CSR and CE indicate the preferential adsorption and competitive effects, respectively, and the black dotted line indicates that no such effect occurred, i.e., the value for the CSR was 1 and for the CE was 0. Interestingly, more preferential sorption of arsenate than phosphate at pH 4 was observed, but the CSRAs(V)/P(V) gradually decreased with increasing pH. The CSRAs(V)/P(V) values were 1.50, 1.27, 1.08, and 1.21 for goethite, hematite, maghemite, and magnetite, respectively, at pH 4 and decreased to 0.98, 0.53, 0.54, and 0.61 at pH 10, respectively. Based on the observation, it was confirmed that the arsenate was more preferentially adsorbed onto the iron (hydr)oxides at the low pH conditions, but phosphate was preferred at the high pH conditions for all the iron (hydr)oxides except goethite.

Line and scatter plot of the pH and competitive sorption ratio (CSR) (a), and the pH and competitive effect (CE) (b) calculated to interpret the competitive sorption of arsenate and phosphate. CSR and CE were calculated with Γmax from the Langmuir isotherm fitting result. A CSRAs(V)/(P) greater than 1 indicates that arsenate is more predominant than phosphate, and vice versa. A CEAs(V),P(V) close to zero implies that no competitive effect was observed, and positive and negative values show competitive or promotive effects, respectively
As noted above, previous studies reported controversial results (Fig. S1). Based on the previous studies, the CSRAs(V)/P(V) was calculated. Several studies confirmed the increase in the CSRAs(V)/P(V) with increasing pH, such as 0.95–1.02 at pH 3–5 on goethite according to Hongshao and Stanforth (2001) and 1.0–1.45 at pH 3.7–9.9 on ferrihydrite according to Jain and Loeppert (2000). In contrast, opposite patterns have also been found; Gao and Mucci (2001) reported CSRAs(V)/P(V) values of 0.9–1.8 at pH 3–8.7 on goethite, Violante and Pigna (2002) reported 1.0–1.2 at pH 4–7 on ferrihydrite, and Manning and Goldberg (1996) found 0.88–1.18 at pH 2.5–11.2 on goethite. No significant correlation among the CSRAs(V)/P(V) pattern, type of iron (hydr)oxide, and experimental condition was found from the previous studies; however, we observed an interesting pattern for interpreting the relationship between surface loading and CSRAs(V)/P(V) (Fig. S4). The CSR of each pH was close to 1 at low surface loading for all iron (hydr)oxides, and the pattern of the CSR with increasing pH was distinctive at the high surface loading condition. In addition, the maghemite and magnetite showed similar patterns, possibly due to the transformation of the magnetite surface to magnetite. The arsenate was more preferentially adsorbed on the iron (hydr)oxide surface at low pH, and phosphate was dominant at high pH, as shown by our observation and previous studies. The surface loading also showed different CSRAs(V)/P(V) patterns for each of the iron (hydr)oxides.
Competitive effect
We observed a relatively uniform pattern in the CSRAs(V)/P(V) results, but the CEAs(V),P(V) results showed a more ambiguous pattern. Γmax was applied in Eq. (3) to interpret the results (Fig. 2b), and the surface loading effect determined using qs is illustrated in Fig. 3. The CEAs(V),P(V) values at pH 4, 7, and 10 were 0.06, 0.12, and 0.20, respectively for goethite; − 0.05, − 0.02, and − 0.17, respectively, for hematite; − 0.13, 0.15, and 0.20, respectively, for maghemite; and 0.16, 0.42, and 0.12, respectively, for magnetite. We hypothesized that an increase in pH would lead to an increase in the CEAs(V),P(V) because more deprotonated sorption sites would be available due to the increased amount of hydroxyl ion, which does not apply to the adsorption sites of oxyanions. In our results, the pattern of the CEAs(V),P(V) agreed with the hypothesis for the goethite and maghemite treatments, but we observed all negative CEAs(V),P(V) values in the hematite treatment and a maximum at pH 7 in the magnetite treatment, which caused the unsaturated curve described above. To explain the results, we considered the surface loading effect because we also hypothesized that increased surface loading would lead to competition for adsorption sites. The relationship between the surface density and CEAs(V),P(V) is illustrated in Fig. 3 to interpret the surface loading effect.

Line and scatter plot of the surface density (molecule nm−2) and competitive effect (CE) of arsenate and phosphate on goethite, hematite, maghemite, and magnetite at pH 4 (a), 7 (b), and 10 (c). CE was calculated with experimental sorption data, and the dotted line indicates zero CEAs(V),P(V), which implies that no competitive effect is observed. We defined the crossing point of the dotted line and solid line as the surface density without competitive or promotive effects (ΓCE = 0)
We found an interesting phenomenon in the goethite, maghemite, and magnetite in which the CEAs(V),P(V) was close to zero at low surface density and became negative with increasing surface density. In addition, a dramatic increase was observed at certain points except for hematite; the crossing point of the dotted line and solid line was defined as ΓCE = 0, where the competitive and promotive effects were offset (Fig. 3). This phenomenon was also observed in previous studies by calculating the CEAs(V),P(V) and was explained by the competition for limited sorption sites. At low surface density, excess adsorption sites were available for both oxyanions; thus, no competitive or promotive effects occurred. However, an increase in surface density led to more adsorption capacity in the mixed treatment than the sum of the single treatments for arsenate and phosphate, which could be explained by the difference in the adsorption mechanisms of both oxyanions on the iron (hydr)oxide surface.
Previous studies confirmed that each oxyanion has a different thermodynamic stability on the plane of the crystal structure. For example, Kubicki et al. (2012) identified the adsorption energies of phosphate as monodentate and bidentate configurations through molecular dynamic simulation, and the results suggested that the bidentate configuration was favorable in the (101), (010), and (100) planes, whereas the monodentate configuration was more stable in the (210) and (001) planes. Kubicki et al. (2007) also showed that the Gibbs free energy of adsorption (ΔGads) for monoprotonated and biprotonated arsenate on bioctahedral ferric hydroxide and biprotonated arsenate showed lower ΔGads than the monoprotonated arsenate complex. Based on the simulation results, it was inferred that the speciation and structural configuration of each oxyanion would lead to a thermodynamically preferential adsorption mechanism on each plane of the iron (hydr)oxide surface, and the overlaid result was only observable from the various adsorption mechanisms. Based on the inference, a sudden decrease in CEAs(V),P(V) could be explained as a result of the difference in the thermodynamic stabilities of the structural configuration for each oxyanion on each iron (hydr)oxide surface, i.e., the promotive effect derived from the difference in the sorption mechanisms of each oxyanion. The promotive and competitive effects are illustrated in Fig. S5. In the figure, chemicals A and B are the oxyanions, which are mono- and divalent ions and have two and three sorption mechanisms via inner- and outer-sphere complexations. At low surface loading (CT = 2), there is unoccupied space in the sorbent; thus, no competitive effect (CEA,B = 0) or preferential sorption (CSRA/B = 1) was observed. At CT = 4, we observed two types of sorption mechanisms for chemical A, three types for chemical B, and three types for the A+B mixture. It was assumed that the IS complex of chemical A (ISA) is not thermodynamically favorable when competing with the IS complex of chemical B with the bidentate configuration (ISB); thus, only the OSA complex was found in the A+B mixture, but both the IS and OS complexes were found in chemical B. In addition, chemical B has two OS complex types, the electrostatic pair interaction with the sorbent (OSB-type 1) and the electrostatic pair with the sorbent and Na+ ion (OSB-type 2). In the A+B mixture, four chemicals were observed at the surface, and 7 chemicals were observed in the A and B treatments; thus, the CEA,B was calculated as − 0.07, which indicates the promotive effect of changing the adsorption mechanism of chemical B. There was no preferential sorption because all chemicals were adsorbed.
In our experiment, a dramatic increase in the CEAs(V),P(V) was observed with the promotive effect, and ΓCE = 0 was defined as where the degrees of the promotive effect and competitive effect are equal. In Fig. S5, CT = 6 describes ΓCE = 0, but the surface loading was used instead of the surface density because the difference in surface loading could not be shown in this example. At this point, the promotive effect is equal to the CT = 4 condition; however, competition was introduced, and the competitive effect counterbalanced the promotive effect. The CSRA/B decreased to 0.33 because more chemical B was adsorbed on the surface than chemical A. After ΓCE = 0, an increase in the CEA,B was also observed due to the change in the OS complex in chemical B. With increasing concentration, OSB-type 2 is more favorable for all OS complexes; thus, three OSB-type 2 were formed, and nine chemicals were adsorbed in the single treatments of A and B, whereas only four chemicals were adsorbed in the A+B mixed treatment. As a result, CEA,B increased to 0.06.
With the interpretation of the example, it is possible to explain the experimental results. Similar patterns were observed in goethite and magnetite at all pH conditions: CEAs(V),P(V) was zero at low surface density and decreased with increasing surface density; then, it increased after ΓCE = 0. The ΓCE = 0 values of goethite at pH 4, 7, and 10 were 1.8, 1.7, and 1.2 molecule nm−2, respectively, and the ΓCE = 0 values of magnetite were 4.9, 1.9, and 1.7 molecule nm−2, respectively. The ΓCE = 0 values of maghemite at pH 7 and 10 were 2.3 and 1.4, respectively. An increase in pH led to a decrease in ΓCE = 0, and we observed that the degree of the decrease significantly differed depending on the iron (hydr)oxide and pH. The results show that there are no promotive or competitive effects when the surface is unsaturated with a low concentration of chemicals and that increasing the surface loading from the low concentration leads to a greater increase in the total adsorbed concentration than the sum of the individually adsorbed concentrations. This phenomenon is called a promotive effect because there is an increase in the adsorbed concentration. Then, the ΓCE = 0 point appears, where the promotive effect and competitive effect are counterbalanced. At high surface loading, the total adsorbed concentration is less than the sum of the individually adsorbed concentrations, and the competitive effect would be greater than the promotive effect; thus, a positive value should be obtained.
Iron oxide stability with the presence of oxyanions
The adsorption characteristics and the stability of the sorbent are crucial to estimate the bioavailability and leachability of oxyanions in the soil environment. There are three main types of iron in the aqueous phase after the centrifugation and filtration, colloidal iron oxide less than 200 nm (because a 0.2-μm filter was used), aqueous iron-oxyanion complexes, and iron ions. However, quantification of their abundance was not possible due to difficulty in obtaining measurements. Thus, the results were interpreted using three rules from previous studies. Numerous studies reported the effect of pH on iron dissolution and revealed that more ferric ions would be released at a low pH condition (Schwertmann 1991; Panias et al. 1996; Cornell and Schwertmann 2003). If the solution pH was close to the PNZC of the colloidal particle, then the precipitation would increase; thus, the concentration of colloidal iron oxide would decrease. In contrast, if the solution pH was not close to the PNZC, then the colloidal particles would increase. Ferric arsenate or the ferric phosphate complex has been reported as a pathway of interaction between iron oxide and aqueous phosphate in the soil environment (Jia and Demopoulos 2008; Du et al. 2014; Wang et al. 2016). Yang et al. (2015) revealed that increasing pH led to the ferric arsenate complex above pH 2.38, and the formation of the FeAsO4 complex from Fe3+ and H2AsO4− ions yielded − 448.2 kJ mol−1 of Gibbs free energy, indicating the thermodynamic stability of the ferric arsenate complex. For this reason, the iron dissolution rate at a high pH condition was mainly caused by the ferric arsenate complex.
The iron dissolution rate is demonstrated in Fig. 4 for the single arsenate or phosphate treatments and the mixed treatment in goethite, hematite, maghemite, and magnetite at a 10-mM concentration. The iron dissolution rates at different concentrations were also measured, but illustration of the concentration effect was too complex; thus, the pH and oxyanion input types were focused on. To summarize the concentration effect, in most conditions, a higher concentration of the arsenate, phosphate, or mixed treatments led to a decrease in the iron dissolution rate, except for goethite (data not shown). Regarding the effect of the pH and oxyanion, the maximum iron dissolution rate was observed at pH 4, except for goethite, and was 0.021, 0.020, and 0.042 μmol m−2 h−1 for hematite, maghemite, and magnetite, respectively, under phosphate treatment. Interestingly, goethite showed a maximum iron dissolution rate at pH 10 with the mixed treatment, and bernalite formation was observed with the presence of arsenate or phosphate, which was identified in previous studies (Han and Ro, 2018). From the HRTEM measurement, we identified the size of the bernalite formation as approximately 30 nm at the high pH condition with high concentrations of arsenate, phosphate, or the mixture, which was the reason for the high iron dissolution rate in goethite at pH 10.

The average iron dissolution rates (nmol m−2 h−1) at 10 mM arsenate (red), phosphate (blue), and mixed (black) treatments as a function of pH on goethite (a), hematite (b), maghemite (c), and magnetite (d). The error bar indicates the standard deviation of the iron dissolution rates at a 10-mM oxyanion concentration
In most conditions, except for the goethite treatment with a high pH, the iron dissolution rates of the arsenate treatments were lower than or similar to those of the phosphate or mixed treatments. The phosphate and mixed treatments showed no significant difference for hematite and magnetite, and the minimum iron dissolution rate occurred at pH 7. A general pattern was observed, showing a decrease in the iron dissolution rate with increasing pH, mainly caused by the reduction in ferric ion dissolution from the iron (hydr)oxide surface. An increase in the iron dissolution rate with increasing pH was observed in the hematite and magnetite, and it was inferred that the formation of ferric arsenate or ferric phosphate resulted in the increase in the iron dissolution rate with increasing pH. The iron dissolution rate of the mixture in maghemite and magnetite showed similar patterns that were distinctive from that in the presence of arsenate. The arsenate treatments showed a lower iron dissolution rate than the phosphate or mixture treatments, but the iron dissolution rates of the arsenate and phosphate treatments were similar for magnetite. Similar results were found in previous studies. Khoe and Robins (1988) measured the formation constant of ferric arsenate and ferric phosphate as − 1.34 and 0.78, respectively. Kuzin et al. (2013) observed an increase in the iron dissolution rate due to the addition of phosphate, whereas Paige et al. (1997) observed a decrease in the iron dissolution rate with the presence of arsenate, caused by surface precipitation. Based on previous studies, the combination of the rate of ferric-oxyanion formation and surface precipitation controls the iron dissolution rate. In addition, both magnetite and maghemite did not show significant differences, and it was presumed that the reduction of arsenate and oxidation of the magnetite surface led to such an effect.
An extremely distinctive pattern of the iron dissolution rate was observed as a function of pH, iron (hydr)oxide, and surface loading, and the individual patterns were difficult to explain based on the current study. However, bernalite as a colloidal iron hydroxide has been found, which would cause the previously reported colloidal mobilization of arsenate and phosphate (Zhang and Selim 2007), and a high level of phosphate would facilitate the iron dissolution compared with the arsenate-rich conditions in most treatments. Hematite showed relatively superior stability with the change in pH and oxyanion than the other iron (hydr)oxides. In addition, our results suggest that the oxyanion could work as a protector or a destroyer of nanosized iron (hydr)oxide in the soil environment. Environmental problems, such as soil acidification or nutrient depletion, would decrease the stability of iron (hydr)oxide in the soil, which would thus accelerate soil degradation by releasing the iron (hydr)oxide from the soil.
Conclusion
In this study, adsorption isotherms were determined by changing the pH of four synthetic nanosized iron (hydr)oxides, and the results were compared by employing the CSR and newly proposed CE to better interpret the competitive adsorption in this study and previous studies. Single or mixed treatments of arsenate and phosphate yielded different results as a function of the pH and the type of iron hydroxide. Arsenate was preferentially adsorbed at a low pH, and phosphate was dominant at a high pH. The CEAs(V),P(V) was close to zero at low surface density and sequentially changed to a negative then positive value with increasing surface density due to promotive and competitive effects. Transformation from goethite to bernalite was identified at high pH with the presence of an oxyanion, and the iron (hydr)oxides showed resistance upon the addition of an oxyanion and a change in pH. In the presence of phosphate, arsenate, or both and at low pH, the stability of iron (hydr)oxide decreased.
In the real environment, oxyanions always compete with each other at different concentrations, pH values, and adsorbents, but the detailed mechanisms of competition are not fully understood. However, our investigation revealed that the environmental variables are interconnected, and this significantly affects the adsorption characteristics of oxyanions in the soil environment. The hematite showed a significant promotive effect regardless of the pH, and it could more effectively reduce problems caused by overfertilization compared to the other iron (hydr)oxides. We focused on the competitive adsorption at equal concentrations of arsenate and phosphate; in the future, the competitive adsorption at nonequal concentrations with a more complex composition must be described to interpret the natural phenomenon in the soil environment.
References
Abdala DB, Northrup PA, Arai Y, Sparks DL (2015) Surface loading effects on orthophosphate surface complexation at the goethite/water interface as examined by extended X-ray absorption fine structure (EXAFS) spectroscopy. J Colloid Interface Sci 437:297–303. https://doi.org/10.1016/j.jcis.2014.09.057
Antelo J, Avena M, Fiol S, López R, Arce F (2005) Effects of pH and ionic strength on the adsorption of phosphate and arsenate at the goethite-water interface. J Colloid Interface Sci 285:476–486. https://doi.org/10.1016/j.jcis.2004.12.032
Barberis E, Ajmone Marsan F, Scalenghe R, Lammers A, Schwertmann U, Edwards AC, Maguire R, Wilson MJ, Delgado A, Torrent J (1995) European soils overfertilized with phosphorus: part 1. Basic properties. Fertil Res 45:199–207. https://doi.org/10.1007/BF00748590
Bolanz RM, Wierzbicka-Wieczorek M, Čaplovičová M, Uhlík P, Göttlicher J, Steininger R, Majzlan J (2013) Structural incorporation of As 5+ into hematite. Environ Sci Technol 47:9140–9147. https://doi.org/10.1021/es305182c
Brechbühl Y, Christl I (2012) Competitive sorption of carbonate and arsenic to hematite: combined ATR-FTIR and batch experiments. J Colloid Interface Sci 377:313–321. https://doi.org/10.1016/j.jcis.2012.03.025
Carabante I, Grahn M (2010) In situ ATR–FTIR studies on the competitive adsorption of arsenate and phosphate on ferrihydrite. J Colloid Interface Sci 351:523–531. https://doi.org/10.1016/j.jcis.2010.07.064
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurrences, and uses. Wiley-VCH
Dai C, Liu J, Hu Y (2018) Impurity-bearing ferrihydrite nanoparticle precipitation/deposition on quartz and corundum. Environ Sci Nano 5:141–149. https://doi.org/10.1039/C7EN00835J
Dar MI, Shivashankar SA (2014) Single crystalline magnetite, maghemite, and hematite nanoparticles with rich coercivity. RSC Adv 4:4105–4113. https://doi.org/10.1039/C3RA45457F
Dixit S, Hering JG (2003) Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ Sci Technol 37:4182–4189. https://doi.org/10.1021/es030309t
Du J, Jing C, Duan J et al (2014) Removal of arsenate with hydrous ferric oxide coprecipitation: effect of humic acid. J Environ Sci (China) 26:240–247. https://doi.org/10.1016/S1001-0742(13)60437-4
Elser JJ, Bracken MES, Cleland EE, Gruner DS, Harpole WS, Hillebrand H, Ngai JT, Seabloom EW, Shurin JB, Smith JE (2007) Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol Lett 10:1135–1142. https://doi.org/10.1111/j.1461-0248.2007.01113.x
Farquhar ML, Charnock JM, Livens FR, Vaughan DJ (2002) Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: an X-ray absorption spectroscopy study. Environ Sci Technol 36:1757–1762. https://doi.org/10.1021/es010216g
Fendorf S, Eick MJ, Grossl P, Sparks DL (1997) Arsenate and chromate retention mechanisms on goethite. 1. Surface structure. Environ Sci Technol 31:315–320. https://doi.org/10.1021/es950653t
Frau F, Addari D, Atzei D, Biddau R (2010) Influence of major anions on As (V) adsorption by synthetic 2-line ferrihydrite. Kinetic investigation and XPS study of the competitive effect of bicarbonate. Water Air & Soil Pollut 205:25–41. https://doi.org/10.1007/s11270-009-0054-4
Gao Y, Mucci A (2001) Acid base reactions, phosphate and arsenate complexation, and their competitive adsorption at the surface of goethite in 0.7 M NaCl solution. Geochim Cosmochim Acta 65:2361–2378. https://doi.org/10.1016/S0016-7037(01)00589-0
Goh KH, Lim TT (2010) Influences of co-existing species on the sorption of toxic oxyanions from aqueous solution by nanocrystalline Mg/Al layered double hydroxide. J Hazard Mater 180:401–408. https://doi.org/10.1016/j.jhazmat.2010.04.045
Han J, Ro H-M (2018) Identification of bernalite transformation and tridentate arsenate complex at nano-goethite under effects of drying, pH and surface loading. Sci Rep 8:8369. https://doi.org/10.1038/s41598-018-26808-4
Hongshao Z, Stanforth R (2001) Competitive adsorption of phosphate and arsenate on goethite. Environ Sci Technol 35:4753–4757
Jain A, Loeppert RH (2000) Effect of competing anions on the adsorption of arsenate and arsenite by ferrihydrite. J Environ Qual 29:1422–1430. https://doi.org/10.2134/jeq2000.00472425002900050008x
Jain C, Ali I (2000) Arsenic: occurrence, toxicity and speciation techniques. Water Res 34:4304–4312
Jia Y, Demopoulos GP (2008) Coprecipitation of arsenate with iron(III) in aqueous sulfate media: effect of time, lime as base and co-ions on arsenic retention. Water Res 42:661–668. https://doi.org/10.1016/j.watres.2007.08.017
Khoe GH, Robins RG (1988) The complexation of iron(III) with sulphate, phosphate, or arsenate ion in sodium nitrate medium at 25 °C. J Chem Soc Dalt Trans 8:2015–2021. https://doi.org/10.1039/DT9880002015
Kim SS, Min JH, Lee JK, Baik MH, Choi JW, Shin HS (2012) Effects of pH and anions on the sorption of selenium ions onto magnetite. J Environ Radioact 104:1–6. https://doi.org/10.1016/j.jenvrad.2011.09.013
Kubicki J, Paul K, Kabalan L, Zhu Q (2012) ATR–FTIR and density functional theory study of the structures, energetics, and vibrational spectra of phosphate adsorbed onto goethite. Langmuir 28:14573–14587. https://doi.org/10.1021/la303111a
Kubicki JDJ, Kwon KD, Paul KW, Sparks DL (2007) Surface complex structures modelled with quantum chemical calculations: carbonate, phosphate, sulphate, arsenate and arsenite. Eur J Soil Sci 58:932–944. https://doi.org/10.1111/j.1365-2389.2007.00931.x
Kuzin A V, Gorichev IG, Lainer YA (2013) Stimulating effect of phosphate ions on the dissolution kinetics of iron oxides in an acidic medium. Russ Metall 9:652–657. https://doi.org/10.1134/S0036029513090073
Kwon JS, Yun ST, Lee JH, Kim SO, Jo HY (2010) Removal of divalent heavy metals (Cd, Cu, Pb, and Zn) and arsenic(III) from aqueous solutions using scoria: kinetics and equilibria of sorption. J Hazard Mater 174:307–313. https://doi.org/10.1016/j.jhazmat.2009.09.052
Leermakers M, Baeyens W, De Gieter M et al (2006) Toxic arsenic compounds in environmental samples: speciation and validation. TrAC - Trends Anal Chem 25:1–10. https://doi.org/10.1016/j.trac.2005.06.004
Li F, Geng D, Cao Q (2015) Adsorption of As(V) on aluminum-, iron-, and manganese-(oxyhydr)oxides: equilibrium and kinetics. Desalin Water Treat 56:1829–1838. https://doi.org/10.1080/19443994.2014.954146
Liu CH, Chuang YH, Chen TY, Tian Y, Li H, Wang MK, Zhang W (2015) Mechanism of arsenic adsorption on magnetite nanoparticles from water: thermodynamic and spectroscopic studies. Environ Sci Technol 49:7726–7734. https://doi.org/10.1021/acs.est.5b00381
Liu X, Li J, Huang Y, Wang X, Zhang X, Wang X (2017) Adsorption, aggregation, and deposition behaviors of carbon dots on minerals. Environ Sci Technol 51:6156–6164. https://doi.org/10.1021/acs.est.6b06558
Loring JS, Sandström MH, Norén K, Persson P (2009) Rethinking arsenate coordination at the surface of goethite. Chem - A Eur J 15:5063–5072. https://doi.org/10.1002/chem.200900284
Lu P, Zhu C (2010) Arsenic Eh–pH diagrams at 25°C and 1 bar. Environ Earth Sci 62:1673–1683. https://doi.org/10.1007/s12665-010-0652-x
Luengo C, Brigante M, Avena M (2007) Adsorption kinetics of phosphate and arsenate on goethite. A comparative study. J Colloid Interface Sci 311:354–360. https://doi.org/10.1016/j.jcis.2007.03.027
Mahmood T, Din SU, Naeem A, Tasleem S, Alum A, Mustafa S (2014) Kinetics, equilibrium and thermodynamics studies of arsenate adsorption from aqueous solutions onto iron hydroxide. J Ind Eng Chem 20:3234–3242. https://doi.org/10.1016/j.jiec.2013.12.004
Manning BA, Goldberg S (1996) Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci Soc Am J 60:121. https://doi.org/10.2136/sssaj1996.03615995006000010020x
Masscheleyn PPH, Delaune RD, Patrick WH (1991) Effect of redox potential and pH on arsenic speciation and solubility in a contaminated soil. Environ Sci Technol 25:1414–1419. https://doi.org/10.1021/es00020a008
Michael Bolanz R, Bläss U, Ackermann S et al (2013) The effect of antimonate, arsenate, and phosphate on the transformation of ferrihydrite to goethite, hematite, feroxyhyte, and tripuhyite. Clay Clay Miner 61:11–25. https://doi.org/10.1346/CCMN.2013.0610102
Neupane G, Donahoe RJ, Arai Y (2014) Kinetics of competitive adsorption/desorption of arsenate and phosphate at the ferrihydrite-water interface. Chem Geol 368:31–38. https://doi.org/10.1016/j.chemgeo.2013.12.020
Paige CR, Snodgrass WJ, Nicholson RV, Scharer JM (1997) An arsenate effect on ferrihydrite dissolution kinetics under acidic oxic conditions. Water Res 31:2370–2382. https://doi.org/10.1016/S0043-1354(97)00107-3
Panias D, Taxiarchou M, Paspaliaris I, Kontopoulos a. (1996) Mechanisms of dissolution of iron oxides in aqueous oxalic acid solutions. Hydrometallurgy 42:257–265. https://doi.org/10.1016/0304-386X(95)00104-O
Puls RW, Powell RM, Clark D, Eldred CJ (1991) Effects of pH, solid/solution ratio, ionic strength, and organic adds. Water Air and Soil Poll 57:423–430
Roberts LC, Hug SJ, Ruettimann T, Billah MM, Khan AW, Rahman MT (2004) Arsenic removal with iron(II) and iron(III) in waters with high silicate and phosphate concentrations. Environ Sci Technol 38:307–315. https://doi.org/10.1021/es0343205
Ruíz-Baltazar A, Esparza R, Rosas G, Pérez R (2015) Effect of the surfactant on the growth and oxidation of iron nanoparticles. J Nanomater 2015:1–8. https://doi.org/10.1155/2015/240948
Scheckel KG, Chaney RL, Basta NT, Ryan JA (2009) Chapter 1 advances in assessing bioavailability of metal(loid)s in contaminated soils. Adv Agron 104:1–52. https://doi.org/10.1016/S0065-2113(09)04001-2
Schwaminger SP, Bauer D, Fraga-García P, Wagner FE, Berensmeier S (2017) Oxidation of magnetite nanoparticles: impact on surface and crystal properties. CrystEngComm 19:246–255. https://doi.org/10.1039/C6CE02421A
Schwertmann U (1991) Solubility and dissolution of iron-oxides. Plant Soil 130:1–25. https://doi.org/10.1007/BF00011851
Sherman DM, Randall SR (2003) Surface complexation of arsenic(V) to iron(III) (hydr)oxides: structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochim Cosmochim Acta 67:4223–4230. https://doi.org/10.1016/S0016-7037(03)00237-0
Tejedor-Tejedor M, Anderson M (1990) The protonation of phosphate on the surface of goethite as studied by CIR-FTIR and electrophoretic mobility. Langmuir 6:602–611. https://doi.org/10.1021/la00093a015
Tewari PH, Campbell AB (1976) Temperature dependence of point of zero charge of cobalt and nickel oxides and hydroxides. J Colloid Interface Sci 55:531–539. https://doi.org/10.1016/0021-9797(76)90063-1
Truog E (1930) The determination of the readily available phosphorus of soils. J Am Soc Agron 22:874–882. https://doi.org/10.2134/agronj1930.00021962002200100008x
Vahidnia A, Van Der Voet GB, De Wolff FA (2007) Arsenic neurotoxicity - a review. Hum Exp Toxicol 26:823–832. https://doi.org/10.1177/0960327107084539
Violante A (2013) Elucidating mechanisms of competitive sorption at the mineral/water interface. Adv Agron 118:111–176. https://doi.org/10.1016/B978-0-12-405942-9.00003-7
Violante A, Pigna M (2002) Competitive sorption of arsenate and phosphate on different clay minerals and soils. Soil Sci Soc Am J 66:1788–1796. https://doi.org/10.2136/sssaj2002.1788
Wang S, Ma X, Zhang G, Jia Y, Hatada K (2016) New insight into the local structure of hydrous ferric arsenate using full-potential multiple scattering analysis, density functional theory calculations, and vibrational spectroscopy. Environ Sci Technol 50:12114–12121. https://doi.org/10.1021/acs.est.6b02703
Waychunas GA, Rea BA, Fuller CC, Davis JA (1993) Surface chemistry of ferrihydrite: part 1. EXAFS studies on geometry of coprecipitated and adsorbed arsenate. Geochim Cosmochim Acta 57:2251–2269. https://doi.org/10.1016/0016-7037(93)90567-G
Woolson EA, Axley JH, Kearney PC (1971) The chemistry and phytotoxicity of arsenic in soils: 1. Contaminated field soils. Soil Sci Soc Am Proc 35:938–944
Yang J, Chai L, Yue M, Li Q (2015) Complexation of arsenate with ferric ion in aqueous solutions. RSC Adv 5:103936–103942. https://doi.org/10.1039/C5RA21836E
Zhang H, Selim HM (2008) Competitive sorption-desorption kinetics of arsenate and phosphate in soils. Soil Sci 173:3–12. https://doi.org/10.1097/ss.0b013e31815ce750
Zhang H, Selim HM (2007) Colloid mobilization and arsenite transport in soil columns: effect of ionic strength. J Environ Qual 36:1273–1280. https://doi.org/10.2134/jeq2006.0373
Zhu J, Pigna M, Cozzolino V, Caporale AG, Violante A (2011) Sorption of arsenite and arsenate on ferrihydrite: effect of organic and inorganic ligands. J Hazard Mater 189:564–571. https://doi.org/10.1016/j.jhazmat.2011.02.071
Funding
This research was supported by the Basic Science Research Program (NRF-2014R1A1A2059196) and Global Ph.D. Fellowship Program (NRF-2015H1A2A1034068) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education of the Republic of Korea and by the Brain Korea 21 Plus Program funded by the Ministry of Education of the Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Responsible editor: Tito Roberto Cadaval Jr
Electronic supplementary material
ESM 1
(DOCX 1516 kb)
Rights and permissions
About this article

Cite this article
Han, J., Ro, HM. Interpreting competitive adsorption of arsenate and phosphate on nanosized iron (hydr)oxides: effects of pH and surface loading. Environ Sci Pollut Res 25, 28572–28582 (2018). https://doi.org/10.1007/s11356-018-2897-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-2897-y