
Abstract
Pharmaceuticals such as nonsteroidal anti-inflammatory drugs (NSAIDs) and lipid regulators are being repeatedly detected at low concentrations (pg · mL−1–ng · mL−1) in the environment. A large fraction of these compounds are ionizable. Ionized compounds show different physico-chemical properties and environmental behavior in comparison to their neutral analogs; as a consequence, the quantification methods currently available, based on the neutral molecules, might not be suitable to detect the corresponding ionized compounds. To overcome this problem, we developed a specific analytical method to quantify NSAIDs and lipid regulators (i.e., ibuprofen, diclofenac, naproxen, and clofibric acid) and their ionized compounds. This method is based on three steps: (1) the extraction of the organic compounds with an organic solvent assisted with an ultrasonic probe, (2) the cleaning of the extracts with a dispersive SPE with C18, and (3) the determination of the chemical compounds by GC-MS (prior derivatization of the analytes). We demonstrated that the proposed method can successfully quantify the pharmaceuticals and their ionized compounds in aqueous samples, lumpfish eggs, and zebrafish eleutheroembryos. Additionally, it allows the extraction and the cleanup of extracts from small samples (0.010 g of wet weight in pools of 20 larvae) and complex matrixes (due to high lipid content) and can be used as a basis for bioaccumulation assays performed with zebrafish eleutheroembryos in alternative to OECD test 305.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
NSAIDs together with lipid regulators are among the most commonly applied analgesics and anti-inflammatory drugs worldwide (Macia et al. 2007). Within those, ibuprofen (IBU), diclofenac (DC), naproxen (NP), and clofibric acid (CA) are some of the most frequently used. As a result of their widespread use, these products and their metabolites and products of degradation can enter the water supply from domestic or industrial waste water discharges, commercial food treatment procedures, and the ground application of manure, and in recent years, they are being detected at low concentrations levels (range of pg- or ng · mL−1) in various environmental media such as surface water and sediments (Azzouz et al. 2010; Azzouz et al. 2015; Gentili et al. 2012; Arroyo et al. 2011; Dubreil-Chéneau et al. 2011; Grueiro Noche et al. 2011; Zhang H et al. 2013; Sadat Hasheiminasab et al. 2013; Coimbra et al. 2015; Corcoran et al. 2015).
There is evidence that NSAIDs and CA can induce ecotoxicological effects and bioaccumulation in aquatic organism (Corcoran et al. 2010). Coimbra et al. found a significant reduction in growth, a lower muscle triglyceride, impacts in male gonad development, and a decrease in the fecundity in adult zebrafish exposed at 10 mg · g−1 as well as abnormalities in the offspring of fish when exposed at low CA doses (1 mg · g−1) (Coimbra et al. 2015). Saravanan et al. demonstrated that red blood cells, plasma ions such as Na+ or K+ and enzymatic levels decreased in common carp exposed to 1, 10, and 100 ng · mL−1 of clofibric acid and diclofenac (Saravanan et al. 2011). In another study, these authors found a decrease of erythrocyte (RBC), of mean cellular hemoglobin concentration (MCHC), of mean cellular volume (MCV) and an increase in the level of plasma protein, glucose, and enzymes when fishes were exposed to 14.2 μg · mL−1 of ibuprofen (24 h LC50 IBU = 142 μg · mL−1) (Saravanan et al. 2012).
As a consequence of the high use of these and other drugs, a public, regulatory, and scientific concern of the environmental impacts of pharmaceuticals has appeared, considering them as emerging contaminants (Zenker et al. 2014). According to the European Regulation concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), chemicals have to be registered by the responsible manufacturers or importers, supplying information/data (persistency, bioaccumulation potential, and toxicity) to assess the risk for humans and the environment. Standard information (bioaccumulation in aquatic species) is required for substances imported or manufactured ≥100 t/a. The decision whether a bioaccumulation test is necessary is mainly based on the octanol-water partitioning coefficient (K OW), which correlates well with the sorption of organic substances. However, some waiving reasons can be considered such as low potential for bioaccumulation due to log K OW <3. A normal screening criterion is established for log K OW >4.5, as in those cases the substance is potentially bioaccumulable or very bioaccumulable. However, 49 % of the chemicals registered under REACH are not in their neutral status, and most of the time they are charged. Pharmaceuticals are no exception and are actually found mainly as ionized molecules (Franco et al. 2010).
Available methods for the quantification and the evaluation of bioaccumulation potential of chemicals don’t seem to be appropriate for ionized organic compounds (IOC). A recent study concerning the compliance of registration dossiers (above 1000 t/a) in regard to data requirements within REACH, screened 1932 dossiers (German Federal Environment Agency (UBA) 2015). The project revealed that for about 18 % of the registered substances, the bioaccumulation data calculated was waived because the organic substances were ionized or hydrolytically unstable. It is unclear if these waiving is justified, because screening approaches and criteria are lacking for these cases. Therefore, the knowledge gathered around the description of neutral compounds cannot be transferred one-to-one to the description of charged compounds, and considering the scarcity of physico-chemical data of ions, mechanistic models are of high interest (Bittermann et al. 2016).
One of the methods proposed for quantifying charged compounds is the calculation of the D OW, a correction of the K OW for the fraction of the neutral form for IOC (European Chemicals Agency 2014). A recent study highlighted that this method also fails in the approach to reality, mainly because the D OW parameter is highly dependent on the type and concentration of the counter ions present as an ion changes from one bulk phase (e.g., water) to the other (e.g., octanol) it has to be accompanied by a counter ion either as an ion pair or separately. Depending on the salt concentration (i.e., the concentration of the counter ion), the D OW can therefore differ by more than two orders of magnitude (Bittermann et al. 2016). Other methods are being used to estimate partitioning between water and membranes such as COSMOmic (COnductor-like Screening MOdel for MICells) that is based on quantum chemistry and fluid-phase thermodynamics (Bittermann et al. 2014).
Although much effort is being made to find realistic screening methods, it is clear that to validate these screening strategies real data is necessary. The official method proposed by the REACH for evaluating the bioaccumulation potential of drugs is the Organization for Economic Cooperation and Development (OECD) bioconcentration test 305 (OECD 2012). This method is based on the evaluation of bioaccumulation factors (BCF) of a chemical in adult fish by measuring its final concentration in the fish and the surrounding media after a steady state is reached. This method requires high number of fishes and large exposure time to the chemical compounds (up to 60 days) which implies high costs of the experiments. The European legislation proposes fish embryos and larvae as animal-free approaches and in vitro systems (European Parliament and of the Council 2010). For this purpose, the development of alternative methods to determine acute fish toxicity and other ecotoxicology parameters using zebrafish or other fish embryos and larvae is gaining interest in ecotoxicology. Taking all this into account, an alternative to the test OECD 305 has recently been developed (Sanz-Landaluze et al. 2015). Promising results have been obtained, reducing dramatically the time, reagents, and animal suffering during experiments. When working with embryos and larvae, it is important to consider that even when the experiments are carried out at high concentration levels, only a small amount of each compound may be accumulated; additionally, samples have a very small size (0.44 mg/eleutheroembryo). Therefore, the detecting method must be extremely sensitive.
The analysis of complex matrix such as food and the quantification of analytes at trace levels are two analytical challenges because they require expensive and larger sample treatment and lower detection limits. Several techniques used to extract and preconcentrate the analytes in food products, aqueous, and biological samples have been reported. Solid-phase extraction (SPE) and liquid–liquid extraction are the main techniques used to extract organic compounds in several matrix samples but these techniques use large sample volume and are environmentally unfriendly, expensive, and time-consuming (extraction time is large) (Paíga et al. 2015; Caban et al. 2015; Migowska et al. 2012; Hu et al. 2012). For this reason, other “green methods” may solve the problems of classic LLE and SPE as solid-phase microextraction (SPME) and liquid-phase microextraction (LPME) (Hoon Lee et al. 2014; Sagristá et al. 2010; Arroyo et al. 2011).
The final step of the analytical procedure is the identification and quantification of the tested analytes. Chromatographic techniques are widely used in combination with different detectors for their determination. Techniques as gas chromatography (GC) equipped with FID or mass detector and liquid chromatography (LC) coupled to mass spectrometry (LC-MS or LC-MS/MS) or even UV and DAD detectors have been frequently used to determine pharmaceuticals (CFB, IBU, NP, DC) (Dubreil-Chéneau et al. 2011; Larsson et al. 2014; Grueiro Noche et al. 2011; Zhang et al. 2013; Debska et al. 2005; Kang et al. 2014; Jedziniak et al. 2010).
The aim of this work is the development of a miniaturized method specific for quantifying NSAIDs and lipid regulators (IBU, NP, DC, and CFB) in their neutral and in their ionic form in different types of samples including small and complex matrixes such as fish roe and zebrafish embryos. The analytical protocol we devised is based on three steps: (i) the extraction of the organic compounds with an organic solvent assisted with an ultrasonic probe, (ii) the use of a dispersive SPE with C18 to clean up the extracts, and (iii) the determination of the compounds by GC-MS (prior derivatization of the analytes). To set up the method, we used a variety of samples, namely aqueous samples, fish roe from lumpfish (Cyclopterus lumpus) and zebrafish (Danio rerio) eleutheroembryos. We finally applied it by conducting bioconcentration assays with zebrafish eleutheroembryos for BCF determination using an alternative methods to the OECD guideline 305 developed previously (Sanz-Landaluze et al. 2015).
Materials and methods
Reagents and standard solutions
The pharmaceutical compounds studied in this work were purchased from Sigma–Aldrich (Alcobendas, Spain). They are as follows: clofibric acid (CFB), 2-(p-chlorophenoxy)-2-methylpropioniac acid (CAS number 882-09-7), ibuprofen (IBU), (±)-2-(4-isobutylphenyl)propanoic acid (CAS number 15687-27-1), naproxen (NP), (S)-(+)-2-(6-methoxy-2-naphthyl)propionic acid (CAS number 22204-53-1), diclofenac sodium salt (DC), and 2-[(2,6-dichlorophenyl)amino]benceneacetic acid sodium salt (CAS number 15307-79-6). Table 1 summarizes some relevant physico-chemical properties of those pharmaceuticals. Triclosan (TRI), 5-chloro-2-(2,4-dichlorophenoxy)phenol (CAS number 3380-34-5), purchased from Sigma–Aldrich, Chlorpyrifos, O,O-diethyl O-3, 5, 6-trichlropyridin-2-yl phosporothioate (CAS number 2921-88-2) and triclosan-methyl, 2, 4, 4′- Trichloro-2′-methoxydiphenyl ether (CAS number 4640-01-1) from Sigma–Aldrich (Alcobendas, Spain) were used as internal standards. The derivatization reagent used were N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) + 1 % tert-butyldimethylchlorosilane (TBDMCS) and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) + 1 % trimethylchlorosilane (TMCS) and were purchased from Sigma–Aldrich (Alcobendas, Madrid).
Analytical-grade solvents were used; acetonitrile (AcN) and ethyl acetate (EtOAc) were supplied by LAB-SCAN (Gliwice, Poland), toluene and hexane were supplied by Panreac (Barcelona, Spain), and methanol (MeOH) and acetone were purchased from Scharlau (Barcelona, Spain). Silica gel, C18, and PSA sorbent were purchased from Agilent Technologies (Madrid, Spain), Florisil from Sigma–Aldrich (MO, USA), and AFFINILUTE SPE MIP for extraction of NSAIDs from Symta (Madrid, Spain). Finally, ultrapure water with a resistivity of 18.0 M Ω · cm was provided by a Millipore ZMFQ 23004 Milli-Q water system (Bedford, MA, USA).
Stock solutions containing 1000 μg · mL−1 of each compound were prepared in methanol and stored at 4 °C in the dark. Working solutions were prepared daily by dilution of each standard in acetonitrile or high-purity water. Stock solutions were stable for at least 6 months.
Instrumentation
Gas chromatographic analysis was performed by Agilent GC instrument Mod. 7890A Series (Agilent Technologies, Madrid, Spain) equipped with a HP 7683B Series autoinjector and mass spectrometry detection was carry out using an HP 5975C VL MSD detector (Agilent Technologies S.A., Madrid, Spain) operated by a computer running ChemStation software (Agilent Technologies SA, Madrid, Spain). The GC system was equipped with a polydimethylsiloxan (95 %) cross-linked ZB-5 capillary column (30 m × 0.25 mm I.D., 0.25-μm film thickness) from Phenomenex (Madrid, Spain), using Helium (purity >99.999 %) as carrier gas at 1 mL · min−1. The oven temperature was held at 70 °C for 1 min, and then, a temperature gradient was applied at a rate of 20 °C/min to 120 °C and increased at a rate of 8 °C/min to 200 °C. After the second transition, the temperature was increased from 200 to 280 °C at 15 °C/min, and finally, this temperature was maintained for 3 min. The running time for each chromatogram was 22 min. In this work, the mass spectrometer was used under the following conditions: ion source temperatures, 230 and 150 °C for the quadrupole and 280 °C for the transfer line; scan range was m/z 50–550; and the time for solvent delay was 10 min. For quantification of the analytes, ion monitoring (SIM) mode was used and the retention time and the mass to charge ratio (m/z) values of the compounds are summarized in Table 1.
For the extraction procedures a vortex mixer Genie-2 from Scientific Industries (NY, USA) and a Vibra cell VCx130 ultrasonic processor (Connecticut, USA) equipped with a titanium 2-mm-diameter microtip and fitted with a high-frequency generator of 130 W at 20 KHz were used. Solvent evaporation was carried out with VacElut 20-place vacuum manifold, coupling pieces supplied by Varian (Harbor Citi, CA, USA). Centrifugation of the samples was carried out in a Microcentrifuge 5415R from Eppendorf (Hamburgo, Germany).
Samples, spiking, and bioaccumulation experiment
Commercial fish roes from lumpfish (Cyclopterus lumpus) were purchased from local market. Zebrafish eleutheroembryos were obtained from wild-type adult zebrafish bred and maintained in the AZTI Zebrafish Facility (REGA ES489010006105) under standard conditions (Westerfield 2007). They were exposed at 72 h post fertilization (hpf). At this developmental stage and up to 120 hpf, they are not considered laboratory animals and can be considered non-feeding for further 24 h. Embryos were maintained in a specific aqueous media named embryo water (294 mg of CaCl2 · 2H20, 123.3 mg of MgSO4 · 7H2O, 63 mg of NaHCO3, and 5.5 mg of KCl in 1000 mL of distilled water). Embryo water was used as a basis for the exposure to the pharmaceuticals tested in this work and was also tested as aqueous media in which to quantify the pharmaceuticals.
A mixture of the four pharmaceuticals was used for spiking the three types of samples at different concentrations depending on the sample. Specifically, 10 and 50 ng · mL−1 of the mixture were used to spike aqueous media, 200 and 500 ng · g−1 were used for fish roe samples, and 200 ng · g−1 for eleutheroembryo samples. Samples were maintained in the dark at 4 °C for 30 min to allow the interaction and stabilization with the matrix.
A bioaccumulation experiment was also performed on zebrafish embryos. This experiment was carried out in two phases according to test 305 (OECD 2012) and an alternative protocol described in a previous study (Sanz-Landaluze et al. 2015). Thereby, zebrafish eleutheroembryos were exposed to a mixture of the four compounds studied during 48 h (uptake phase). Subsequently, they were exposed during 24 h to the medium in the absence of the compounds (depuration phase). The nominal exposure solution concentration of each compound was established according to OECD Test 305, which determines that the highest concentration of the test substance was at 1 % and 0.1 % of the LC50 value of each compound (when detection limits allows the determination of the analytes) and the lower concentration should differ by a factor of 10 from the first concentration. Different studies have stablished LC50 values for analytes tested in this work: 318.2–526.2 μg · mL−1 for clofibric acid was obtained in experiments carried out in algae and fish at 48 h of the assay or 87.22 μg · mL−1 in crustaceans (Nunes et al. 2005). For diclofenac, values of 166.6 μg · mL−1 in juvenile Danio rerio and 6.11 in embryonic stages of D. rerio were achieved in acute toxicity tests (Praskova et al. 2011). Other LC50 values in experiments with fish at 96 h were 37.66, 41.56, and 193.34 μg · mL−1, in crustacean at 48 h were 25.75, 27.85, and 121.55 μg · mL−1 and 41.41, 41.13, and 137.94 μg · mL−1 in algae from diclofenac, ibuprofen, and naproxen, respectively (Li et al. 2015). So finally taking all these values into account, bioconcentration factors were calculated using a mixture assay with four pharmaceuticals at 10 and 50 ng · mL−1. Exposure media was refreshed every 24 h to ensure the nominal exposure concentration to be constant.
Exposures were performed at 27 °C on a 12-h light/12-h dark cycle. The conditions maintained for these experiments were the following: dissolved oxygen ≥60 %, 27 °C and pH 6–8.5, according to the OECD 305 guideline. During the exposure, 20 eleutheroembryos were removed from the tanks at different times (0, 6, 24, 45, 48 50, 54, 69, and 72 h) to determine the absorbed and accumulated concentration of each compound. Three replicates of contaminated samples were collected for each time evaluated. According to the OECD 305 Guideline, the loading rate of fish at the beginning of the experiments ranged between 0.7 and 0.8 mg · mL−1 (wet weight) and the mortality of fish was lower of 20 % at the end of the test.
The bioconcentration factor (BCF) is defined by the OECD as the ratio between the concentration of a compound in an organism and in the exposure media, once the equilibrium is reached (OECD 2012). BCF has been calculated employing two different approaches: (a) calculating the ratio between the concentrations of the compound measured at the maximum exposure time and the average in the exposure media or (b) calculating the BCF from a first order model if the steady state is not reached. Both approaches have been described in more detail in a previous paper (Sanz-Landaluze et al. 2015).
Sample preparation for the detection of pharmaceuticals
Embryo water was prepared as follows: 500 μL of the exposed solution was extracted with 1000 μL of ethyl acetate and mixed for 30 s in the vortex. The organic phase was separated and evaporated to dryness under vacuum conditions. Next, the extract was re-dissolved in 80 μL of acetone and 20 μL of MTBSTFA for derivatization heating the vials at 70 °C for 30 min. Finally, 1 μL of the silylated derivatives were analyzed by GC-MS in the SIM mode with previously described parameters (Xu et al. 2008; Yu and Wu 2011 and 2012).
Fish roe samples (10 mg) were placed in an Eppendorf tube and extracted with 500 μL of acetonitrile and sonicated during 1 min at 40 % of amplitude. Extracts were centrifuged for 15 min at 15 °C and 14 rcf. The organic phase was separated and the extract was purified through dispersive SPE with 0.1 g of C18. Then, the extract was centrifuged and filtered through 0.22-μm PTFE syringe filters from Bonna-Agela Technologies Inc. (Wilmington, DE, USA) and the extract was dried under vacuum and re-dissolved in 80 μL of acetone and 20 μL of MTBSTFA. Next, the vials were placed in an oven at 70 °C for 30 min for derivatization, and finally, 1 μL was injected in the GC-MS.
Zebrafish eleutheroembryos in group of 20, with an overall wet weight of approximately 10 mg, were treated with 500 μL of AcN and sonicated for 1 min at 40 % of amplitude. Extracts were centrifuged and purified through dispersive SPE with 0.1 g of C18, centrifuged and filtered, and dried to dryness under vacuum. Finally, 80 μL of acetone and 20 μL of MTBSTFA were added and the vials were placed in the oven for derivatization at 70 °C for 30 min. One microliter of the extract was injected in the GC-MS.
Validation of the analytical procedure
The developed method was assessed to estimate the linearity, precision, recovery, and limits of detection and quantification in blank samples using exposure solution, commercial fish roe from lumpfish, and zebrafish eleutheroembryos. Matrix effect was evaluated for comparing external calibration curves and matrix-matched calibration.
Finally, analytes were extracted with the method described above. Linearity was studied from matrix-matched calibration with dissolutions prepared daily as follows: in the case of exposure solutions, the calibration was carried out in the range of 5–200 ng · mL−1 (using 30 ng · mL−1 of triclosan as internal standard) and 50–1500 ng · g−1 (using 300 ng · g−1 of triclosan as IS) for fish roe and blank eleutheroembryos samples for all analytes.
Recovery experiments (n = 6) were carried out using spiked samples at the concentration of 10 and 50 ng · mL−1 for aqueous samples, 200 and 500 ng · g−1 for fish roe samples and 200 ng · g−1 for eleutheroembryo samples. The results were obtained considering matrix-matched calibration using relative peak areas to IS. Limits of detection and quantification were calculated as a signal to noise ratio of 3 and 10, respectively, and from blank samples spiked at lowest concentration of all analytes. These values were corrected according to the matrix effect over each compound in the different samples types.
The precision of the method was determined by injecting six matrix-matched extracts (n = 6) in the same day (intra-day) and six daily samples in three different days (n = 18) (inter-day) with extracts obtained from samples fortified with all analytes at concentration of 200 and 500 ng · g−1 for fish roe and 200 ng · g−1 for zebrafish eleutheroembryos. Results were calculated as relative standard deviation (RSD, %). Recoveries were calculated from blank samples spiked at 50 ng · mL−1 for aqueous samples, 200 and 500 ng · g−1 for fish roe and 200 ng · g−1 for eleutheroembryo samples. The recoveries values were evaluated from calibration graphs obtained from blank samples fortified after extraction procedure and were compared with theoretical values added.
Results and discussion
Determination of analytes: GC-MS conditions
The lipid regulator (CFB) and NSAIDs (IBU, NP and DC) were separated and determined using GC-MS system. Their quantification was corrected with the use of an internal standard. Different internal standards with similar functional groups and properties as parental compounds were studied such as chlorpyrifos, methyl-triclosan, and triclosan. Triclosan was selected because it offered high sensitivity and good resolutions. Different temperature programs were evaluated to get a good separation of the analytes and the IS from its interferences. The temperature gradient selected was described in the section 2.2. For the quantification of analytes, the m/z values used were obtained from mass spectra of a standard solution of each analyte at a concentration of 1 μg · mL−1 in SCAN mode, determining the retention time of each compound and the proper m/z values (Table 1). With these data, a SIM method was used for quantification, using the sum of the area for the three selected ions for each of the compounds. The m/z values obtained were in accordance with the literature (Yu and Wu 2012).
Optimization of the derivatization reaction
Chemical derivatization is a practice widely employed for the determination and separation of low-volatile compounds by gas chromatography. There are various types of derivatization reactions, but the most widely employed for pharmaceuticals is sylilation (Azzouz et al. 2010). In this work, we evaluated two different sylilation reagents: N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA), with the aim of selecting the most appropriate for our derivatization reaction. Additionally, we established its optimal concentration and reaction time.
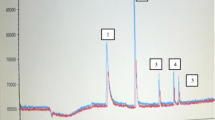
To select the derivatization reagent, 10 μL of each reagent was added separately to 90 μL of solution containing 40 ng · mL−1 of each analyte in acetone. The reaction was incubated at 70 °C for 30 min according to literature (Yu and Wu 2012). The best results were obtained with MTBSTFA because good sensibility and resolution were obtained for all the analytes studied allowing their determinations at environmental concentrations. In the case of BSTFA, lower signals were detected for all analytes. Therefore, MTBSTFA was chosen as derivatization reagent for this work. The optimum amount of reagent to be used in derivatization reaction was evaluated testing different proportions of MTBSTFA: sample (10:90 and 20:80). The best results were achieved using a proportion of 20:80 of MTBSTFA. In both cases, the results obtained were the same for clofibric acid, ibuprofen, and naproxen but, in the case of diclofenac, area values obtained were lower using proportion 10:90; so, the highest concentration of MTBSTFA (20 μL) was chosen. Finally, the reaction time was evaluated—20 μL of MTBSTFA was added to 80 μL of a solution containing 1 μg · mL−1 of pharmaceuticals, and the vials were heated for 30 min and 1 h. The extracts were injected in GC-MS under the conditions described in the 2.2 section and the results obtained showed that clofibric acid, ibuprofen, and naproxen gave the same peak signals for the two reaction times studied; however, the signal of diclofenac decreased considerably when the reaction time was increased to 1 h (ESM 1: Fig. S1 of Supporting Information). Hence, the optimum reaction time chosen for this work was 30 min. Figure 1 shows the typical SIM chromatogram of a medium solution containing 10 ng · mL−1 of all analytes studied as N-trimethylsilyl derivatives. As shown in the figure, the four analytes were separated in approximately 22 min in a quite clean chromatogram obtained in SIM mode.

GC-MS chromatogram obtained from culture media spiked with 20 ng · mL−1 of the four analytes and triclosan as internal standard at 30 ng · mL−1
Optimization of sample preparation and of the pharmaceuticals detection
The sample preparation consists of two steps: a) extraction of the organic compounds from the sample matrices and b) clean-up of the extracts of the extracted samples to reduce the presence of interference and matrix effects on the quantification of the analytes.
Aqueous samples
For the extraction of pharmaceuticals from aqueous samples, a liquid-liquid extraction was employed and different solvents such as EtOAc, toluene, hexane, and several mixtures with hexane: EtOAc (50:50 and 25:75 v/v) were evaluated. Five hundred microliters of exposure solutions (blank samples namely, the salt mixture of embryo water and samples spiked with 10 ng · mL−1 of each analyte and 30 ng · mL−1 of the IS) were extracted with 1 mL of solvents as described above. Table 2 shows the recoveries obtained for all the analytes tested in the aqueous sample spiked at a 10 ng · mL−1 (and 30 ng · mL−1 as IS), using different extraction solvents after the developed procedure. These recoveries were calculated in total percent with the peak area ratio of the analyte to the internal standard related to the same areas of standards in acetone. The best results were obtained with EtOAc as the extracts were cleaner and the recoveries higher. A repetition with a second extraction was evaluated but the improvements on the recoveries obtained were below 7 % for all analytes. For this reason, a single extraction step was established for the determination of all analytes.
Fish roe samples
To perform the extraction of analytes in fish roe samples, a focused ultrasonic probe (USP) was used and different solvents (hexane, ethyl acetate, acetonitrile, AcN/citric acid 0.1 M, and acetic acid/AcN (1:99 v/v)) previously found as adequate were tested (Subedi et al. 2011; Gentili et al. 2012). Extraction conditions had been optimized previously (Gonzalo-Lumbreras et al. 2012; El-Amrani et al. 2012): ultrasonic probe amplitude, 1 min of extraction time and sample temperature 25 °C. Fish roe samples were spiked with 300 ng · g−1 for each analyte and internal standard. As shown in Table 3, the analytes were not recovered when hexane and ethyl acetate were used. On the other hand, AcN and AcN/citric acid 0.1 M allowed obtaining higher areas in the chromatogram for all analytes tested. Also, the chromatograms were cleaned-up. All these results were achieved considering the ratio between peak area of the analyte and the internal standard. C18 was initially fixed as clean-up step to evaluate recoveries with each solvent. Finally, AcN was chosen as optimal solvent as the recoveries were higher and the chromatograms showed less interferences than AcN/citric acid 0.1 M.
To optimize clean-up procedure and removal, the co-extracted lipid material present in the matrix (normally the fat content in zebrafish embryos is approximately 15 % of wet weight), solid-phase extraction (SPE) procedures to clean-up the extracts were necessary. For this purpose, different commercial sorbent using these analytes have been reported (Sousa et al. 2011; Subedi et al. 2011; Huerta et al. 2013; Azzouz et al. 2010; Guitart and Readman 2010; Matamoros et al. 2012). In this work, C18, PSA, Florisil, and commercial MIP (Affinilute MIP) and different combinations of among them were studied to evaluate the highest lipid removal. Fish roe samples spiked with 300 ng · g−1 of pharmaceuticals and internal standard were evaluated to select the best sorbent. Florisil improved the cleanliness of the extracts but the analytes were extracted with lower recoveries. When C18 was used, the recoveries obtained were higher and cleaner chromatograms were obtained (Table 4). In the case of PSA, the analytes were retained in the sorbent so this sorbent was discarded. Clean chromatograms were obtained using commercial MIP in the SPE procedure but the peak areas of each analyte were widely variable, so also this sorbent was rejected. Also, when combinations of sorbents are used with C18, PSA, and Florisil, the results were not significantly improved. In conclusion, only C18 allowed obtaining good recoveries with cleaner extracts.
Zebrafish eleutheroembryos
The optimized method in fish roe samples was applied to determine CFB, IBU, NP, and DC in eleutheroembryo samples. Each sample consisted of a pool of 15–20 eleutheroembryo (8–10 mg/sample) non-spiked (controls) and sample spiked with 300 ng · g−1 of NSAIDs and CFB and 300 ng · g−1 of TRI. The results showed the effectiveness of the optimized method in the final extract by comparison of blank and spiked eleutheroembryo samples (Table 5). Non-interfering peaks at retention times of the target compounds were detected. Also, quantitative recoveries of analytes studied were obtained.
Method validation
Linear regression and matrix-matched calibration using area ratios were used to evaluate recoveries and matrix effects. Matrix effects in the quantification of the four pharmaceuticals by GC-MS were observed in aqueous, fish roe, and eleutheroembryo samples when standard calibration was used. For this reason, calibration curves with spiked samples at nine different concentrations were prepared in the range of 10–1000 ng · mL−1 before extraction or after extraction (only to calculate recoveries of the analytes). For this reason, it is necessary to use matrix-matched solutions prepared fresh daily as follows: 10–1500 ng · g−1 (using 300 ng · g−1 as internal standard) for blank samples of fish roe and eleutheroembryos spiked with an increasing amount of each stock solution. Good correlations were obtained for all analytes (r >0.995). Limits of detection (LODs) and quantification (LOQs) were calculated from calibration graphs by signal to noise ratios (3 and 10 for LOD and LOQ, respectively) at 10 ng · mL−1 for aqueous samples and 100 ng · g−1 for fish roe and eleutheroembryo samples. LODs were between 0.1–1.9 ng · mL−1, 13–26 and 3–15 ng · g−1 for aqueous, fish roe, and eleutheroembryo samples. LOQs were 0.4-6.3 ng · mL−1, 43–86 ng · g−1 and 10–51 ng · g−1 for aqueous, fish roe, and eleutheroembryo, respectively. Intra-day and inter-day precisions were lower than 10 and 8 % for the three types of samples (ESM 1: Table S1 in Supporting Information)
Bioaccumulation assays
Exposure solution
CFB, IBU, NP, and DC were measured in the exposure solution at different times to control their stability and determine the bioaccumulation factors (Table 6). Two concentrations were tested; namely, 10 and 50 ng · mL−1. At the 10 ng · mL−1, the concentration of each compound was constant during the 48 h of the uptake period. However, at 50 ng · mL−1, a clear decrease in the concentration of all the pharmaceuticals was observed after 6 h. However, at 24 h the concentration increased considerably and remained constant. These data are in agreement with the application of the OECD 305 test which requires that the concentration of analytes in the media remain constant. Also, the control and the depuration solutions were measured and no peaks were found at the retention time of the analytes.
Eleutheroembryos
Figure 2 shows a chromatogram obtained by GC-MS of eleutheroembryo samples exposed 48 h to 50 ng · mL−1 of CFB, IBU, NP, and DC in comparison with other samples spiked with 20 ng · mL−1 of CFB, IBU, NP, and DC. The results obtained at different concentrations showed that eleutheroembryo do not bioaccumulate these analytes (Table 7).

Chromatograms obtained by GC-MS of eleutheroembryos samples after 48 h of exposure to 50 ng · mL−1 of CFB, IBU, NP and DC (in blue) and eleutheroembryos spiked with 20 ng · mL−1 (in black) of each compound
Bioaccumulation factors
Bioconcentration factors were calculated using the experimental data obtained in this work adjusted to a first order model (Sanz-Landaluze et al. 2015). The BCF data obtained were as follows: 7.4 and 2.3 for clofibric acid, 4.9 and 1.1 for ibuprofen, 9.3 and 1.9 for naproxen, and 7.9 and 2.0 for diclofenac at 10 and 50 ng · mL−1 of nominal concentration, respectively.
According to the OECD the main criteria for accumulation is a log K ow >3, meaning there is a tendency for accumulation, which is the case of three of the four compounds studied, and for the clofibric acid, the value is very close (Table 1, OECD 2012). As mentioned above, this criterion can be erroneous when dealing with organic compounds. Even other more sophisticated models as the one proposed by Arnot and Gobas (Arnot and Gobas 2006) that uses a mechanistic QSAR approach (log K ow and a number of correction factors but does not consider a chemical’s molecular weight and size related parameters) or modifications of this model introducing biotransformation processes (Arnot et al. 2008) seem to overestimate the BCF values (Table 8), probably because the pharmaceuticals studied all have a pKa <5 (Table 1), thus would be primarily ionized at pH∼7.5, typical of these field exposures. This is probably an important component in the model generally overestimating the BCFs for naproxen, diclofenac, ibuprofen, and clofibric acid because ionic species typically show much lower bioconcentration than non-ionized compounds (Brown et al. 2007). Nallani et al., also found very low BCF levels (0.08–1.4) in a study exposing fathead minnow (Pimephales promelas) and channel catfish (Ictalurus punctatus) to 250 ng mL−1 of ibuprofen for 28 days followed by a 14-day depuration phase (Nallani et al. 2011). Nevertheless, other authors indicate that chemical uptake rates change only slightly for weak acids in fish mainly because the reduced pH of the gill surface and the fact that ionized molecules contribute to the uptake by maintaining high gradients of neutral molecules across membranes (Erickson et al. 2006). However, this statement does not fit very well with the experimental data obtained in this and other studies (Table 8).
The EPI Suite software from US Environmental Protection Agency (EPI Suite BCFWIN™, 2012) uses the BCFBAF program, based on the Arnot–Gobas model. This system takes into account the D OW, (the correction of the K OW for the fraction of the neutral form for IOC), leading predictions much closer to experimental values (Table 8) and making this prediction methodology a tough tool to estimate real values. The BCFs of diclofenac, naproxen, and ibuprofen in the bile (Brozinski et al. 2013) was roughly 1000 times higher than other values; it is evident that bile is an organ much more prone to bioaccumulate and not very representative of the whole body burden, but it can be used for screening purposes.
Another issue to take into account in further studies is the metabolization of these products and the environmental behavior of these metabolites, because some studies have demonstrated the presence of metabolites in concentrations clearly exceeding those of the unmetabolized compounds in rainbow trout (Oncorhynchus mykiss) exposed for 10 days to mixtures of five pharmaceuticals (diclofenac, naproxen, ibuprofen, bisoprolol, and carbamazepine) (Lathi et al. 2011). A QSAR study performed to assess the possible adverse effects of 96 PPCPs and metabolites and to establish a ranking of concern showed that a high number of metabolites has a concern score equal to or greater than their parent compounds (Ortiz de García et al. 2013).
Conclusions
A large fraction of commercially used chemicals is ionizable. Approximately 50 % of the nearly 150,000 preregistered compounds under REACH are ionogenic, i.e., they are acids, bases or zwitterions. There is an increasing interest in risk assessment for ionogenic organic chemicals from different stakeholders such as industry and authorities. Experimental measurement of bioconcentration factors is time-consuming and expensive, and is not feasible for a large number of chemicals of regulatory concern. In this respect, REACH also highlights the convenience of reducing the number of tests involving animals and suggests the need of developing ecotoxicological tests based on animal-free approaches alternative to existing in vivo test methods using laboratory animals. To continue in the knowledge of the risk assessments, in the past few years, a great effort has been made following mainly two types of approaches: (i) non-testing methods involving mechanistic models that aim to predict a chemical property or biological activity for a test substance on the base of its (known) physico-chemical properties, as QSAR models and (ii) experimental alternatives involving, e.g., cells or very early life-stages of fish.
Here, a miniaturized analytical methodology for fish roe samples has been developed and applied and validated to zebrafish eleutheroembryo samples, demonstrating the suitability of the method to determine CFB, IBU, NP, and DC in small samples and study bioaccumulation factors (BCF) using zebrafish eleutheroembryos, as an alternative method to the OECD guideline 305. Bioconcentration values obtained were quite low, ranging from 1.1 to 9.3; however, they were in agreement with those reported in the literature. Results show, on one side, the feasibility of this alternative method for the estimation of toxicokinetic parameters and bioconcentration factors using zebrafish eleutheroembryos instead of adult fishes, reducing considerable animal testing, as suggested by the European legislation. On the other side, the obtained values are far from the values expected attending to classical QSAR linear relationship between octanol-water partition coefficient and bioconcentration values used for screening in neutral compounds, encouraging new studies to deepen the knowledge of the ionogenic organic chemicals and their environmental behavior.
References
Arnot JA, Gobas FAPC (2006) A review of bioconcentration factor (BCF) and bioaccumulation factor (BAF) assessments for organic chemicals in aquatic organisms. Env Rev 14(4):257–297
Arnot JA, Mackay D, Parkerton TF, Bonnell M (2008) A database of fish biotransformation rates for organic chemicals. Environ Toxicol Chem 27(11):2263–2270
Arroyo D, Ortiz MC, Sarabia LA (2011) Optimization of the derivatization reaction and the solid-phase microextraction conditions using a D-optimal design and three-way calibration in the determination of non-steroidal anti-inflammatory drugs in bovine milk by gas chromatography–mass spectrometry. J Chromatogr A 1218:4487–4497
Azzouz A, Ballesteros E (2015) Multiresidue method for the determination of pharmacologically active substances in egg and honey using a continuous solid-phase extraction system and gas chromatography–mass spectrometry. Food Chem 178:63–69
Azzouz A, Souhail B, Ballesteros E (2010) Continuous solid-phase extraction and gas chromatography –mass spectrometry determination of pharmaceuticals and hormones in water samples. J Chromatogr A 1217:2956–2963
Biterrmann K, Spycher S, Endo S, Pohler L, Huniar U, Goss KU, Klamt A (2014) Prediction of phospholipide-water partition coefficients of ionic organic chemicals using the mechanistic model COSMOmic. J Phys Chem B 110:14833–14842
Bittermann K, Spycher S, Goss KU (2016) Comparison of different models predicting the phospholipidmembrane water partition coefficients of charged compounds. Chemosphere 144:382–391
Brown JN, Paxéus N, Förlin L, Larsson DGL (2007) Variations in bioconcentration of human pharmaceuticals from sewage effluents into fish blood plasma. Environ Toxicol Phar 24:267–274
Brozinski JM, Lahti M, Meierjohann A, Oikari A, Kronberg L (2013) The anti-inflammatory drugs diclofenac, naproxen and ibuprofen are found in the bile of wild fish caught downstream of a wastewater treatment plant. Environ Sci Technol 47:342–348
Caban M, Lis E, Kumirska J, Stepnowski P (2015) Determination of pharmaceutical residues in drinking water in Poland using a new SPE-GC-MS(SIM) method based on Speedisk extraction disks and DIMETRIS derivatization. Sci Total Environ 538:402–411
Coimbra AM, Peixoto MJ, Coelho I, Lacerda R, Carvalho AP, Gesto M, Lyssimachou A, Lima D, Soares J, André A, Capitão A, Castro LFC, Santos MM (2015) Chronic effects of clofibric acid in zebrafish (Danio rerio): a multigenerational study. Aquat Toxicol 160:76–86
Corcoran J, Winter MJ, Tyler CR (2010) Pharmaceuticals in the aquatic environment: a critical review of the evidence for health effects in fish. Crit Rev Toxicol 40:287–304
Corcoran J, Winter MJ, Lange A, Cumming R, Owen SF, Tyler CR (2015) Effects of the lipid regulating drug clofibric acid on PPAR-regulated gene transcript levels in common carp (Cyprinus carpio) at pharmacological and environmental exposure levels. Aquat Toxicol 161:127–137
Debska J, Kot-Wasik A, Namiesnik J (2005) Determination of nonsteroidal anti-inflammatory drugs in water samples using liquid chromatography coupled with diode-array detector and mass spectrometry. J Sep Sci 28:2419–2426
Dubreil-Chéneau E, Pirotais Y, Bessiral M, Roudaut B, Verdon E (2011) Development and validation of a confirmatory method for the determination of 12 non steroidal anti-inflammatory drugs in milk using liquid chromatography–tandem mass spectrometry. J Chromatogr A 1218:6292–6301
El-Amrani S, Pena-Abaurrea M, Sanz-Landaluze J, Ramos L, Guinea J, Cámara C (2012) Bioconcentration of pesticides in zebrafish eleutheroembryos (Danio rerio). Sci Total Environ 425:184–190
EPI Suite BCFWIN™ (2012) Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11 or insert version used. United States Environmental Protection Agency, Washington, DC, USA
Erickson RJ, Mckim JM, Lien GJ, Hoffman AD, Batterman SL (2006) Uptake and elimination of ionizable organic chemicals at fish gills: II. Observed and predicted effects of pH, alkalinity, and chemical properties. Environ Toxicol Chem 25:1522–1532
European Chemicals Agency (2014) Guidance on Information requirements and chemical safety assessment. Endpoint specific guidance, chapters R.7a. European Chemicals Agency, Helsinki, Finland
European Parliament and of the Council (2010) Legislation for the protection of animals used for scientific purposes (Directive 2010/63/EU)
Franco A, Ferranti A, Davidsen C, Trapp S (2010) An unexpected challenge: ionizable compounds in the REACH chemical space. Int J Life Cycle Assess 15:321–325
Gentili A, Caretti F, Bellante S, Mainero Rocca L, Curini R, Venditti A (2012) Development and validation of two multiresidue liquid chromatography tandem mass spectrometry methods based on a versatile extraction procedure for isolating non-steroidal anti-inflammatory drugs from bovine milk and muscle tissue. Anal Bional Chem 404:1375–1388
German Federal Environment Agency (UBA) (2015) REACH Compliance: Data Availability of REACH Registrations, Project No. (FKZ) 3714 67 4200. Report No. (UBA-FB) 002111
Gonzalo-Lumbreras R, Sanz-Landaluze J, Guinea J, Cámara C (2012) Miniaturized extraction methods of triclosan from aqueous and fish roe samples. Bioconcentration studies in zebrafish larvae (Danio rerio). Anal Bioanal Chem 403:927–937
Grueiro Noche G, Fernández Laespada ME, Pérez Pavón JL, Moreno Cordero B, Muniategui Lorenzo S (2011) In situ aqueous derivatization and determination of non-steroidal anti-inflammatory drugs by salting-out-assisted liquid–liquid extraction and gas chromatography–mass spectrometry. J Chromatogr A 1218:6240–6247
Guitart C, Readman JW (2010) Critical evaluation of the determination of pharmaceuticals, personal care products, phenolic endocrine disrupters and faecal steroids by GC/MS and PTV-GC/MS in environmental waters. Anal Chim Acta 658:32–40
Hilal SH, Saravanaraj AN, Whiteside T, Carreira LA (2007) Calculating physical properties of organic compounds for environmental modeling from molecular structure. J Comput Aided Mol Des 21:693–708
Hoon Lee C, Shin Y, Woo Nam M, Yeong KM, Lee J (2014) A new analytical method to determine non-steroidal anti-inflammatory drugs in surface water using in situ derivatization combined with ultrasound-assisted emulsification microextraction followed by gas chromatography–mass spectrometry. Talanta 129:552–559
Hu T, Peng T, Li X, Chen D, Dai H, Deng X, Yue Z, Wang G, Shen J, Xia X, Ding S, Zhou Y, Zhu A, Jiang H (2012) Simultaneous determination of thirty non-steroidal anti-inflammatory drug residues in swine muscle by ultra-high-performance liquid chromatography with tandem mass spectrometry. J Chromatogr A 1219:104–113
Huerta B, Jakimska A, Gros M, Rodríguez-Mozaz S, Barceló D (2013) Analysis of multi-class pharmaceuticals in fish tissues by ultra-high-performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1288:63–72
Jedziniak P, Szprengier-Juszkiewicz T, Olejnik M, Zmudzki J (2010) Determination of non-steroidal anti-inflammatory drugs residues in animal muscles by liquid chromatography–tandem mass spectrometry. Anal Chim Acta 672:85–92
Kang J, Park S, Park H, Gedi V, So B, Lee K (2014) Multiresidue determination of ten nonsteroidal anti-inflammatory drugs in bovine, porcine, and chicken liver tissues by HPLC-MS/MS. Appl Biochem Biotechnol 174:1–5
Lahti M, Brozinski J-M, Jylhä A, Kronberg L, Oikari A (2011) Uptake from water, biotransformation, and biliary excretion of pharmaceuticals by rainbow trout. Environ Toxicol Chem 30:403–1411
Larsson E, Al-Hamimi S, Áke Jönsson J (2014) Behaviour of nonsteroidal anti-inflammatory drugs and eight of their metabolites during wastewater treatment studied by hollow fibre liquid phase microextraction and liquid chromatography mass spectrometry. Sci Total Environ 485–486:300–308
Li J, Xu L, Shi Z, Hu M (2015) A novel two-dimensional liquid chromatographic system for the online toxicity prediction of pharmaceuticals and related substances. J Hazard Mater 293:15–20
Macia A, Borrul F, Calul M, Aguilar C (2007) Capillary electrophoresis for the analysis of non-steroidal anti-inflammatory drugs. Trends Anals Chem 26(2):133–153
Matamoros V, Calderón-Preciado D, Domínguez C, Bayona JM (2012) Analytical procedures for the determination of emerging organic contaminants in plant material: a review. Anal Chim Acta 722:8–20
Migowska N, Caban M, Stepnowski P, Kumirska J (2012) Simultaneous analysis of non-steroidal anti-inflammatory drugs and estrogenic hormones in water and wastewater samples using gas chromatography–mass spectrometry and gas chromatography with electron capture detection. Sci Total Environ 441:77–88
Nallani GC, Paulos PM, Constantine LA, Venables BJ, Huggett DB (2011) Bioconcentration of ibuprofen in fathead minnow (Pimephales promelas) and channel catfish (Ictalurus punctatus). Chemosphere 84:1371–1377
Nunes B, Carvalho F, Guilhermino L (2005) Acute toxicity of widely used pharmaceuticals in aquatic species: Gambusia holbrooki, Artemia parthenogenetica and Tetraselmis chuii. Ecotox Environ Safe 61:413–419
OECD 2012. Test No. 305: Bioaccumulation in Fish: Aqueous and Dietary Exposure, OECD Guidelines for the Testing of Chemicals, Section 3, O.P., 2012. OECD guidelines for testing of chemicals. Bioaccumulation in Fish : Aqueous and Dietary Exposure doi:10.1787/2074577x
Ortiz de García S, Pinto Pinto G, García-Encina PA, Irusta Mata R (2013) Ranking of concern, based on environmental indexes, for pharmaceutical and personal care products: an application to the Spanish case. J Environ Manag 129:384–397
Paíga P, Lolic A, Hellebuyck F, Santos Lúcia HMLM, Correia M, Delerue-Matos C (2015) Development of a SPE–UHPLC–MS/MS methodology for thedetermination of non-steroidal anti-inflammatory and analgesic pharmaceuticals in seawater. J Pharmaceut Biomed 106:61–70
Praskova E, Voslarova E, Siroka Z, Plhalova L, Mocova S (2011) Assessment of diclofenac LC50 reference values in juvenile and embryonic stages of the zebrafish (Danio rerio). Pol J Vet Sci 14(4):545–549
Sadat Hasheiminasab K, Reza Fakhari A, Shahsavani A, Ahmar H (2013) A new method for the enhancement of electromembrane extraction efficiency using carbon nanotube reinforced hollow fiber for the determination of acidic drugs in spiked plasma, urine, breast milk and wastewater samples. J Chromatogr A 1285:1–6
Sagristá E, Larsson E, Ezoddin M, Hidalgo M, Salvadó V, Jönsson JA (2010) Determination of non-steroidal anti-inflammatory drugs in sewage sludge by direct hollow fiber supported liquid membrane extraction and liquid chromatography–mass spectrometry. J Chromatogr A 1217:6153–6158
Sanz-Landaluze J, Pena-Abaurrea M, Muñoz-Olivas R, Cámara C, Ramos L (2015) Zebrafish (Danio rerio) eleutheroembryo-based procedure for assessing bioaccumulation. Environ Sci Technol 49:1860–1869
Saravanan M, Karthika S, Malarvizhi A, Ramesh M (2011) Ecotoxicological impacts of clofibric acid and diclofenac in common carp (Cyprinus carpio) fingerlings: hematological, biochemical, ionoregulatory and enzymological responses. J Hazard Mater 195:188–194
Saravanan M, Usha Devi K, Malarvizhi A, Ramesh M (2012) Effects of Ibuprofen on hematological, biochemical and enzymological parameters of blood in an Indian major carp. Environ Toxicol Pharm 34:14–22
Schwaiger J, Ferling H, Mallow U, Wintermayr H, Negele RD (2004) Toxic effects of the non-steroidal anti-inflammatory drug diclofenac part I: histopathological alterations and bioaccumulation in rainbow trout. Aquat Toxicol 68:141–150
Sousa MA, Gonçalves C, Cunha E, Hajšlová J, Alpendurada MF (2011) Cleanup strategies and advantages in the determination of several therapeutic classes of pharmaceuticals in wastewater samples by SPE-LC-MS/MS. Anal Bioanal Chem 399:807–822
Subedi B, Mottaleb MA, Chambliss CK, Usenko S (2011) Simultaneous analysis of select pharmaceuticals and personal care products in fish tissue using pressurized liquid extraction combined with silica gel cleanup. J Chromatogr A 1218:6278–6284
Tanoue R, Nomiyama K, Nakamura H, Hayashi T, Kim JW, Isobe T, Shinohara R, Tanabe S (2014) Simultaneous determination of polar pharmaceuticals and personal care products in biological organs and tissues. J Chromatogr A 1355:193–205
Wang J, Gardinali PR (2013) Uptake and depuration of pharmaceuticals in reclaimed water by mosquito fish (Gambusia holbrooki): a worst-case, multiple-exposure scenario. Environ Toxicol Chem 32(8):1752–1758
Westerfield MA (2007) Guide for the Laboratory Use of Zebrafish (Danio Rerio), 5th Edition. University of Oregon Press, Eugene, USA.
Xu J, Wu L, Chen W, Chang AC (2008) Simultaneous determination of pharmaceuticals, endocrine disrupting compounds and hormone in soils by gas chromatography–mass spectrometry. J Chromatogr A 1202:189–195
Yu Y, Wu L (2011) Comparison of four extraction methods for the analysis of pharmaceuticals in wastewater. J Chromatogr A 1218:2483–2489
Yu Y, Wu L (2012) Analysis of endocrine disrupting compounds, pharmaceuticals and personal care products in sewage sludge by gas chromatography–mass spectrometry. Talanta 89:258–263
Zenker A, Cicero MR, Prestinaci F, Bottoni P, Carere M (2014) Bioaccumulation and biomagnification potential of pharmaceuticals with a focus to the aquatic environment. J Environ Manage 133:378–387
Zhang H, Du Z, Ji Y, Mei M (2013) Simultaneous trace determination of acidic non-steroidal anti-inflammatory drugs in purified water, tap water, juice, soda and energy drink by hollow fiber-based liquid-phase micro extraction and ultra-high pressure liquid chromatography coupled to tandem mass spectrometry. Talanta 109:177–184
Acknowledgments
The authors want to thank the financial support of this work by the Ministry of Economy and Competitivity (Project SAFEFOOD, grant code CTQ2014-54801-C2-1-R), Comunidad Autónoma of Madrid (Project AVANSECAL, code S2013/ABI-3028) and Universidad Complutense for supporting the research group “Determinación de Trazas y Especiación.” Noemí Molina thanks the Spanish Ministry of Education for the FPU predoctoral fellowship (Ref. AP2010-0740). We are grateful to AZTI (Bizkaia, Spain) for providing the eleutheroembryo samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Ester Heath
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Fig. S1 Analyte areas obtained using different derivatization time for a solution of four compounds (CFB, IBU, NP, DC) at 1 μg · mL−1. Table S1 Recoveries and %RSD obtained for the four pharmaceuticals in an intra and inter-day study at two different concentrations for the three different matrices. (DOC 88 kb)
Rights and permissions
About this article

Cite this article
Molina-Fernandez, N., Perez-Conde, C., Rainieri, S. et al. Method for quantifying NSAIDs and clofibric acid in aqueous samples, lumpfish (Cyclopterus lumpus) roe, and zebrafish (Danio rerio) eleutheroembryos and evaluation of their bioconcentration in zebrafish eleutheroembryos. Environ Sci Pollut Res 24, 10907–10918 (2017). https://doi.org/10.1007/s11356-016-6671-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-6671-8