
Abstract
Soil provides a critical environment for microbial community development. However, microorganisms may be sensitive to substances such as heavy metals (HMs), which are common soil contaminants. This study investigated bacterial communities using 16S ribosomal RNA (rRNA) gene fragment sequencing in geographic regions with and without HM pollution to elucidate the effects of soil properties and HMs on bacterial communities. No obvious changes in the richness or diversity of bacterial communities were observed between samples from mining and control areas. Significant differences in bacterial richness and diversity were detected between samples from different geographic regions, indicating that the basic soil characteristics were the most important factors affecting bacterial communities other than HMs. However, the abundances of several phyla and genera differed significantly between mining and control samples, suggesting that Zn and Pb pollution may impact the soil bacterial community composition. Moreover, regression analyses showed that the relative abundances of these phyla and genera were correlated significantly with the soil-available Zn and Pb contents. Redundancy analysis indicated that the soil K, ammoniacal nitrogen (NH4 +–N), total Cu, and available Zn and Cu contents were the most important factors. Our results not only suggested that the soil bacteria were sensitive to HM stresses but also indicated that other soil properties may affect soil microorganisms to a greater extent.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metal (HM) pollution from human activities such as mining, chemical industries, and agriculture has increased greatly over the past century. HMs are non-biodegradable and persist in soil for long periods, even thousands of years (Kabata and Pendias 2001; Fu and Wang 2011). HMs interfere with numerous physiological processes, including alteration of enzyme specificity, disruption of cellular functions, and damage to cell membranes and DNA structure (Bruins et al. 2000). In addition, essential metals can be displaced from their native enzymatic binding sites by contaminant HMs (Bruins et al. 2000). Many studies have reported that excessive exposure to HMs can have deleterious effects on soils, watercourses, the atmosphere, ecosystems, and human health (Wcisło et al. 2002; Pérez-de-Mora et al. 2006; Kasassi et al. 2008; Zhao et al. 2012). Thus, widespread HM pollution is currently one of the most serious environmental problems, especially in mining areas.
Although high HM concentrations have been shown to decrease populations of normal soil microorganisms, such conditions also promote HM-resistant microorganisms in contaminated environments (Bruins et al. 2000; Valls and de Lorenzo 2002). There are several mechanisms underlying HM resistance, including exclusion by barriers, extracellular and intracellular sequestration, reduction in the sensitivity of cellular targets, and enzymatic detoxification (Nies 2003; Hobman et al. 2007). Furthermore, it has been shown that these resistance systems, which may be carried by transposons or plasmids, can be transferred to other community members (Sørensen et al. 2005). Therefore, strong selective pressures on microorganisms living in HM-contaminated soils may have significant consequences for bacterial communities.
Previous studies have revealed that HM accumulation in soils have adverse effects on microbial biomass, activity, and diversity (Frostegård et al. 1993; Macdonald et al. 2011; Margesin et al. 2011). For example, changes in soil microbial community structure and activities have been observed after the addition of HMs (Khan et al. 2010). Li et al. (2015) found that long-term nickel exposure altered the bacterial community composition in agricultural soils. However, some reports have shown only moderate influence of HM pollution on soil bacterial communities and total community richness (Müller et al. 2001). Gans et al. (2005) reported that microbial diversity in heavily polluted soils may constitute only 1 % of that in pristine soils, whereas Niklińska et al. (2005) found that HM pollution weakly influenced the functional structure of microbial communities in long-term polluted forest soils. These studies reveal that different microbial communities responses to HM pollution diversely depending on the soil system.
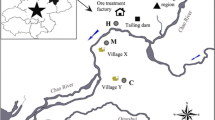
It has been shown that diverse bacterial communities develop similarly when colonizing the same habitat, resulting in similar community compositions (Delmont et al. 2014). This may be explained by the adaptation of bacterial communities to the same selective pressures. The objective of this study was to investigate how different bacterial communities and components of bacterial communities respond to HM selective pressures. We collected soil samples from different geographical regions that varied in HM contents (Fig. 1). Using high throughput sequencing of 16S ribosomal RNA (rRNA) gene fragments, we revealed relationships between HM stresses and microbial components, structure, and diversity.

The study area and sampling site location
Materials and methods
Soil samples
Samples were collected at four sites in Yunnan province, China (Fig. 1). Two lead and zinc enrichment sites were located near a lead and zinc ore enrichment facility (MY, 24° 0′ N, 103° 2′ E) in Jianshui County and an abandoned lead and zinc mine (HZ, 26° 39′ N, 103° 43′ E) in Qujing, while two respective control sites were in the nearby suburbs of Kunming (KM, 24° 49′ N, 102°49′ E) and Shaotong (QJ, 26° 46′ N, 103° 3′ E) (Fig. 1). The lead and zinc ore enrichment facility has been put into use for more than 40 years. The abandoned lead and zinc mine has a hundred years of large-scale mining history and has been abandoned in recent years. Soils closed to these two mines are polluted by human activities like mining and ore washing and hauling. The polluted sampling site is more than 5 km2 for both MY and HZ. The polluted and respective control soils in each region were from same soil types (brown soils for region 1, red soils for region 2) (Fig. 1). All samples were collected around the rhizosphere of Picris divaricata to exclude the influence of vegetation cover. In August 2015, topsoil samples (0–15-cm depth) were collected, mixed, and homogenized by passing them through a 2-mm sieve to remove aboveground plant materials, roots, and stones. Two or three samples were collected at each site, and each sample was collected in triplicate at each sampling location. After packing in sterile Ziploc bags, the soil samples were transported to the laboratory and stored either at 4 °C for further physicochemical properties analysis or at −80 °C for DNA extraction.
Soil chemical analysis and heavy metal determination
All three sample replicates were analyzed independently for their physicochemical properties. The pH, soil total C (TC), soil total N (TN), NH4 +–N, NO3 −–N, soil organic matter (OM), total P, and total K of the samples were measured according to our previous study (Chen et al. 2016). To determine the total HM (Zn, Pb, Cu, and Mn) contents, each soil sample was air-dried, first at room temperature, then at 105 °C for 6 h, and finally passed through a 0.15-mm nylon sieve. After digesting the soils with a mixture of HNO3 and HClO4 (4:1, v/v; Bansal and Kapoor 2000), the Mn, Cu, and Zn concentrations of the resulting solution was determined using inductively coupled plasma optical emission spectrometry (ICP-OES; 710series, Agilent Technologies, Palo Alto, CA, USA). Inductively coupled plasma mass spectrometry (ICP-MS; NexION 300X, PerkinElmer, Norwalk, CT, USA) was used to measure the total Pb concentrations. To determine the available HM concentrations, 20 ml Modified Morgan’s solution (1-M ammonium acetate at pH 4.8) were combined with 4 g soil in a 100-ml Erlenmeyer flask (Chen et al. 2016). After shaking on a rotary shaker at 150 rpm for 15 min, the soil mixture was filtered through filter paper. The extracts were then analyzed for HMs using ICP-MS.
DNA extraction and PCR amplification
Soil DNA was extracted from 0.5 g soil using a FastDNA SPIN Kit following the manufacturer’s instructions (MP Biomedicals, Santa Ana, CA, USA). The extracted DNA was dissolved in 50-μl TE buffer, quantified by spectrophotometry, and then stored at −20 °C. The conserved bacterium-specific primer set, 515F (5′-GTGCCAGCMGCCGCGGTAA-3′)/806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al. 2011), was used to amplify the V4 region of the 16S rRNA gene, which yields accurate taxonomic information and has few biases among various bacterial taxa (Bates et al. 2011). To characterize each sample, a 6-bp error-correcting barcode was included in the reverse primer (Chen et al. 2016). The PCR was carried out in a mixture in a final volume of 20 μl, which contained a 10 ng DNA template, 0.8 μl of each 5 μM primer, 0.4 μl FastPfu Polymerase (TransGen Biotech, Beijing, China), 4 μl 5× FastPfu Buffer, 2 μl 2.5 mM dNTPs, and 0.2 μl bovine serum albumin (BSA; Takara Biotechnology, Dalian, China). The amplification was performed using the following conditions: 95 °C for 3 min; 30 cycles of denaturation at 95 °C for 30 s, primer annealing at 55 °C for 30 s, and extension at 72 °C for 45 s, followed by a final extension period of 10 min at 72 °C. For each sample, all three soil replicates were analyzed independently.
Illumina MiSeq sequencing
Three independent PCR amplification products for each soil sample replicate were combined to construct PCR amplicon libraries to minimize the impact of potential early round PCR errors. After extraction from 2 % agarose gels, the amplicons were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s protocol. Then, the amplicons were quantified using QuantiFluor™-ST (Promega, Madison, WI, USA). After pooling them in equimolar volumes, the purified amplicons were paired-end sequenced (2 × 150) on an Illumina MiSeq platform (Majorbio Bioinformatics Technology Co., Ltd., Shanghai, China) according to standard protocols. In total, 30 sequencing libraries were constructed, and amplicons for each repetition were sequenced independently.
Processing and analyzing the sequencing data
The Illumina raw reads were quality-filtered by QIIME (Caporaso et al. 2010) using the criteria described in Chen et al. (2016). Before any further analysis, resampling was performed based on the minimum sequence numbers across all samples. The sequences were assigned to operational taxonomic units (OTUs) with a 97 % similarity cutoff, and the OTUs were chosen using the UPARSE pipeline (Edgar 2013). Chimeric sequences were identified and removed using UCHIME (Edgar et al. 2011). Representative sequences for each OTU were selected and assigned to taxonomic data at the 70 % threshold using the RDP classifier (Caporaso et al. 2011). The rarefaction curves and indices of Chao1, ACE richness, and Shannon diversity were generated by the Mothur software (Schloss et al. 2009). The beta-diversity estimates (Bray–Curtis distances) were calculated by the QIIME pipeline (Caporaso et al. 2010). The principal coordinate analysis (PCoA) based on Bray–Curtis distances was performed using R (http://www.r-project.org/). The differences and similarities among the communities were characterized using a Venn diagram with unique and shared OTUs. Redundancy analysis (RDA) was carried out using CANOCO for Windows (Etten 2005) to examine the relationships among samples, environmental variables, and frequencies of phyla.
Statistical analyses
Statistical analyses were performed using SPSS version 19.0 (SPSS Inc., Chicago, IL, USA). Parameters were analyzed by one-way analysis of variance, and Tukey’s test was used for multiple comparisons (P < 0.05).
Results
Soil chemical characteristics
The soil characteristics and HM contents are presented in Table 1. The pH changed slightly from 7.58 to 8.52. There were no obvious differences in organic matter (OM) contents among different sampling sites. The high TN and NO3 −–N contents at QJ1 and 2 were likely the result of fertilization. The higher K content at KM and MY compared with HZ and QJ was likely due to geography, revealed by the similar values among samples in the same geographical region. The total Pb concentrations ranged from 3.706 g/kg at MY2 to 36.586 g/kg at HZ1. Similar results were found for Zn (3.168–31.082 g/kg). Both the available and total Zn and Pb concentrations in the control samples (KM and QJ) were significantly lower than those in soils in the mining area (HZ and MY). The Pb and Zn concentrations of samples in the mining area exceeded the Chinese national background values (29.8 and 94.6 mg/kg, respectively, lithosol in Southwest China; Chen et al. 1991). The control areas which surround the mining areas were not polluted, which suggested that Pb and Zn concentrations in the control areas were less than or similar to the background values. Compared to Pb and Zn, much lower Cu and Mn concentrations were detected in mining area (1–4 times of the background values), while no Cu or Mn pollution was found in control areas (Online Resource 1).
Richness and diversity indices
After filtration, 1,465,650 paired-end sequences were obtained from the 30 libraries using the 515F/806R primer sets. These paired-end sequences were overlapped to obtain high-quality tag sequences, the average length of which was 276 bp. The details of the tag sequences obtained from each of the 30 samples are provided in Online Resource 2. All of the rarefaction curves tended to reach saturation (Fig. 2), revealing that the data volume of sequenced reads was sufficient to detect the majority of sequence types. This rarefaction curve indicated that the total number of OTUs varied greatly among the different samples (Fig. 2). We found 7475 OTUs in the bacterial communities of the four sample sites based on 97 % species identity; 2233 (30 %) of the OTUs were shared among the four sample sites (Fig. 3). Fewer site-specific OTUs were found in the samples from the mining area (304 for KM; 574 for MY) than from the control area (656 for HZ; 642 for QJ) (Fig. 3).

Rarefaction curves of the OTU number at 3 % dissimilarity level

Comparison of OTUs in the bacterial communities of soils. The Venn diagram depicts OTUs that are shared or unique for different soil samples
The bacterial richness and diversity were calculated based on randomly selected sequences according to the minimum sequence numbers across all samples (Table 2). We found significantly different numbers of OTUs between the two different sampling regions (region 1: 2791 for HZ and 2981 for QJ on average; region 2: 2335 for KM and 2428 for MY on average). However, no differences were detected between the samples in mining and control areas. The richness indices, Chao1 and ACE, showed that the KM and MY soils consistently had the lowest number of bacterial OTUs, while the HZ and QJ soils had the highest number, with significant differences between regions. No significant differences in richness indices were detected between the mining and control samples. Similarly, the HZ and QJ soils had a significantly higher Shannon diversity, except for HZ2. All samples had a high Good’s query coverage (98 %).
The PCoA based on the OTU composition showed that the samples within each site clustered tightly and differentiated the sites (Fig. 4). That is, there were similarities among samples from the same sites and variations among samples from different sites. A total of 41.6 and 25.4 % of the variations in the bacterial communities could be explained by the first and second principal components, respectively. The difference in community composition revealed by tightly clustering these samples was small between MY and KM compared with other samples.

Principal coordinate analysis (PCoA) of soil samples based on Bray–Curtis distances of bacterial communities
Taxonomy composition
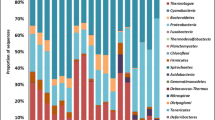
The compositions of the bacterial communities from mining and control samples were similar (Fig. 5). Although similar compositions were observed, variations in the relative contents of phyla were detected among these samples. The classified sample sequences were affiliated with 12 bacterial phyla. The sequences that could not be classified into any known group were assigned as unclassified bacteria (0.6–2.4 %) and others (0.3–1.4 %). Proteobacteria, Actinobacteria, Chloroflexi, Acidobacteria, and Bacteroidetes were the most abundant phyla in all samples. However, more bacteria belonging to Proteobacteria, Chloroflexi, and Acidobacteria were found in region 1 (40, 12, and 9 % respectively) compared with region 2 (36, 8, and 5 % respectively) while region 2 was more abundant in Acidobacteria (37 %) than that of region 1 (20 %) (Fig. 5). The relative abundances of these four main phyla were very similar between MY and KM in region 2. Between HZ and QJ in region 1, variations in contents of Actinobacteria and Chloroflexi were detected, while Proteobacteria and Acidobacteria showed similar relative abundances (Fig. 5). Four phyla (Verrucomicrobia, Nitrospirae, Firmicutes, and Saccharibacteria) were found at relative abundances of <1 %. Abundances of Actinobacteria, Acidobacteria, and Planctomycetes differed significantly among geographical regions, while Verrucomicrobia abundance varied significantly between the mining and control samples (Fig. 6). At the genus level, the classified sequences for each sample were affiliated with 102 bacterial genera (Online Resource 3). The most abundant genera (Arthrobacter, Sphingomonas, Anaerolineaceae_uncultured, Nitrosomonadaceae_uncultured, and Nocardioides) were found in all samples with average abundances >2 %. Abundance differences in 20 genera were found between the two regions, while only seven genera were common between the mining and control areas (Fig. 6). Decreased abundances of Nocardioides, Gaiella, Comamonadaceae_unclassified, Acidimicrobiaceae_uncultured, Actinobacteria_norank, and Skermanella and increased abundances of Bradyrhizobium were detected in mining samples compared with the control samples (Fig. 6).

The abundance percentages of the bacterial phyla for soil samples

The relative abundances of the bacterial phyla and genera: the phyla (a) and genera (c) that varied significantly among samples from mining and control areas and the phyla (b) and genera (d) that varied significantly among samples from different regions. Bars represent the standard errors
Relationship between bacterial community structure and soil parameters
RDA was used to assess a significant correlation between soil chemical properties and bacterial community structure. The first two RDA components explained 93.5 % of the total variation (Fig. 7). The first component (RDA1) separated samples from different regions and explained 65.7 % of the variation. The soil K, NH4 +–N, total Cu, and available Zn and Cu contents had a significant (P < 0.05) correlation between each variable and the ordination scores and were the most important along the RDA1 axes.

Redundancy analysis (RDA) of bacterial communities and soil properties. Zn/Pb/Cu/Mn(a), available Zn/Pb/Cu/Mn; Zn/Pb/Cu/Mn(t), total Zn/Pb/Cu/Mn; **P < 0.01; *P < 0.05
Regression analyses showed that the relative abundances of Nocardioides, Gaiella, Comamonadaceae_unclassified, Acidimicrobiaceae_uncultured, Actinobacteria_norank, and Skermanella were correlated negatively (Spearman’s rank correlation, P < 0.05) with the available soil Zn/Pb content (Table 3). The Verrucomicrobia and Bradyrhizobium abundances had significant and positive correlations (Table 3).
Discussion
In soil ecosystems, soil provides the basic nutritional environment for microbial community development (Ulrich and Becker 2006). Meanwhile, soil microorganisms play important roles in nutrient cycling and energy flow in soil ecosystems (Frische and Hoper 2003; Falkowski et al. 2008; Shen et al. 2014), which are key regulators of soil organic matter dynamics and nutrient availability (Chen et al. 2014). Thus, soil type has been a known key determinant of microbial communities for a long time (Bardgett et al. 1999; Chen et al. 2010). Any discrepancies in basic soil properties can effect changes in microbial assemblages. Many microorganisms are sensitive to harmful substances in soil, such as HMs, which can lead to different bacterial community structures.
To elucidate the effects of soil properties and harmful substances on bacterial communities, we evaluated the evolution of bacterial communities in different geographic regions under the selective pressure of HMs. Significant differences in both richness and diversity indices were found among communities from different sampling regions, while communities in the same sampling region showed no significant differences between the mine and control areas. These results indicated that the basic soil characteristics were the most important factors determining diversity and species richness. Previous studies have shown that despite high levels of Pb and Zn soil contamination, the species diversity and richness of bacterial communities are similar to or only slightly lower than those found in unpolluted soils (Will et al. 2010; Nacke et al. 2011). Similarly, no clear impacts on bacterial diversity or abundance have been identified for nickel (Li et al. 2015) or copper (Berg et al. 2012) soil pollution. Chodak et al. (2013) showed that the structure and diversity of soil microbial communities depends mainly on soil pH, while the energy source and nutrient availability were the most essential factors for microbial biomass and basal respiration. Such results suggest that the effect of high HM contents on soil microbial properties is weaker than that on other soil properties.
Many soil properties have been shown to influence bacterial communities, including pH and OM (Lorenz et al. 2006; Lauber et al. 2009; Hu et al. 2013; Landa et al. 2013, 2014). Soil pH has been reported to be a dominant determinant factor (Lauber et al. 2009; Hu et al. 2013). OM, which provides the carbon source for bacteria, is also considered important (Landa et al. 2013, 2014). In our study, however, pH and OM in all samples were similar. Rather, RDA showed that soil K, NH4 +–N, and Cu contents were the most important factors in our samples. K concentration has also been indicated as an important factor that affects bacterial composition (Lanzén et al. 2013; Pereira et al. 2014). This may be because bacteria can affect the solubility and availability of K and thus affect the growth of plants and the selection of specific bacteria associated with K (Miransari 2013). The effects of N on rhizosphere bacterial communities have been demonstrated (Doran 1980; Li et al. 2015). Similarly, Cu has also been detected to be the important environment factors to influence the bacterial communities (Chodak et al. 2013; Hong et al. 2015).
This study showed that available Zn contents were important to discriminate samples at the phylum level, despite no significant differences in bacterial community structure between mining and control samples. The phylum level may be inadequate for assessing differences in soil bacterial communities. The specific effects can be seen at lower taxonomic levels (Gołębiewski et al. 2014), indicating that these data may be critical to demonstrate the effect of HMs. Indeed, different abundances of seven bacterial genera were detected between samples from the mining and control areas. Furthermore, all seven abundances were correlated significantly with the available Zn and Pb contents in soil. These results suggest that Zn and Pb altered the bacterial community compositions in both mining areas, with less impact on the bacterial abundance and diversity. Long-term copper (Berg et al. 2012) and nickel (Li et al. 2015) exposure has been shown to change bacterial community structures but not diversity. Other similar effects of HMs on bacterial communities can be found in many other studies (Macdonald et al. 2011; Singh et al. 2014), suggesting that taxonomic diversity is relatively insensitive to HM stresses compared with bacterial community composition. These results demonstrate not only that soil bacteria are sensitive to HMs but also that other soil properties may affect microorganisms to a greater extent.
Our results revealed that stress on bacteria exposed to HMs in polluted soils induced shifts in bacterial community compositions. It also showed that bacterial groups responded differently to HM pollution. For example, Verrucomicrobia were more abundant in all mining samples than in all control samples, suggesting their potential in tolerance to Zn and Pb. Verrucomicrobia have also been shown to be tolerant to mercury (Vishnivetskaya et al. 2011). At the genus level, Nocardioides, Gaiella, Comamonadaceae_unclassified, Acidimicrobiaceae_uncultured, Actinobacteria_norank, and Skermanella were sensitive to Zn and/or Pb contamination, whereas Bradyrhizobium appeared potential tolerance. Indeed, the tolerance of Bradyrhizobium to HMs such as nickel and zinc has been reported previously (Wani et al. 2007). Additionally, Bradyrhizobium promotes plant growth in nickel- and zinc-contaminated soils by reducing HM uptake by plant organs, suggesting that it may be useful for remediating nickel- and zinc-contaminated sites (Wani et al. 2007). However, the tolerance of Verrucomicrobia and Bradyrhizobium to Zn and Pb should be verified by experimental evidence in future.
In this study, we analyzed the difference of bacterial communities from two aspects: (1) geographic distance (regions 1 and 2) and (2) HM pollution (mining and control areas). The effect of geographic distance on bacterial communities might result from the different soil types (brown soils for region 1, red soils for region 2) with different soil properties, leading to diverse bacterial communities. It has been proved that diverse bacterial communities could develop similarly when colonizing the same habitat (Delmont et al. 2014). The HM pollution could be another stress on bacteria exposed to HMs in polluted soils, which may result in similar development of diverse bacterial communities. Our results showed the significant differences of bacterial communities in different regions, while bacterial communities were similar in mining and control samples. These results suggest that bacterial communities in different geographic regions can be separated first by soil parental materials and then by HMs, which means although soil bacteria were sensitive to HM stresses, soil properties may affect soil microorganisms to a greater extent.
Conclusion
Bacterial communities with and without HM stress in different sampling regions were evaluated. The richness and diversity of bacterial communities were similar between mining and control samples within the same regions. Significant differences in richness and diversity were observed in samples from different regions. However, we found that Zn and Pb pollution significantly impacted soil bacterial community composition. Our results suggest that bacterial communities in different soils can be separated first by parental materials and then by HMs. Furthermore, bacteria in the phylum Verrucomicrobia and genus Bradyrhizobium were shown to be Zn- and Pb-tolerant and may be useful for bioremediation in HM-contaminated sites.
References
Bansal S, Kapoor KK (2000) Vermicomposting of crop residues and cattle dung with Eisenia foetida. Bioresour Technol 73:95–98
Bardgett RD, Lovell RD, Hobbs PJ, Jarvis SC (1999) Seasonal changes in soil microbial communities along a fertility gradient of temperate grasslands. Soil Biol Biochem 31:1021–1030
Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N (2011) Examining the global distribution of dominant archaeal populations in soil. ISME J 5:908–917
Berg J, Brandt KK, Al-Soud WA, Holm PE, Hansen LH, Sørensen SJ, Nybroe O (2012) Selection for Cu-tolerant bacterial communities with altered composition, but unaltered richness, via long-term Cu exposure. Appl Environ Microbiol 78:7438–7446
Bruins MR, Kapil S, Oehme FW (2000) Microbial resistance to metals in the environment. Ecotox Environ Safe 45:198–207
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 1:4516–4522
Chen J, Wei F, Zheng C, Wu Y, Adriano DC (1991) Background concentrations of elements in soils of China. Water Air Soil Pollut 57:699–712
Chen X, Zhang LM, Shen JP, Xu ZH, He JZ (2010) Soil type determines the abundance and community structure of ammonia-oxidizing bacteria and archaea in flooded paddy soils. J Soils Sediments 10:1510–1516
Chen JH, He F, Zhang XH, Sun X, Zheng JF, Zheng JW (2014) Heavy metal pollution decreases microbial abundance, diversity and activity within particle-size fractions of a paddy soil. FEMS Microbiol Ecol 87:164–181
Chen C, Zhang J, Lu M, Qin C, Chen Y, Yang L, Huang Q, Wang J, Shen Z, Shen Q (2016) Microbial communities of an arable soil treated for 8 years with organic and inorganic fertilizers. Biol Fertil Soils 52:455–467
Chodak M, Gołębiewski M, Morawska-Płoskonka J, Kuduk K, Niklińska M (2013) Diversity of microorganisms from forest soils differently polluted with heavy metals. Appl Soil Ecol 64:7–14
Delmont TO, Francioli D, Jacquesson S, Laoudi S, Mathieu A, Nesme J, Ceccherini MT, Nannipieri P, Simonet P, Vogel TM (2014) Microbial community development and unseen diversity recovery in inoculated sterile soil. Biol Fertil Soils 50:1069–1076
Doran JW (1980) Soil microbial and biochemical changes associated with reduced tillage. Soil Sci Soc Am J 44:765–771
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Etten EV (2005) Multivariate analysis of ecological data using CANOCO. Austral Eco 30:486–487
Falkowski PG, Fenchel T, Delong EF (2008) The microbial engines that drive Earth’s biogeochemical cycles. Science 320:1034–1039
Frische T, Hoper H (2003) Soil microbial parameters and luminescent bacteria assays as indicators for in situ bioremediation of TNTcontaminated soils. Chemosphere 50:415–427
Frostegård Å, Tunlid A, Bååth E (1993) Phospholipid fatty acid composition, biomass, and activity of microbial communities from two soil types experimentally exposed to different heavy metals. Appl Environ Microbiol 59:3605–3617
Fu F, Wang Q (2011) Removal of heavy metal ions from wastewaters: a review. J Environ Manag 92:407–418
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and highmetal toxicity in soil. Science 309:1387–1390
Gołębiewski M, Deja-Sikora E, Cichosz M, Tretyn A, Wróbel B (2014) 16S rDNA pyrosequencing analysis of bacterial community in heavy metals polluted soils. Microb Ecol 67:635–647
Hobman J, Yamamoto K, Oshima T (2007) Transcriptomic responses of bacterial cells to sublethal metal ion stress. In: Nies D, Silver S (eds) Molecular microbiology of heavy metals. Microbiology monographs. Springer, Berlin, pp. 73–115
Hong C, Si Y, Xing Y, Li Y (2015) Illumina MiSeq sequencing investigation on the contrasting soil bacterial community structures in different iron mining areas. Environ Sci Pollut Res 22:10788–10799
Hu HW, Zhang LM, Dai Y, Di HJ, He JZ (2013) pH-dependent distribution of soil ammonia oxidizers across a large geographical scale as revealed by high-throughput pyrosequencing. J Soils Sediments 13:1439–1449
Kabata A, Pendias H (2001) Trace elements in soils and plants. CRC, Washington DC
Kasassi A, Rakimbei P, Karagiannidis A, Zabaniotou A, Tsiouvaras K, Nastis A, Tzafeiropoulou K (2008) Soil contamination by heavy metals: measurements from a closed unlined landfill. Biores Technol 99:8578–8584
Khan S, Hesham AE, Qiao M, Rehman S, He JZ (2010) Effects of Cd and Pb on soil microbial community structure and activities. Environ Sci Pollut Res 17:288–296
Landa M, Cottrell MT, Kirchman DL, Blain S, Obernosterer I (2013) Changes in bacterial diversity in response to dissolved organic matter supply in a continuous culture experiment. Aquat Microb Ecol 69:157–168
Landa M, Cottrell MT, Kirchman DL, Kaiser K, Medeiros PM, Tremblay L, Batailler N, Caparros J, Catala P, Escoubeyrou K, Oriol L, Blain S, Obernosterer I (2014) Phylogenetic and structural response of heterotrophic bacteria to dissolved organic matter of different chemical composition in a continuous culture study. Environ Microbiol 16:1668–1681
Lanzén A, Simachew A, Gessesse A, Chmolowska D, Jonassen I, Øvreås L (2013) Surprising prokaryotic and eukaryotic diversity, community structure and biogeography of Ethiopian soda lakes. PLoS One 8:e72577
Lauber CL, Hamady M, Knight R, Fierer N (2009) Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120
Li J, Hu HW, Ma YB, Wang JT, Liu YR, He JZ (2015) Long-term nickel exposure altered the bacterial community composition but not diversity in two contrasting agricultural soils. Environ Sci Pollut Res 22:10496–10505
Lorenz N, Hintemann T, Kramarewa T, Katayama A, Yasuta T, Marschner P, Kandeler E (2006) Response of microbial activity and microbial community composition in soils to long-term arsenic and cadmium exposure. Soil Biol Biochem 38:1430–1437
Macdonald CA, Clark IM, Zhao FJ, Hirsch PR, Singh BK, McGrath SP (2011) Long-term impacts of zinc and copper enriched sewage sludge additions on bacterial, archaeal and fungal communities in arable and grassland soils. Soil Biol Biochem 43:932–941
Margesin R, Płaza GA, Kasenbacher S (2011) Characterization of bacterial communities at heavy-metal-contaminated sites. Chemosphere 82:1583–1588
Miransari M (2013) Soil microbes and the availability of soil nutrients. Acta Physiol Plant 5:3075–3084
Müller AK, Westergaard K, Christensen S, Sørensen SJ (2001) The effect of long-term mercury pollution on the soil microbial community. FEMS Microbiol Ecol 36:11–19
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 6:e17000
Nies DH (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27:313–339
Niklińska M, Chodak M, Laskowski R (2005) Characterization of the forest humus microbial community in a heavy metal polluted area. Soil Biol Biochem 37:2185–2194
Pereira LB, Vicentini R, Ottoboni LM (2014) Changes in the bacterial community of soil from a neutral mine drainage channel. PLoS One 9:e96605
Pérez-de-Mora A, Burgos P, Madejón E, Cabrera F, Jaeckel P, Schloter M (2006) Microbial community structure and function in a soil contaminated by heavy metals: effects of plant growth and different amendments. Soil Biol Biochem 38:327–341
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75:7537–7541
Shen JP, Xu ZH, He JZ (2014) Frontiers in the microbial processes of ammonia oxidation in soils and sediments. J Soils Sediments 14:1023–1029
Singh BK, Quince C, Macdonald CA, Khachane A, Thomas N, Al-Soud WA, Sørensen SJ, He ZL, White D, Sinclair A, Crooks B, Zhou JZ, Campbell CD (2014) Loss of microbial diversity in soils is coincident with reductions in some specialized functions. Environ Microbiol 16:2408–2420
Sørensen SJ, Bailey M, Hansen LH, Kroer N, Wuertz S (2005) Studying plasmid horizontal transfer in situ: a critical review. Nat Rev Microbiol 3:700–710
Ulrich A, Becker R (2006) Soil parent material is a key determinant of the bacterial community structure in arable soils. FEMS Microbiol Ecol 56:430–443
Valls M, de Lorenzo V (2002) Exploiting the genetic and biochemical capacities of bacteria for the remediation of heavy metal pollution. FEMS Microbiol Rev 26:327–338
Vishnivetskaya TA, Mosher JJ, Palumbo AV, Yang ZK, Podar M, Brown SD, Brooks SC, Gu B, Southworth GR, Drake MM, Brandt CC, Elias DA (2011) Mercury and other heavy metals influence bacterial community structure in contaminated Tennessee streams. Appl Environ Microbiol 77:302–311
Wani PA, Khan MS, Zaidi A (2007) Effect of metal tolerant plant growth promoting Bradyrhizobium sp. (vigna) on growth, symbiosis, seed yield and metal uptake by greengram plants. Chemosphere 70:36–45
Wcisło E, Ioven D, Kucharski R, Szdzuj J (2002) Human health risk assessment case study an abandoned metal smelter site in Poland. Chemosphere 47:507–515
Will C, Thürmer A, Wollherr A, Nacke H, Herold N, Schrumpf M, Gutknecht J, Wubet T, Buscot F, Daniel R (2010) Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76:6751–6759
Zhao H, Xia B, Fan C, Zhao P, Shen S (2012) Human health risk from soil heavy metal contamination under different land uses near Dabaoshan mine, southern China. Sci Total Environ 417:45–54
Acknowledgments
The research was funded by the National Natural Science Foundation of China (31400328 and 31501689) and the Science Foundation of Jiangsu Province, China (BK20140697 and BK20150670).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible Editor: Zhihong Xu
Electronic supplementary material
ESM 1
(PDF 307 kb)
Rights and permissions
About this article

Cite this article
Xu, X., Zhang, Z., Hu, S. et al. Response of soil bacterial communities to lead and zinc pollution revealed by Illumina MiSeq sequencing investigation. Environ Sci Pollut Res 24, 666–675 (2017). https://doi.org/10.1007/s11356-016-7826-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-7826-3