
Abstract
The air quality of three different microenvironments (school, dwelling, and coffee bar) located in the city of Rome, Italy, was assessed. Indoor and outdoor concentrations of polycyclic aromatic hydrocarbons (PAHs) associated with PM2.5 particles were determined during an intensive 3-week sampling campaign conducted in March 2013. In interiors, total particulate PAHs ranged from 1.53 to 4.96 ng/m3 while outdoor air contained from 2.75 to 3.48 ng/m3. In addition, gaseous toxicants, i.e., NO2, NO x , SO2, O3, and BTEX (benzene, toluene, ethyl-benzene, and xylene isomers), were determined both in internal and external air. To solve the origin of indoor and outdoor PAHs, several source apportionment methods were applied. Multivariate analysis revealed that emissions from motor vehicles, biomass burning for heating purposes, and soil resuspension were the major sources of PAHs in the city. No linear correlation was established between indoor and outdoor values for PM2.5 and BTEX; the respective indoor/outdoor concentration ratios exceed unity except for PM2.5 in the no smoking home and benzene in all school floors. This suggests that important internal sources such as tobacco smoking, cleaning products, and resuspension dust contributed to indoor pollution. Using the monitoring stations of ARPA Lazio regional network as reference, the percentage within PAH group of benzo[a]pyrene, which is the WHO marker for the carcinogenic risk estimates, was ca. 50 % higher in all locations investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Though most investigations dealing with ambient pollution are focused on work places or open air, the majority of humans spend over 85 % of their time in non-industrial internal locations (i.e., life environments), e.g., schools, dwellings, offices, shops, restaurants, sporting centers, and churches. Thus, in recent years, increasing attention has been paid, with regards to indoor air quality, to chemically characterize the peculiarities of these environments compared to the external air (Menichini et al. 2007; Delgado-Saborit et al. 2011; Zhang et al. 2012). Many studies were aimed at assessing the impact of indoor air quality on health of both the whole population and special segments of it, selected according to age (children, elderlies) or job (employees, workers). Identification and source apportionment of specific substances in indoor air (e.g., depending to acute or long-term toxicity) is of great importance in developing regulatory/technological controls for the indoor air quality management (Zhou and Zhao 2012).
Particulate matter (PM) in ambient air was the subject of a number of large-scale studies. PM levels were routinely monitored over long time in Europe and abroad, through ACES and MedParticles projects (Fischer et al. 2000; Slezakova et al. 2009; Lepeule et al 2012). Recently, fine particulate (PM2.5) concentrations were regulated in Europe (Directive 2008/50/EC); the adverse effects induced by PM2.5 penetrating into the respiratory system, which causes the reduction of lung function, alterations of lung tissue and structure, were the focus of a rich scientific literature (Huang and Ghio 2006; Ruckerl et al 2011; Chalbot et al. 2012). The association of carcinogenic and mutagenic compounds (WHO 1998) such as polycyclic aromatic hydrocarbons (PAHs) with fine particles (Kameda et al. 2005) has been found to increase their adverse impact and result into long-term risks for health (Forastiere et al. 2005, 2008; Daresta et al. 2010; Liuzzi et al. 2011). PAHs generally occur as complex mixtures. They are released by incomplete combustion of organic substances and are introduced into the environment by both natural processes and anthropogenic activities, e.g., forest fires and energy production (Albinet et al. 2007; Martuzevicius et al 2011). By consequence, PAHs are ubiquitous in ambient air, both in interiors and outdoors. On the other hand, many of them display carcinogenic and/or mutagenic properties (IARC 2010). The importance of internal sources (e.g., tobacco smoke, cooking, domestic heating, decorative candles, etc.) with regards to the PAH occurrence in interiors is well documented (Gustafson et al. 2008; Castro et al. 2011; Orecchio 2011); concurrently, vehicles and industries act as external sources (Chang et al. 2004; Ravindra et al. 2008; Hanedar et al. 2014) overall in urban areas. The percent distributions of PAHs and the concentration ratios between pairs of compounds have been fruitfully used to apportion the different source categories (Manoli et al. 2004; Ravindra et al. 2008; Cecinato et al. 2014a, b, c).
The interaction of other pollutants with PAHs is also of great environmental concern. For instance, nitrogen dioxide, ozone, and free radicals (OH, NO3) are known to easily react with PAHs and give rise to more toxic products including nitro- and keto-PAHs, PAH-quinones, and lactones (Valerio et al. 2000; Tham et al. 2008). On the other hand, sulfur dioxide shares its emission sources with PAHs (Nguyen and Kim 2006; Stracquadanio et al. 2007; Rao et al. 2008). Monitoring these toxicants can help in understanding the atmospheric processes involving particulate PAHs, the corresponding differences observed in emission exhausts, and the ways to mitigate their impact on the environment.
Volatile organic compounds such as benzene, toluene, and xylenes (BTEX) are another important issue in assessing the air quality of interiors (Kliucininkas et al. 2011; De Gennaro et al. 2014). BTEX are released by a variety of internal sources, including solvent evaporation, office and household products, tobacco smoking, and any combustion process of fossil fuels or wood materials, which combine with intrusion from outside of vehicular, industrial, and agricultural exhausts (Hodgson et al. 2000; Sarigiannis et al. 2011).
Measurements of benzene, nitrogen dioxide and, in some cases, of by-products triggered by typical indoor pollutants such as nitrous acid (Gligorovski 2016) were carried out in interiors by means of both diffusive (Bertoni et al. 2001; Topp et al. 2004) and active sampling (Poupard et al. 2005; Triantafyllou et al. 2008; Stranger et al. 2008). Analogously, diffusive (Bernard et al. 1999; Helaleh et al. 2002) and active sampling techniques (Barrese et al. 2014) have been applied to investigate ozone in interiors. Since the assessment of personal exposure to toxicants was the purpose of most of these investigations, the monitoring plan was implemented in different environments like schools and homes (Helaleh et al. 2002; Lee et al. 2004) or workplaces and homes (Mi et al. 2006).
Dealing with indoor air quality, the features of environments chosen as target should be considered, comprising the following: (i) the room occupancy; (ii) the building air tightness and the kind of air exchange (natural or forced ventilation); (iii) the emission factors of internal sources, if any; and (iv) the complex homogeneous and heterogeneous reactions influencing the concentration of compounds under study. For instance, principal components analysis (PCA) revealed that NO2 gives rise to nitrous acid formation in environments hosting combustion devices and high moisture (Poupard et al. 2005).
Our team carried out a series of measurements both inside and outside of ten private houses, six schools, and two offices across Rome, starting from winter 2011 up to summer 2012. This research, partly reported in previous works (Romagnoli et al. 2014), was undertaken within the LIFE + EXPAH project funded by the European Commission. Its goal was to better understand differences among site typologies with regard to pollution of interiors, keeping constant the macro-environmental features (external air). Besides, the links of indoor concentrations with those measured outdoors (i.e., the infiltration models) and with nature and strength of internal emission sources merited to be better understood.
The concentration profiles of the polycyclic aromatic hydrocarbons and possible correlations with other pollutants were examined for distinct internal environments and were compared with those observed outdoors at the same sites and at urban stations of the Air Pollution Control Network run by ARPA Agency of Regione Lazio (ARPA network).
Materials and methods
Sampling sites
PM2.5 and particulate PAHs were measured from 7 to 24 March 2013, at two dwellings, one coffee bar, and one school in Rome. All sites were naturally ventilated and domestic heating, methane fueled, was running during the whole period (see Table 1). Besides, PAHs were measured at three ARPA Lazio stations [namely, Cipro (CYP, residential site), Francia (FRA, kerb site), and Villa Ada (VAD, urban background)]. All sites as a whole could be regarded as representative of typical examples of air pollution in the city (see Fig 1). The average traffic volume at the sites ranged from ca. 35,000 to 60,000 vehicles per day and comprised mainly of gasoline-fueled passenger cars and secondarily diesel-fueled vehicles (buses, taxis). The dwellings under study were located in medium-sized buildings, at the first (HCB home) and the third floor (HPR home), respectively. During the sampling campaign, the occupants lived at home and normal daily activities such as cooking and dusting were carried out.

Map of Rome showing location of sampling sites. Gray circle, ARPA Lazio Network Stations; white triangle, school (IGM); white circle, homes (HCB and HPR); white square, coffee bar
Low-volume samplers were placed at the following: (i) at HCB, in the bedroom (HCB-B) and dining room (HCB-D) as well as in respective balconies (the site was expected to undergo the impact of tobacco smoking); (ii) at HPR, in the dining room and corresponding balcony (dwelling occupied by non-smokers); (iii) in primary school G. Massaia (IGM), where the sampling system for airborne particulate was deployed in a corridor to minimize the noise impact and risk of children accidents; and (iv) in coffee bar (BAR); all samplers were deployed at ca. 2 m above the ground and 1 m from walls. The school remained closed on weekends and normally hosted ca. 350 students (6–14 children) in weekdays. The coffee bar was visited by ca. 500 people per day on weekdays and was closed on Sundays; none attendants or visitors smoked inside the bar.
The air samples were collected simultaneously indoors and outdoors to estimate the intrusion capability of PAHs. HCB, school, and coffee bar lied within a circle of 100 m; therefore, all differences observed in the behavior and concentration levels of compounds investigated could be associated with the site typology rather than the characteristics of the city district.
In the dwellings, PM2.5 and associated PAHs were collected by means of pumping systems, while mono-aromatic hydrocarbons (BTEX), nitrogen oxides (NO x ) and dioxide (NO2), sulfur dioxide (SO2), and ozone (O3) were monitored by applying diffusive sampling to assess the specific impacts of these toxicants. In the school, the diffusive samplers were deployed at each level of the building. At ground level, the devices were located in the teachers’ room and at the first floor in the proximity of the particulate sampler.
Sampling and chemical analysis procedures
The sampling devices and analytical procedures adopted to measure airborne PAHs were described in details elsewhere (Cecinato et al. 2012a, b; Romagnoli et al 2014). Airborne particulate was collected on polytetrafluoroethylene (PTFE) membranes each day over 24 h, starting at 08:00 a.m. The PM2.5 amount collected was determined gravimetrically (162 PTFE samples in total), then the samples were gathered to form 2-or 5-day pools (weekends and weekdays, respectively) before performing chemical characterizations. To minimize the micro-environment perturbation, internal and external samplings were performed using dedicated low-volume instruments (6–10 L/min; Asbesto purchased from Tecora, Fontenay sous Bois, France or Silent from FAI, Fonte Nuova RM, Italy), equipped with PM2.5-selective inlets in compliance with EN14907:2005 norm. For external reference of school and coffee bar, samplings were performed also at the Belloni-Cinecittà ARPA Lazio station located in the close neighborhood, using a medium-volume system (Skypost TCT from Tecora). The comparability of measurements conducted using low- and medium volume systems was previously verified successfully (EXPAH 2012).
The analytes were extracted from samples by means of an accelerated solvent extractor (ASE 150 Dionex, from Thermo Scientific, Rodano MI, Italy) using the mixture of acetone, n-hexane, and toluene in the 60:30:10 ratios. After solvent evaporation, the extracts were cleaned through alumina column chromatography and chemically characterized by means of gas chromatography coupled with mass spectrometric detection (GC–MSD). PAHs were identified according to retention times and ion trace intensity ratios compared to mass spectra of authentic standards. Compounds were quantified vs. the respective reference substances (perdeuterated PAH mixture OSI No. 7-3, purchased from Chemical Research, Rome, Italy), spiked at the beginning of the analytical procedure. All samples were analyzed in triplicate; the percent standard deviations ranged from 4 to 10 % for all compounds. Our study was focused on PAH compounds classified by IARC (2010) as possible (2B), probable (2A), or certain (1) carcinogens, namely benz[a]anthracene (BaA), benzo[b]fluoranthene (BbF), benzo[j]fluoranthene (BjF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), indeno[1,2,3-cd]pyrene (IP), and dibenz[ah]anthracene (DBA), on two mutagenic congeners, i.e., benzo[ghi]perylene (BPE) and chrysene (CH), and two further compounds, benzo[e]pyrene (BeP) and perylene (PE).
As for diffusive sampling, at the end of collection time, the devices (all Analyst type, and specific for the target gases, purchased from Marbaglass, Rome, Italy) were sealed, stored in the dark, and analyzed according to procedures reported elsewhere (De Santis et al. 1997, 2002; Bertoni et al. 2001). In particular, the Analyst devices for NO2 and O3 were extracted by adding a Na2CO3/NaHCO3 buffer solution directly in the sampling vessel and stirred with a VIBROMIX 203 EVT (from Tehtnica, Železniki, Poland) adapted to this purpose; then, the solution was analyzed through ion chromatography (IC) (Dionex ICS 1000 equipped with AS12A column). In the case of NO x , the absorbing pad was removed from the body of the sampler and analyzed separately in a suitable vial. For SO2, 0.03 % of H2O2 was added to the extracting solution to quantitatively oxidize sulfur products to sulfate. The concentrations of analytes such as nitrate, nitrite, and sulfate were determined referring to calibration curves constructed with water solutions prepared by opportune dilution of stock standards (Certipur from Merck, Milan, Italy) containing 1000 mg/L of each analyte.
The Analyst devices for mono-aromatic hydrocarbons (BTEX) were extracted in the vessel with carbon disulfide fortified with chlorobenzene (internal standard); after 1 h, the solution was analyzed by GC-FID (Ultra-GC from Thermo) using a J&W DB-wax column (L = 60 m, i.d. = 0.32 mm, film = 1.2 μm) provided by CPS, Milan, Italy.
The limit of detection (LOD) of diffusive samplers was set equal to three times the standard deviation (3σ) of values obtained with distinct blanks. According to the exposure period of devices (20 days, from 6 to 25 March 2013), LODs corresponded roughly to an equivalent concentration in air of 0.25 ppb for NO2 and NO x , and to 0.50 ppb in the case of O3 and SO2. About 60 devices were exposed in total to monitor inorganic species at 13 locations, including duplicates and blanks.
Statistical differences among the concentrations detected were analyzed applying the one-way ANOVA method and the two-sided t test at the 95 % confidence level. Moreover, the air quality of interiors was compared with that of external air using the indoor-to-outdoor concentration ratios (R I/O) and linear regression modeling.
Results and discussion
PM2.5
Table 2 shows the mean concentrations of PM2.5 and individual PAHs (± standard deviations, SD), observed indoors and outdoors at the target sites over the whole campaign; besides, the corresponding values recorded at ARPA stations and the rates of PAH concentration ratios commonly adopted to identify the pollution sources are reported.
PM2.5 was usually higher indoors except for one dwelling (HPR). On the average (N = 18), it ranged from 11 ± 3 to 46 ± 13 μg/m3 (gross average = 29 ± 14 μg/m3), exceeding the annual guideline value proposed for Europe (25 μg/m3). Concurrently, the outdoor PM2.5 concentration ranged between 15 ± 6 and 17 ± 4 μg/m3 (mean = 16 ± 1 μg/m3) and the values were less spread than indoors. Higher concentrations were observed at ARPA network stations (22 ± 6 μg/m3). According to outdoor values of all sites, no significant differences were found and the relative standard deviation resulted ∼10 %. Wide statistical variations were observed site by site (p < 0.05) at indoor locations, which suggest that important internal sources influenced them. PM2.5 concentrations were statistically higher in the three internal sites frequented by smokers (i.e., HCB home and coffee bar) and exceeded by two to three times the rates recorded outdoors and in the non-smokers’ sites (i.e., HPR and IGM). The PM enrichment in the air was similar to that found by other authors (Stranger et al. 2007; Castro et al. 2011). Breysse et al. (2005) reported that ca. 1.0 μg/m3 is added to indoor PM concentration by each smoked cigarette; again, Lai et al. (2004) mentioned a 100 % increase of indoor PM2.5 as associated to environmental tobacco smoke (ETS). The maximum indoor values (87 μg/m3 at HCB-D and 72 μg/m3 at HCB-B) were recorded on 18 March, which was attributed to increased tobacco smoking and scarce room ventilation on that day. The mean PM2.5 concentrations over 18–22 March reached 63 ± 17 and 53 ± 14 μg/m3, respectively, i.e., much more than the mean values over the whole period.
Looking at the two interiors studied at HCB home, none of indoor PM2.5 correlated with the respective outdoor value (R 2 = 0.030, at p < 0.05 confidence level); therefore, differences can likely be explained by internal sources. By contrast, the means calculated at dining and bed rooms were not significantly different (p < 0.05) (R 2 = 0.85), showing a good agreement between the two datasets. In general, tobacco smoking was confirmed as major source of indoor toxicants (Hoh et al. 2012). Cecinato et al. (2014a, b, c) reported in a previous work the presence of nicotine, at the coffee bar, HPR, and IGM, despite that no one smoked inside during the in-field campaign. Therefore, the tobacco smoking by-products were introduced into internal locations through desorption from dresses and hair, and through diffusion from outside (high local ventilation). Further factors contributing to particulates were presumably equipment, cleaning products, dust and soil resuspension from surfaces, and condensation of vapors.
No important linear correlation was found between internal and external PM2.5 concentrations; besides, differences between interiors and external air were not significant (p < 0.05) only at school. Different results were observed by Raysoni et al. (2011). They found fine correlations between internal and external concentrations at schools, suggesting that indoor particles were mainly of external origin.
The ratio of indoor vs. outdoor concentrations of PM2.5 (R I/O see Table 3), which is a critical indicator of internal sources, ranged from 0.6 to 2.9, respectively, at HPR and HCB-D (mean = 1.9 ± 1.0). On average, R I/O exceeded 1.0 at all sites except for HPR. The HCB dining room was already investigated in January and July 2012 (Romagnoli et al. 2014), when I/O ratios equal to 1.6 and 1.2, respectively, were calculated. The presence of smoking guests during the March 2013 campaign, combined with scarce room ventilation typical of the cold period, likely explains this difference. At school, internal PM2.5 concentration reached 17 ± 4 μg/m3 and R I/O was equal to ∼1.1. PM2.5 levels indoors were influenced by the presence of people and intensity of their activities, as well as by infiltration of external air. Though this kind of environment is normally free of typical PM sources such as smoking and cooking, many children occupy a limited space over several hours, and insufficient ventilation is likely present during the winter. The cleaning products usage, floor polishing, and release from surfaces could give rise to the presence of toxicants in the air, together with the dust resuspension due to children and school workers activities. Our results are in line with findings of studies on schools of other countries, though small differences could depend on sampling methodologies applied. The indoor PM2.5 concentrations were comparable with those observed in Lisbon (Portugal), Ohio (USA), and Queensland, Australia (Lin and Peng 2010; Guo et al. 2010; Almeida et al. 2011), but much lower than in Wroclaw (Poland), Antwerp (Belgium), and Athens (Greece) (Diapouli et al. 2008; Zwozdzihk et al. 2013). In the current work, the I/O ratio of PM2.5 (∼1.1) was similar to those calculated in Stockholm (∼1.0) and Barcelona (∼1.2) (Wichmann et al 2010; Rivas et al 2014). Moreover, a study conducted in Agra, India, reported I/O ratios from 0.92 to 1.11 (Wichmann et al 2010). Higher I/O ratios (1.3–1.7) were found in the city center and rural schools in Aveiro, Portugal (Alves et al. 2013).
PAHs
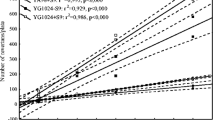
In total, 67 indoor and outdoor composite samples were processed for PAH determination. The concentrations at the sites are summarized in Table 2. Total PAHs (∑PAHs) refer to the sum of the 11 PAHs analyzed without computing further congeners. ∑PAHs ranged from ca. 1.53 ± 0.72 to 4.96 ± 3.57 ng/m3 indoors, and from 2.75 ± 1.48 to 3.48 ± 1.82 outdoors (mean concentrations are pictured in Fig. 2 with other statistical parameters). Outdoors, PAHs did not exhibit concentration rates statistically different (p < 0.05 arithmetic mean = 3.12 ± 0.30 ng/m3), which could depend on similarity of environmental contour and vehicular fleets running at the sites. Very similar PAH percent profiles were found outdoors, suggesting that the same mix of sources affected the sites and air reactivity run at analogous extents. Previous studies indicated that in Rome the air is subjected to concurrent impact of vehicle exhausts and domestic heating emissions (Cecinato et al. 1998; Dimitriou and Kassomenos 2014). Indoors, PAHs were more variable, depending on the internal activities and the degree of pollutant intrusion from outside; nonetheless, on the average, internal and external concentrations did not differ significantly (p < 0.05). Analogously to PM2.5, the PAH maximums were measured in HCB dining room (mean 4.96 ± 3.57 ng/m3). Higher levels of PAHs were recorded in IGM than in the coffee bar. The concentrations measured there were consistent with those previously reported for homes and public indoor spaces in Europe and America (Delgado-Saborit et al. 2011). The difference between dining rooms of non-smokers’ (HPR) and smokers’ (HCB-D) homes was statistically significant (p < 0.05) only when Fisher’s test was applied. The behavior of BaP was object of special concern, since this toxicant is widely used as an indicator of pollution-related human health risk due to its carcinogenic potency (WHO 2000). Indoors, its concentrations ranged from 0.15 ± 0.08 to 0.52 ± 0.30 ng/m3; meanwhile, outdoors it reached 0.25 ± 0.15 up to 0.33 ± 0.16 ng/m3, in line with rates typical of urban areas in Italy and Europe (Menichini et al. 2007; Pietrogrande et al. 2011; Martellini et al. 2012). BaP never exceeded the Italian guideline (i.e., 1 ng/m3 as annual average) in accordance with what recently found by ARPA Lazio (Sozzi et al. 2012). Good agreement existed at external locations between BaP and other PAHs, while internal values of BaP were significantly different (p < 0.05). In fact, where ETS and other combustion sources were absent, BaP in interiors (e.g., 0.15 ± 0.08 at HPR) was less concentrated than outdoors (p < 0.05), while the reverse was observed in the presence of ETS (e.g., 0.52 ± 0.30 ng/m3 in HCB-D, with peak at 1.07 ng/m3). Meaningful differences were observed between the two HCB rooms. In the dining room, both PM2.5 and PAHs loads exceeded those affecting the bedroom, with the concentration ratios between the two rooms reaching 1.4 and 1.7, respectively; also nicotine was higher in HCB-D (Cecinato et al. 2014a, b, c), with a concentration ratio between the rooms equal to 1.6. This confirms that tobacco smoking was the principal source of pollution in HCB, though other contributions could come from cooking fumes and vehicle exhausts.

Total PAH concentrations measured in all sites. Boxes show 25th–75th percentiles, lower and upper asterisks show minimum and maximum values, and lines inside the box show the median values. Indoor concentrations are shown in white and outdoor concentrations in gray. Symbols of sites: HCB-B = CB home (bedroom); HBC-D = CB home (dining room); HPR = PR home; IGM = Massaia Institute (school); BAR = (coffee bar)
The impact of internal sources with regards to PAH occurrence was investigated through applying two complementary methods, namely (i) evaluating R I/Os and statistical relationships between indoor and outdoor data sets; and (ii) analyzing the PAH fingerprints through comparison with those typical of indoor and outdoor sources.
Ohura et al. (2004a) reported that indoor PAHs are regulated by intrusion from outside and concentrations in interiors do not exceed those in open air, provided ETS is not important. In our study, PAHs peaked at HCB and especially in HCB-D, confirming that tobacco smoking is a major source of indoor pollution (Gee et al. 2005; Lu and Zhu 2007). Fine linear correlation between indoor and outdoor PAHs at almost all locations could be found (R 2 = 0.82–0.97), but it failed at HBC-D and coffee bar (R 2 = 0.09). Aside from the two sites, high R 2 coefficients indicated that 88–97 % of the indoor PAH variance depended on external concentrations. The indoor vs. outdoor concentration plots showed that at HPR and IGM the intercept values were close to zero and slopes were indicative of how much of PAHs detected indoors came from outside (∼50 and 78 % of the total, respectively).
The indoor/outdoor concentration ratio rates of PAHs were widely variable (see Table 3) and changed with location and compound examined, from 0.59 ± 0.14 up to 2.02 ± 1.33. In the absence of strong internal sources, e.g., at HPR, R I/O was ∼0.6, and at IGM and BAR, they were somehow higher (∼0.8–0.9). This means that cooking or fuel burning did not affect appreciably the sites. Looking at individual PAHs, most of R I/Os were <1 at HPR, IGM, and BAR (minimum, 0.46; maximum, 1.34), while at HCB, they ranged from 0.78 up to 2.50, confirming the presence of an internal PAH source. The wide variability of R I/Os could depend on distinct penetration rates of airborne particulates, air exchange rates, and building features. In particular, the concentration levels were heavily influenced by human activity and ventilation regime at each room.
Looking at the percent distribution of PAH congeners (Fig. 3), fingerprints changed with sites. Benzo[b]fluoranthene, benzo[g,h,i]perylene, and indeno[1,2,3-cd]pyrene exhibited the highest percentage in most samples, while benzo[e]pyrene was high at school and coffee bar. The distinct origin of PM2.5 and PAHs could be put in evidence by plotting ∑PAH vs. PM2.5 at the sites. No correlation was found, indicating that the respective sources were uncoupled; besides, the PAH percentages could be modified by reactivity in the presence of light, OH radicals, and NO2 (Marr et al. 2006).

Mean percent distribution of PAH compounds at all study sites. For comparison, the average PAHs composition at ARPA Lazio stations (outdoors) is also provided. Error bars represent ± standard deviation. PAH symbols: see Table 1
When mean concentrations at ARPA stations (Francia, Cipro, and Ada; 22 ± 6 μg/m3 and 3.77 ± 0.95 ng/m3, respectively, for PM2.5 and PAHs) were compared with those of internal locations, a fine accordance was found. The means did not differ significantly (p < 0.05) with the exception of HPR. The ARPA values exceeded both internal and external concentrations at most sites, but did not at HCB. Noticeably, the BaP percentage at ARPA stations was lower than at all our locations (ca. 50 % less). Thus, the true exposure of citizens to BaP exceeded that estimated according to official databases. Unlike our sampling points, PAHs could be brought to ARPA stations from quite far sources, so their actual concentrations did not take into account the sink processes to which toxicants are subject in the air. For instance, chrysene accounted for 16 % of ∑PAH at ARPA stations, which was much more than at all target sites.
Among the PAH diagnostic ratios commonly examined for source identification, our concern was focused on the following series: BaA/CH; IP/BPE; BaP/BPE. Besides, the ratio of BaP vs. BeP was adopted to assess the air parcel ageing because BaP is degraded sooner than BeP. The calculated values (average 0.89 ± 0.30, ranging from 0.57 at ARPA stations to 1.68 at HBC-B) were indicative of fresh emission at the sites. Concurrently, the BaP/BPE ratio (mean = 0.70 ± 0.10) suggested the influence of vehicles and other PAH sources, including domestic heating and soil resuspension. The small variability detected could derive from the distances of sites from sources. The IP/BPE ratio ranged at our sites from 0.85 to 0.95. According to literature data (Tobiszewski and Namieśnik 2012), these values could be associated with mixed combustion processes, confirming the impact of vehicular traffic and biomass burning. BPE, tracing petrogenic vehicular emission, occurred always at relatively high concentrations, while the IP/BPE ratio values (∼1) indicated the prevalent contribution of diesel engines (Caricchia et al. 1999; Cecinato et al. 2014a, b, c). Light and heavy-duty trucks and buses fueled with oil account for ca. 20 % of total vehicles in Rome; in spite of that, diesel engines can emit up to 200 times more particulate matter than gasoline cars, heavily impacting on air pollution. Meanwhile, the contribution of gasoline-fueled vehicles and street dust could be detected according to BaA/CH ratio (mean = 0.32 ± 0.06). As for the importance of chrysene at ARPA stations, the CH/BaP ratio seemed to underline the impact of biomass combustion. In conclusion, vehicles (both diesels and gasoline cars) were the primary emission source of PAHs, with contributions coming from domestic heating (i.e., biomass burning) and soil resuspension. This result was in agreement with a previous study undertaken in Rome and Mediterranean region (Cecinato et al. 2014a, b, c; Romagnoli et al. 2014).
The samples collected in weekends were examined distinctly from those of weekdays, to investigate the weekly modulations. Figure 4 shows the mean PAH concentrations inside and outside at the sites. At homes and school, PAHs were similar and without drastic changes from weekends to weekdays, both indoors and outdoors (weekend/weekday ratio Rw ∼ 1). At school, internal concentrations on weekdays were mainly due to occupants’ activities, while during weekends PAHs could depend on poor air exchange with external air. Only at the dining room of HCB we found Rw ∼1.9 indoors and ∼0.7 outdoors. At the coffee bar, Rw was as low as 0.5, clearly indicating the lack of pollution associated to customers’ and workers’ activity; thus, the low ratio rate could be explained on the basis of closure on Sundays. On the other hand, at ARPA stations, all compounds were ∼1.4 times more concentrated on weekends, when people move for shopping, sport, recreation, or family meetings.

Weekday/weekend concentration profile of PAHs observed indoors (a) and outdoors (b). Error bars represent ± standard deviation. Symbols of sites: see Fig. 2
BTEX
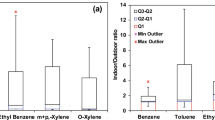
Twenty-nine BTEX samples were collected in total indoors and outdoors at the study sites. The results are summarized in Table S1 (see Supplementary Material). Total BTEX concentrations in interiors ranged from 6.4 to 15.6 μg/m3 with the maximum found in the coffee bar; outdoors, concentrations ranged between 6.5 and 10.7 μg/m3. Both total BTEX and compound percentages changed with the sites. In the school, an important difference was established among the internal BTEX levels by applying the ANOVA test (p < 0.05), depending on the floor level; in fact, 6.4 and 10.2 μg/m3, respectively, were recorded at the first and the third floor. As a confirmation that indoors aromatic compounds can have internal sources and be more than outdoors (Gallego et al. 2008; Stranger et al. 2008), we found that benzene was less concentrated than at the external reference site only at the school. Apart from specific internal sources, building conditions and inadequate ventilation rates could likely promote the accumulation of pollutants. In the two HCB rooms, the BTEX concentrations were finely correlated (R 2 = 0.93).
The benzene concentrations detected in Rome were similar to those observed in Germany, France, and USA, but much lower than those of African and Asian cities (Pankow et al 2003; Gallego et al. 2008). Anyway, all data here reported were below the EU limit value (5 μg/m3, with a long-term target equal to 1 μg/m3). No significant differences among benzene concentrations (average ∼1 μg/m3) were observed in external air at all monitoring sites. Being benzene under restrictions for use in household products and fuels, its occurrence looks as indicative of vehicle emission. In interiors, benzene ranged between 0.89 and 2.57 μg/m3, the maximum occurring in HCB-D. Smoking was identified as a major benzene source indoors, it was found to account for up to +50 % of levels typical of nonsmoker homes (for instance, 1.1 μg/m3 was detected in HPR), and contributed as far as 2–3 μg/m3 to the total indoor concentration. (Wallace et al. 1987; Ilgen et al. 2001). Other internal sources included cooking, heating, combustion, and cleaning products.
Internal concentrations of toluene, known as ubiquitous indoor pollutant (Bruno et al. 2008), were higher than outdoors and ranged from 3.26 to 4.85 μg/m3; only at the coffee bar that the two rates were similar (4.62 and 4.95 μg/m3, respectively). These data were consistent with concentrations recorded at homes in various European cities (∼4–30 μg/m3, Sarigiannis et al. 2011), and much lower than the WHO guidelines (260 μg/m3 over 1 week; WHO 2000). They were also in agreement with concentrations measured in the schools of Lisbon (Portugal) and in homes of Kaunas (Lithuania) and Gothenburg (Sweden), but far below those of non-residential indoor environments, such as at a coffee bar in Porto (Portugal) and in Bari (Italy). (Strandberg et al. 2006; Pegas et al. 2011; Kliucininkas et al. 2011; Sousa et al. 2011). Total xylenes ranged indoors from 2.20 to 8.64 μg/m3 with individual compounds exhibiting similar percentages in all samples; only in the bar (5.63 μg/m3) and at the third floor of the school (3.69 μg/m3) ortho-xylene was particularly abundant. No linear correlations could be established between the internal and external concentrations of any among benzene, toluene, ethyl-benzene, and the xylene isomers.
The indoor/outdoor concentration ratios (R I/O) were examined also for each of BTEX compounds to detect internal sources (see Table 4). The observations made before were confirmed. The principals sources of BTEX were internal (R I/O = 1.26, 1.46, 1.75, and 1.16, respectively, for IGM, BAR, HCB, and HPR sites). Nevertheless, at school and coffee bar, the benzene and toluene R I/O were as low as ∼0.9. Thus, in these cases, hydrocarbons were associated to air entering from outside. This is in agreement with Stranger et al. (2007), which studied indoor air quality in Antwerp (Belgium) and found that at schools R I/O values exceeded 1 for all compounds except for benzene (R I/O ∼0.95). It should be considered that during the sampling the doors of coffee bar faced a subway construction yard where thinners, coatings, rubber, and resins were hugely used. Besides, the o-xylene R I/O reached 6.0, indicating that a peculiar indoor source occurred. At this regard, o-xylene is contained in furniture cleaners, disinfectants, paints, inks, dyes, and adhesives.
Inorganic pollutants
The internal environments survey was completed by monitoring of inorganic pollutants such as NO2, NO x , SO2, and O3 (see Table S2). Their presence indoors can be attributed to inner combustion sources (NO2 and NO x ) or rather to infiltration from outdoors (SO2 and O3). According to detected values, whereas organic and inorganic regulated pollutants were generally more abundant indoors, the opposite was found for O3 and SO2. Since, O3 not only lacks of internal sources, but also decomposes on surfaces.
Outdoors, ozone was spread quite homogeneously (average, 52 μg/m3, individual values at the different locations ranging from 48 to 57 μg/m3). R I/O values calculated at all locations (see Table 4) show that indoor O3 was 6–30 % of outdoors. According to Triantafyllou et al. (2008), the ozone levels found in schools of Greece and Italy are quite similar, ranging from 2 to 20 μg/m3 (average ≈ 7 μg/m3). A meaningful O3 variability was observed in the school, where ozone ranged from 7.2 μg/m3 (teachers’ room) to 15.1 μg/m3 (corridor, second floor). Possibly, an internal source, such as photocopier machine, intensively running there, could explain this finding.
Sulphur dioxide reached externally 2.1–4.4 μg/m3, the minimum recorded at HPR and the maximum at bar and HCB. Indoors, SO2 was as much as 1.8 μg/m3, though a slight difference could be seen in the smokers’ dwelling (HCB, ∼2.2 μg/m3) and the coffee bar (∼2.0 μg/m3). This small increase in concentration could depend on high infiltration from outside, since the external surrounding was the same. Coffee bar looked as the site most influenced by external air, all pollutants apart from O3 and SO2 being characterized by R I/O < 1. By contrast, whenever internal combustion sources were present, e.g., in HCB, NO x , and NO2, indoor values were higher than outdoor ones. In general, interiors were less polluted at school than elsewhere.
Statistical analysis of the dataset
Given the dataset available, comprehensive of about 20 variables measured at five different locations, the statistical treatment based upon PCA was chosen as possible approach. This multivariate technique is widely employed in the atmospheric sciences (Dallarosa et al. 2005; Dimitriou and Kassomenos 2014) and provides data reduction by finding linear combinations of the original variables accounting, as much as possible, for the original total variation.
The analysis of the whole data set including PM2.5 and PAHs was performed to identify similarities/dissimilarities between different environments and to highlight the contribution of each pollutant to the overall variance. Component 1 explained most of the variance (76 % as reported in Table S3; see Supplementary Materials) giving an equal loading to almost all of the pollutants with the exception of xylenes, ethylbenzene, and ozone. In particular, the former pollutant had no influence, whereas ethylbenzene and ozone affected the component at a lesser extent than other pollutants, showing an opposite effect on it. Components 2 and 3 explained about the same proportion (respectively 9 and 8 %) of the variance of the dataset. In particular, component 2 was characterized by similar loadings for total PAHs and PM2.5 and by a greater impact of xylenes and at a lesser extent of toluene, ethylbenzene, and ozone. Component 3 was affected especially by PM2.5 which had an opposite effect if compared to ozone. An equal influence on this component was shown by benzene, toluene, and NO x (see Table S3a).
The components scores for each site are reported in Table S3 (b, c) and plotted in Fig. 5 (a, b). Similarities could be found among three internal locations at the school (teachers room at ground floor, second and third floors), while the location hosting the PM2.5 sampler showed a quite different profile. In particular, it can be noticed that the former locations of the school showed high values of ozone (high I/O ratios); therefore, they made up a distinct group in the plot, since ozone concentration was positively related to component 1. The other location, showing the highest values of ozone, was the bar. This site was apart from the above-mentioned group, since it showed higher values of organics such as ethylbenzene and xylene. These two pollutants had a small or no effect on component 1. We could conclude that two public places such as the school and the local, both naturally ventilated, had some similarities in air exchange mode.

Graphical representation of samples scores (a) and of variable loadings (b) according to principal component analysis (PCA)
All other indoor locations inside the dwellings, chosen to represent smokers and no smokers homes, comprising the two HCB rooms, differed quite a lot. It is possible to notice that the values of ozone concentrations were rather different and generally lower than at the two public places, with a difference between HPR and HCB, being the former characterized by higher values of ozone. Their positions in the score plot were, indeed, quite far from each other and it was not possible to group them together.
In conclusion, there were large differences among the indoor environments studied. This finding can possibly be explained by a different ventilation rate of the locations considered and by the activities carried out indoors (smoking, use of cleaning products, etc.). The school, according to the PCA, appeared to be the environment where the distributions of concentrations were more homogeneous, except for ozone.
Conclusion
Internal and external air concentrations of PM2.5 and aerosol-associated PAHs were determined in three types of environments, i.e., a school, two homes, and a coffee bar during March 2013. Attention was paid also to potential health triggers such as inorganic gases NO2, SO2, O3, and BTEX. Our observation was in agreement with those previously reported for homes and public indoor spaces in Italy and abroad. The study confirmed that the air in these environments is affected by carcinogenic pollutants. It was demonstrated that PAHs were generally less indoors, with the exception of locations exposed to ETS. Tobacco smoking, when present, regulated the PAHs and PM2.5 occurrence in interiors. Indoor toxicants were originated overall outdoors, and infiltration rates depended on compounds and sites. Looking to human health, special care would be paid to indoor locations to assess the citizen exposure. In particular, in interiors, BaP was more than as resulting from regional network records, which means that the global exposure could be much more than expected according to official data archives. The indoor/outdoor concentration ratio of PM2.5 exceeded 1.0 everywhere aside of HPR (no-smoking home), while the principal sources of BTEX were presumably internal (R I/O = 1.26, 1.46, 1.75, and 1.16, respectively, for IGM, BAR, HCB, and HPR sites).
Abbreviations
- BaA:
-
Benz[a]anthracene
- CH:
-
Chrysene
- BbF:
-
Benzo[b]fluoranthene
- BjF:
-
Benzo[j]fluoranthene
- BkF:
-
Benzo[k]fluoranthene
- BeP:
-
Benzo[e]pyrene
- BaP:
-
Benzo[a]pyrene
- PE:
-
Perylene
- IP:
-
Indeno[1,2,3-cd]pyrene
- DBA:
-
Dibenz[ah]anthracene
- BPE:
-
Benzo[ghi]perylene
- ∑PAHs:
-
Total PAH
- HCB-B:
-
CB home (bedroom)
- HBC-D:
-
CB home (dining room)
- HPR:
-
PR home
- IGM:
-
Massaia Institute (school)
- BAR:
-
(Coffee bar)
- BEL:
-
Belloni-Cinecittà ARPA Lazio station
- ARPA:
-
ARPA Lazio Region Network, average three urban stations
References
Albinet A, Leoz-Garziandia E, Budzinski H, ViIlenave E (2007) Polycyclic aromatic hydrocarbons (PAHs), nitrated PAHs and oxygenated PAHs in ambient air of the Marseilles area (South of France): concentrations and sources. Sci Total Environ 384:280–292
Almeida SM, Canha N, Silva A, Freitas M, Pegas P, Alves C, Evtyugina M, Pio CA (2011) Children exposure to atmospheric particles in indoor of Lisbon primary schools. Atmos Environ 45:7594–7599
Alves C, Nunes T, Silva J, Duarte M (2013) Comfort parameters and particulate matter (PM10 and PM2.5) in school classrooms and outdoor air. Aerosol Air Qual Res 13:1521–1535
Barrese E, Gioffrè A, Scarpelli M, Turbante D, Trovato R, Iavicoli S (2014) Indoor pollution in work office: VOCs, formaldehyde and ozone by printer. Occup Dis Environ Med 2:49–55
Bernard NL, Gerber MJ, Astre CM, Saintot MJ (1999) Ozone measurement with passive samplers: validation and use for ozone pollution assessment in Montpellier, France. Environ Sci Technol 33:217–222
Bertoni G, Tappa R, Allegrini I (2001) The internal consistency of the ‘Analyst’ diffusive sampler—a long-term field test. Chromatographia 54:653–657
Breysse PN, Buckley TJ et al (2005) Indoor exposures to air pollutants and allergens in the homes of asthmatic children in inner-city Baltimore. Environ Res 98:167–176
Bruno P, Caselli M, De Gennaro G, Iacobellis S, Tutino M (2008) Monitoring of volatile organic compounds in non-residential indoor environments. Indoor Air 18:250–256
Caricchia AM, Chiavarini S, Pezza M (1999) Polycyclic aromatic hydrocarbons in the urban atmospheric particulate matter in the city of Naples (Italy). Atmos Environ 33:3731–3738
Castro D, Slezakova K, Delerue-Matos C, Alvim-Ferraz M, Morais S, do Carmo Pereira M (2011) Polycyclic aromatic hydrocarbons in gas and particulate phases of indoor environments influenced by tobacco smoke: levels, phase distributions and health risk. Atmos Environ 45:1799–1808
Cecinato A, Ciccioli P, Brancaleoni E, Zagari M (1998) PAH and nitro-PAH in the urban atmophere of Rome and Milan. Ann Chim 12:1133–1141
Cecinato A, Romagnoli P et al (2012) EXPAH, Eds. Technical Report on activities carried out by CNR-IIA and INAIL—ex ISPESL in the frame of the (Actions 3.3) http://www.ispesl.it/expah/documenti
Cecinato A, Balducci C, Romagnoli P, Perilli M (2012b) Airborne psychotropic substances in eight Italian big cities: burdens and behaviors. Environ Pollut 171:140–147
Cecinato A, Balducci C, Romagnoli P, Perilli M (2014a) Behaviors of psychotropic substances in indoor and outdoor environments of Rome, Italy. Environ Sci Pollut Res 21:9193–9200
Cecinato A, Romagnoli P, Perilli M, Patriarca C, Balducci C (2014b) Psychotropic substances in indoor environments. Environ Int 71:88–93
Cecinato A, Guerriero E, Balducci C, Muto V (2014c) Use of the PAH fingerprints for identifying pollution sources. Urban Clim 10:630–643
Chalbot MC, Vei IC, Lianou M, Kotronarou A, Karakatsani A, Katsouyanni K, Hoek G, Kavouras IG (2012) Environmental tobacco smoke aerosol in a non-smoking households of patients with chronic respiratory diseases. Atmos Environ 62:82–88
Chang K-F, Fang G-C, Chen J-C, Wu Y-S (2004) Atmospheric polycyclic aromatic hydrocarbons (PAHs) in Asia: a review from 1999 to 2004. Environ Pollut 142:388–396
Dallarosa JB, Teixeira EC, Pires M, Fachel J (2005) Study of the profile of polycyclic aromatic hydrocarbons in atmospheric particles (PM10) using multivariate methods. Atmos Environ 39:6587–6596
Daresta BE, Liuzzi VC, De Gennaro G, De Giorgi C, De Luca F, Caselli M (2010) Evaluation of the toxicity of PAH mixtures and organic extract from Apulian particulate matter by the model system “Caenorhabditis elegans”. Fresenius Environ Bull 19:2002–2005
De Gennaro G, Dambruoso PR, Demarinis LA, Di Gilio A, Giunga P, Tutino M, Marzocca A, Mazzone A, Palmisani J, Porcell F (2014) Indoor air quality in schools. Environ Chem Lett 12:467–482
De Santis F, Allegrini I, Fazio MC, Pasella D, Piredda R (1997) Development of a passive sampling technique for the determination of nitrogen dioxide and sulphur dioxide in ambient air. Anal Chim Acta 346:127–134
De Santis F, Dogeroglu T, Fino A, Menichelli S, Vazzana C, Allegrini I (2002) Laboratory development and field evaluation of a new diffusive sampler to collect nitrogen oxides in the ambient air. Anal Bioanal Chem 373:901–907
Delgado-Saborit JM, Stark C, Harrison RM (2011) Carcinogenic potential, levels and sources of polycyclic aromatic hydrocarbon mixtures in indoor and outdoor environments and their implications for air quality standards. Environ Int 37:383–392
Diapouli E, Chaloulakou A, Mihalopoulos N, Spirellis N (2008) Indoor and outdoor PM mass and number concentrations at schools in the Athenes area. Environ Monit Assess 136:13–20
Dimitriou K, Kassomenos P (2014) Indicators reflecting local and transboundary sources of PM2.5 and PMCOARSE in Rome—impacts in air quality. Atmos Environ 96:154–162
Fischer PH, Hoek G, van Reeuwijk H, Briggs DJ, Lebret EJ, van Wijnen H, Kingham S, Elliott PE (2000) Traffic-related differences in outdoor and indoor concentrations of particles and volatile organic compounds in Amsterdam. Atmos Environ 34:3713–3722
Forastiere F, Stafoggia M, Picciotto S, Bellander T, D’Ippoliti D, Lancki T, von Klot S, Nyberg F, Peters A, Pokkanen J, Sunyer J, Perucci CA (2005) A case-crossover analysis of out of hospital coronary death and air pollution in Rome, Italy. Am J Respir Crit Care Med 172:1549–1555
Forastiere F, Stafoggia M, Berti M, Bisanti L, Cernigliaro A, Chiusolo M, Mallone S, Miglio R, Pandolfi P, Rognoni M, Serinelli M, Tessari R, Vigotti M, Perucci CA, SISTI Group (2008) Particulate matter and daily mortality: a case-crossover analysis of individual effect modifiers. Epidemiology 19:571–580
Gallego E, Roca FX, Guardino X, Rosell MG (2008) Indoor and outdoor BTX levels in Barcelona City metropolitan area and Catalan rural areas. J Environ Sci 20:1063–1069
Gee I-L, Watson AFR, Carrington J (2005) The contribution of environmental tobacco smoke to indoor pollution in pubs and bars. Indoor Built Environ 14:301–306
Gligorovski S (2016) Nitrous acid (HONO): an emerging indoor pollutant. J Photochem Photobiol A Chem 314:1–5
Guo H, Marawwska L, He C, Zhang YL, Ayoko G, Cao M (2010) Characterization of particle number concentrations and PM2.5 in a school: influence of outdoo air pollution on indoor air. Environ Sci 17:1268–1278
Gustafson P, Ostman C, Sallsten G (2008) Indoor levels of polycyclic aromatic hydrocarbons in homes with or without wood burning for heating. Environ Sci Technol 42:5074–5080
Hanedar A, Alp K, Kaynak B, Avşar E (2014) Toxicity evaluation and source apportionment of polycyclic aromatic hydrocarbons (PAHs) at three stations in Istanbul, Turkey. Sci Total Environ 488–489:437–446
Helaleh MIH, Ngudiwaluyo S, Korenaga T, Tanaka K (2002) Development of passive sampler technique for ozone monitoring: estimation of indoor and outdoor ozone concentration. Talanta 58:649–659
Hodgson AT, Rudd AF, Beal D, Chandra S (2000) Volatile organic compound concentrations and emission rates in new manufactured and site-built houses. Indoor Air 10:178–192
Hoh E, Hunt RN, Quintana PJE, Zakarian JM, Chatfield DA, Wittry BC, Rodriguez E, Matt GE (2012) Environmental tabacco smoke as a source of polycyclic aromatic hydrocarbons in settled household dust. Environ Sci Tecnol 46:4174–4183
Huang YC, Ghio AJ (2006) Vascular effects of ambient pollutant particles and metals. Curr Vasc Pharmacol 4:199–208
Ilgen E, Karfich N, Levsen K, Angerer J, Schneider P, Heinrich J, Wichmann H-E, Dunemann L, Begerow J (2001) Aromatic hydrocarbons in the atmospheric environment: part I. Indoor versus outdoor sources, the influence of traffic. Atmos Environ 35:1235–1252
Kameda Y, Shirai J, Komai T, Nakanishi J, Masunaga S (2005) Atmospheric polycyclic aromatic hydrocarbons: size distribution, estimation of their risk and their depositions to human respiratory tract. Sci Total Environ 340:71–80
Kliucininkas L, Martuzevicius D, Krugly E, Prasauskas T, Kauneliene V, Molnar P, Strandberg B (2011) Indoor and outdoor concentrations of fine particles, particle-bound PAHs and volatile organic compounds in Kaunas Lithuania. J Environ Monit 13:182–191
Lai HK, Kendall M, Ferrier H et al (2004) Personal exposures and microenvironment concentrations of PM2.5, VOC, NO2 and CO in Oxford, UK. Atmos Environ 38:6399–6410
Lee¸ K, Parkhurst W, Xue J, Ozkaynak H, Neuberg D, Spengler JD (2004) Outdoor/indoor/personal ozone exposures of children in Nashville, Tennessee. J Air Waste Manage Assoc 54:352–359
Lepeule J, Laden F, Dockery D, Schwartz J (2012) Chronic exposure to fine particle and mortality: an exstended follow-up of the Harvard Six City study from 1974–2009. Environ Health Perspect 120:965–970
Lin C-C, Peng C-K (2010) Characterization of indoor PM10, PM2.5 and ultrafine particles in elementary school classrooms: a review. Environ Eng Sci 27:915–922
Liuzzi VC, Daresta BE, De Gennaro G, De Giorgi C (2011) Different effects of polycyclic aromatic hydrocarbons in artificial and in environmental mixtures on the free living nematode C. elegans. J Appl Toxicol 32:45–50
Lu H, Zhu L (2007) Pollution patterns of polycyclic aromatic hydrocarbons in tobacco smoke. J Hazard Mater A 139:193–198
Manoli E, Kouras A, Samara C (2004) Profile analysis of ambient and source emitted particle-bound polycyclic aromatic hydrocarbons from three sites in northern Greece. Chemosphere 56:867–878
Marr LC, Dzepina K, Jimenez JL, Reisen F, Bethel HL, Arey J, Gaffney JS, Marley NA, Molina LT, Molina MJ (2006) Sources and transformations of particle-bound polycyclic aromatic hydrocarbons in Mexico City. Atmos Chem Phys 6:1733–1745
Martellini T, Giannoni M, Lepri L, Katsoyiannis A, Cincinelli A (2012) One year intensive PM2.5 bound polycyclic aromatic hydrocarbons monitoring in the area of Tuscany, Italy. Concentrations, source understanding and implications. Environ Pollut 164:252–258
Martuzevicius D, Kliucininkas L, Prasauskas T, Krugly E, Kauneliene V, Strandberg B (2011) Resuspension of particulate matter and PAHs from street dust. Atmos Environ 45:310–317
Menichini E, Iacovella N, Turrio-Baldassarri L, Monfredini F (2007) Relationships between indoor and outdoor air pollution by carcinogenic PAHs and PCBs. Atmos Environ 41:9518–9529
Mi Y-H, Norb D, Tao J, Mi Y-L, Ferm M (2006) Current asthma and respiratory symptoms among pupils in Shanghai, China: influence of building ventilation, nitrogen dioxide, ozone, and formaldehyde in classrooms. Indoor Air 16:454–464
Nguyen HT, Kim K-H (2006) Evaluation of SO2 pollution levels between four different types of air quality monitoring stations. Atmos Environ 40:7066–7081
Ohura T, Amagai T, Sugiyama T, Fusaya M, Matsushita H (2004) Characteristics of particle matter and associated polycyclic aromatic hydrocarbons in indoor and outdoor air in two cities in Shizuoka. Jpn Atmos Environ 38:2045–2054
Orecchio S (2011) Polycyclic aromatic hydrocarbons (PAHs) in indoor emission from decorative candles. Atmos Environ 45:1888–1895
Pankow JF, Luo WL et al (2003) Concentrations and co-occurrence correlations of 88 volatile organic compounds (VOCs) in the ambient air of 13 semi-rural to urban locations in the United States. Atmos Environ 37:5023–5046
Pegas PN, Alves CA, Evtyugina MG, Nunes T, Cerqueira M, Franchi M, Pio CA, Almeida SM, Freitas MC (2011) Indoor air quality in elementary schools of Lisbon in spring. Environ Geochem Health 33:455–468
Pietrogrande MC, Abbaszade G, Schenelle-Kreis J, Bacco D, Mercurialli M, Zimmermann R (2011) Seasonal variation and source estimation of organic compounds in urban aerosol of Aygsburg, Germany. Environ Pollut 159:1861–1868
Poupard O, Blondeau P, Iordache V, Allard F (2005) Statistical analysis of parameters influencing the relationship between outdoor and indoor air quality in schools. Atmos Environ 39:2071–2080
Rao PS, Ansari FM, Pipalatkar P, Kumar A, Nema P, Devotta S (2008) Measurement of particulate phase polycyclic aromatic hydrocarbon (PAHs) around a petroleum refinery. Environ Monit Assess 137:387–392
Ravindra K, Sokhi R, Van Grieken R (2008) Atmospheric polycyclic aromatic hydrocarbons: source attribution, emission factors and regulation. Atmos Environ 42:2895–2921
Raysoni AU, Sarnat JA, Sarnat SE, Garcia JH, Holguin F, Luevano SF et al (2011) Binational school-based monitoring of traffic-related air pollutants in El Paso Texas (USA) and Ciudad Juarez, Chihuahua (Mexico). Environ Pollut 159:2476–2486
Rivas I, Viana M, Moreno T, Pandolfi M, Amato F, Reche C et al (2014) Child exposure to indoor and outdoor air pollutants in schools in Barcelona, Spain. Environ Int 69C:200–212
Romagnoli P, Balducci C, Perilli M, Gherardi M, Gordiani A, Gariazzo C, Gatto MP, Cecinato A (2014) Indoor PAHs at schools, homes and offices in Rome, Italy. Atmos Environ 92:51–59
Ruckerl R, Schneider A, Breitner S, Cyrys J, Peters A (2011) Health effects of particulate air pollution: a review of epidemiological evidence. Inhal Toxicol 23:555–592
Sarigiannis DA, Karakitsios SP, Gotti A, Liakos LI, Katsoyiannis A (2011) Exposure to major volatile organic compounds and carbonyls in European indoor environments and associated health risk. Environ Int 37:743–765
Slezakova K, Castro D, Pereira MC, Morais S, Delerue-Matos C, Alvim-Ferraz MC (2009) Influence of tobacco smoke on carcinogenic PAH composition in indoor PM10 and PM2.5. Atmos Environ 38:6376–6382
Sousa J, Domingues VF, Rosas MS, Ribeiro S-O, Alvim-Ferraz CM, Delerue-Matos CF (2011) Outdoor and indoor benzene evaluation by GC-FID and GC-MS/MS. J Environ Sci Health A 46:181–187
Sozzi R, Bolignano A, Barberini S, Di Giosa AD (2012) Rapporto sullo stato della qualità dell’aria nella regione Lazio 2011. Report ARPA Lazio/Aria_01 Report _2012_DTO. DAI_01: Available at http://www.arpalazio.net/main/aria/doc/pubblicazio0ni.php
Stracquadanio M, Apollo G, Trombini C (2007) A study of PM2.5 and PM2.5-associated polycyclic aromatic hydrocarbons at an urban site in the Po Valley (Bologna, Italy). Water Air Soil Pollut 179:227–237
Strandberg B, Sunesson A-L, Sundgren M, Levin J-O, Sallsten G, Barregard L (2006) Field evaluation of two diffusive samplers and two adsorbent media to determine 1,3-butadiene and benzene levels in air. Atmos Environ 40:7686–7695
Stranger M, Potgieter-Vermaak SS, Van Grieken R (2007) Comparative overview of indoor air quality in Antwerp, Belgium. Environ Int 33:789–797
Stranger M, Potgieter-Vermaak SS, Van Grieken R (2008) Characterization of indoor air quality in primary schools in Antwerp, Belgium. Indoor Air 18:454–463
Tham YWF, Takeda K, Sakugawa H (2008) Exploring the correlation of particulate PAHs, sulfur dioxide, nitrogen dioxide and ozone, a preliminary study. Water Air Soil Pollut 194:5–12
Tobiszewski M, Namieśnik J (2012) PAH diagnostic ratios for the identification of pollution emission sources. Environ Pollut 162:110–119
Topp R, Cyrys J, Gebefügi I, Schnelle-Kreis J, Richter K, Wichmann H-E, Heinrich J (2004) Indoor and outdoor air concentrations of BTEX and NO2: correlation of repeated measurements. J Environ Monit 6:807–812
Triantafyllou AG, Zoras S, Evagelopoulos V, Garas S (2008) PM10, O3, CO concentrations and elemental analysis of airborne particles in a school building. Water Air Soil Pollut Focus 8:77–87
Valerio F, Stella A, Munizzi A (2000) Correlations between PAHs and CO, NO, NO2, O3 along an urban street. Polycycl Aromat Compd 20:235–244
Wallace L, Pellizzari E, Hartwell TD, Perritt RMS, Ziegenfus R (1987) Exposures to benzene and other volatile compounds from active and passive smoking. Environ Heal 42:272–279
WHO (1998) Environmental Health Criteria 202: Selected non-heterocyclic polycyclic aromatic hydrocarbons. World Health Organization, Geneva
WHO (2000) WHO Regional Publications, Eur. Series No. 91. World Health Organization, Regional Office for Europe, Copenhagen
Wichmann J, Lind T, Nilsson MA-M, Bellander T (2010) PM2.5 soot and NO2 indoor-outdoor relationship at homes, pre-schools and schools in Stockholm, Sweden. Atmos Environ 44:4536–4544
Zhang K, Zhang B-Z, Li S-M, Wong C-S, Zeng E-Y (2012) Calculated respiratory exposure to indoor size-fractioned polycyclic aromatic hydrocarbons in an urban environment. Sci Total Environ 431:245–251
Zhou B, Zhao B (2012) Population inhalation exposure to polycyclic aromatic hydrocarbons and associated lung cancer risk in Beijing region: contributions of indoor and outdoor sources and exposures. Atmos Environ 62:472–480
Zwozdzihk A, Sowka I, Krupinska B, Zwozdzihk J, Nych A (2013) Infiltration or indoor source as determinants of the elemental composition of particulate matter inside a school in Wroclaw, Poland. Build Environ 66:173–180
Acknowledgments
We would like to thank Dr. Giuliano Fontinovo (National Research Council of Italy, Institute of Atmospheric Pollution Research, CNR-IIA), for processing the map.
Special acknowledgement is for Mr. Fabrizio Sacco (ARPA Lazio, Rome, Italy) who provided excellent technical assistance in PAH sampling operations for Network ARPA Lazio.
We are indebted with G. Massaia Institute for hospitality and technical support; in particular, we want to thank Dr. Andrea Caroni and Prof. Loreta De Vincentis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Constantini Samara
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Overview of the total and individual BTEX concentrations (μg/m3) at the indoor and outdoor monitoring sites. Site symbols: see Table 1. (DOCX 14 kb)
Table S2
Overview of O3, NO x , NO2, SO2 concentrations (μg/m3) at the indoor and outdoor monitoring sites. Site symbols: see Table 1. (DOCX 13 kb)
Table S3
a) Components in PCA analysis. b) Loadings given to the different variables in PCA analysis. c) Components scores of the different sites in PCA analysis. (DOCX 22 kb)
Rights and permissions
About this article

Cite this article
Romagnoli, P., Balducci, C., Perilli, M. et al. Indoor air quality at life and work environments in Rome, Italy. Environ Sci Pollut Res 23, 3503–3516 (2016). https://doi.org/10.1007/s11356-015-5558-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5558-4