
Abstract
The experimental investigation of chloride ion oxidation under the action of ozone and ultraviolet radiation with wavelength 254 nm in the bulk of acid aqueous solution at pH 0–2 has been performed. Processes of chloride oxidation in these conditions are the same as the chemical reactions in the system O3 – OH – Cl−(aq). Despite its importance in the environment and for ozone-based water treatment, this reaction system has not been previously investigated in the bulk solution. The end products are chlorate ion ClO3 − and molecular chlorine Cl2. The ions of trivalent iron have been shown to be catalysts of Cl− oxidation. The dependencies of the products formation rates on the concentrations of O3 and H+ have been studied. The chemical mechanism of Cl− oxidation and Cl2 emission and ClO3 − formation has been proposed. According to the mechanism, the dominant primary process of chloride oxidation represents the complex interaction with hydroxyl radical OH with the formation of Cl2 − anion-radical intermediate. OH radical is generated on ozone photolysis in aqueous solution. The key subsequent processes are the reactions Cl2 − + O3 → ClO + O2 + Cl− and ClO + H2O2 → HOCl + HO2. Until the present time, they have not been taken into consideration on mechanistic description and modelling of Cl− oxidation. The final products are formed via the reactions 2ClO → Cl2O2, Cl2O2 + H2O → 2H+ + Cl− + ClO3 − and HOCl + H+ + Cl− ⇄ H2O + Cl2. Some portion of chloride is oxidized directly by O3 molecule with the formation of molecular chlorine in the end.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The investigation of the complex interaction between ozone and chloride-ion in aqueous solution under ultraviolet illumination is of great importance to environmental chemistry, and for the chemistry of processes of water and wastewater treatment with ozone. Ozone is ubiquitous in the Earth atmosphere, chloride ion is abundant in the nature and represents the main anion of sea water, and ultraviolet radiation with various wavelengths is present in the sunlight. That is why the photochemical reaction between O3 and Cl− can play an important role in the environment, e.g., as a material contributor of active chlorine in the troposphere, and a significant non-anthropogenic source of environmental contaminants, such as chlorate and perchlorate.
Chemical processes involving ozone in aqueous media are closely related to the reactions of more active species, hydroxyl free radical OH in particular, which is an intermediate of ozone self-decomposition reaction in aqueous solution. The effective laboratory method of hydroxyl radical generation is the photolysis of ozone by UV-radiation with wavelength 254 nm in the presence of water. As this takes place, both hydroxyl radical OH and ozone O3 are present in the reaction system. Therefore, the study of the photochemical reaction between ozone and aqueous chloride ion is the same as the investigation of the complex reaction system O3 – OH – Cl−(aq). Besides natural conditions, chemical interactions in this system are of significance for ozone-based water treatment. Chloride ion is the main end product of mineralization of chlorinated organics with ozone. Further interactions of Cl− with O3 and OH under various conditions can lead to the generation of hazardous by-products Cl2, ClO3 −, and ClO4 −. Also, the formation of these by-products should be taken into account on medical use of ozonized physiological saline.
However, in spite of their importance, the photochemical reaction between O3 and Cl−(aq), and the chemical interactions in the system O3 – OH – Cl−(aq) have not been yet investigated in the bulk of aqueous solution.
On the contrary, the thermal (non-photochemical) reaction between ozone and chloride-ion is quite well investigated. The first published study of molecular chlorine formation on interaction of ozone with hydrogen chloride and water was carried out in the nineteenth century (Broek 1862). The kinetics of the reaction of O3 with Cl− in aqueous solution was investigated for the first time by (Yeatts and Taube 1949). Among other things, they found out that the reaction was catalyzed by H+ ions, and determined rate constants of non-catalytic and catalytic pathways at 0 and 9.5 °C. They were unable to determine rate constants at higher temperatures, due to ozone decomposition. For the same reason, only the upper limit of the non-catalytic rate constant at 23 °C was measured by (Hoigné et al. 1985).
The comprehensive studies of the O3 + Cl−(aq) thermal reaction kinetics and mechanism were presented in the series of papers by Levanov et al. In particular, the main products of the reaction in acid (Levanov et al. 2012c; Levanov et al. 2003a) (molecular chlorine Cl2) and basic (Levanov et al. 2008) (chlorate-ion ClO3 −) media were determined; the catalysis by H+ was investigated in more detail (Levanov et al. 2006c; Levanov et al. 2003a), and the non-catalytic and catalytic rate constants were determined at 7–60 °C Levanov et al. (2003a), see also (Levanov et al. 2012b); the inhibiting action of Cl− and acceleration effect of ion strength were discovered (Levanov et al. 2003a); the catalysis by metal ions (Fe3+, Co2+, MnO4 −, and others) were explored (Levanov et al. 2006a, b, c; Levanov et al. 2005); and the mechanism of the reaction was proposed (Levanov et al. 2012a, b).
The emergence of perchlorate-ion ClO4 − as a by-product of the O3 + Cl−(aq) reaction in neutral and basic solutions was discovered in the pioneer works (Dasgupta et al. 2005; Kang et al. 2008; Rao et al. 2010); also, the authors (Kang et al. 2008; Rao et al. 2010) observed the formation of the main product chlorate-ion ClO3 −.
The extensive and many-sided investigations of B.J. Finlayson-Pitts et al. (Knipping and Dabdub 2002; Knipping et al. 2000; Laskin et al. 2006; Nissenson et al. 2008; Oum et al. 1998; Thomas et al. 2006) have been focused on the study of the photochemical interaction of chloride-ion placed in the liquid aqueous aerosol with ozone and hydroxyl radical, formed on ozone photolysis in the gas phase. Under such conditions, reactions on the gas/aerosol interface are of great importance. The enhanced formation of molecular chlorine has been found, and Cl2 concentrations were much more than those calculated on the basis of known chemical reactions. The hypothetical reaction between gas-phase hydroxyl radical and chloride ion on the aerosol surface has been proposed as an efficient additional source of Cl2. This reaction was supposed to proceed with anomalously high velocity due to the conjectured increase in reactivity on the interface (Knipping and Dabdub 2002; Knipping et al. 2000; Nissenson et al. 2008; Oum et al. 1998).
In general, the formation of significant amounts of Cl2 and other chloride ion oxidation products can as well be caused by some chemical reactions, not yet known, proceeding in the bulk of the aqueous solution instead of the interface. However, to the present day, the studies of chloride ion photochemical oxidation by ozone in the bulk solution have not been described in the literature.
The aim of this work is to investigate the kinetics of chloride ion oxidation under the action of ozone and ultraviolet radiation with wavelength 254 nm in acid aqueous solution, and in particular, first, to determine the composition of products formed on photochemical interaction between ozone and acid chloride solutions; second, to study the dependencies of the products formation rates on significant experimental factors, ozone concentration in initial gas mixture, and acidity of reaction solution; third, to propose a justified chemical mechanism of photochemical chloride ion oxidation by ozone and the products formation. The acid medium has been chosen, because in these conditions, the side reaction of ozone self-decomposition is largely suppressed, and it is much easier to follow the formation of chloride ion oxidation products.
Methods of experiment and kinetic calculations
The scheme of the experimental setup is shown in Fig. 1. The interaction between ozone and chloride ion was carried out in a bubble column reactor filled with the investigated solution of NaCl and 0.01–1 M HCl. In all the experiments, the solution volume was 400 ml and chloride ion concentration and ion strength were equal to 1 M. The reactor was composed of three sections connected with ground joints. The central irradiation section constituted a tube (height ~50 cm, inner diameter 2.5 cm) made of optical grade fused quartz transparent for ultraviolet radiation. The lower section represented a short glass tube with a porous glass filter plate at its bottom. Through the filter, initial gases were fed in the reactor to ensure the fragmentation of gas flow to a large number of small bubbles and efficacious gas–liquid contact. The solution was situated over the filter, the height of liquid column measured ~45 cm. The upper section was just a glass head with a tube for gas withdrawal. The gas communications were made of polytetrafluorethylene or medical polyvinylchloride tubes. Special tests have shown that the decomposition of the tube material by the action of ozone and formation of Cl2 is negligible under the conditions of our experiments.

Principal scheme of the experimental setup
The following reagents were used: distilled water, hydrochloric acid (GOST 3118–77, chemically pure grade, or GOST 14261–77, very high purity grade), sodium chloride (GOST 4233–77, chemically pure grade), hydrogen peroxide unstabilized, and iron(III) chloride hexahydrate (GOST 4147–74, pure for analysis grade). Reaction solutions were prepared by mixing more concentrated stock solutions of 3.6 M HCl and 4.5 M NaCl + 0.25 M HCl. The stock solutions were purified from admixtures of bromide ion and other easily oxidized substances by the technique (Levanov et al. 2012c): a stream of ozonized oxygen (ozone concentration 60 g/m3) was passed through the boiling solutions, placed in round bottom flask with reflux condenser, during 1.5 h. The test according to (Levanov et al. 2012c) showed the absence of bromide ion in the purified stock solutions. The exact concentration of HCl in the stock solutions was determined by acid–base titration with borax using methyl orange indicator, and NaCl was measured by weight of dry residue after evaporation.
Ozone was synthesized in a self-made barrier discharge ozonizer from gaseous oxygen (very high purity grade). Oxygen flow rate was 21 L/h (STP) in all the experiments. Ozone concentration was controlled at entry in the reactor by photometric ozonometer Medozon 254/5 and took values 3–30 g/m3.
The reactor was irradiated by ozoneless amalgam low pressure germicidal lamp ANC 100/32 (LIT Company), consumed power 100 W, and nominal UV radiation power 23 W. The main radiated wavelength was 253.7 nm, the shorter wavelengths being absent. The lamp was 40 cm in length and was arranged vertically parallel to the reactor irradiation section at the distance 3–3.5 cm. The lamp and the reactor were placed inside a special cardboard box for protection against UV radiation. During the experiments, the reaction solution was heated by the lamp from the room temperature (~20 °C) to ~25 °C.
The number of photons absorbed into the reactor was determined by the method of chemical actinometry. As an actinometer was employed, aqueous hydrogen peroxide solution with the initial concentration of 0.3 M, which was placed in the reactor and irradiated in the same conditions as in the regular experiments. The decrease in H2O2 concentration was determined by titration with potassium permanganate according to a standard procedure (Alekseev 1969). The decay of hydrogen peroxide was a zero-order reaction with the rate constant (7.2 ± 0.1) × 10−4 mol L−1 min−1. Since one absorbed UV photon leads to the decay of one hydrogen peroxide molecule in aqueous solution (Baxendale and Wilson 1957; Goldstein et al. 2007; Hunt and Taube 1952), the number of photons absorbed per unit volume of reaction solution, NΦ, is the same as the rate constant: NΦ = (7.2 ± 0.1) × 10−4 Einstein L−1 min−1 = (4.34 ± 0.06) × 1020 photons L−1 min−1 in all the experiments of the present work.
Qualitative analysis of the exit gases was carried out by Raman spectroscopy of their low temperature condensate. The gas sample with a volume of ~1.5 L (STP) was admitted in vacuum flow setup (evacuated by a forvacuum pump) and condensed on the “finger” of a low-temperature reactor-cryostat filled with liquid nitrogen. The Raman spectra of this condensate were taken by means of Horiba Jobin Yvon LabRam HR 800 UV spectrometer (diffraction grating 1800 lines/mm; laser radiation wavelength 534.532 nm). Mainly, those gases were present in the condensate whose pressure at 77 K is the same as of ozone (2 × 10−3 mm Hg) or less; the most part of molecular oxygen, the principal component of the gas mixture, did not condense and was evacuated by pumping. The description of the vacuum setup, the scheme of the low temperature reactor-cryostat and the procedure of taking the Raman spectra are given in (Levanov et al. 2011).
Qualitative composition of nonvolatile reaction products present in the reaction solution was determined by infrared spectroscopy. Reaction solution treated with ozone and UV radiation was evaporated to dryness by a rotary evaporator at 90 °C under a vacuum of water-jet pump. From the powder obtained, a pellet was pressed off, and its IR spectra were taken by means of Equinox 55/S IR spectrometer (Bruker).
Quantitative determination of molecular chlorine in the exit gases was performed by the method of direct UV spectrophotometry. In the UV–vis range, molecular chlorine has a single absorption band with the maximum at 329.5 nm (ε329.5 = 66.82 L mol−1 cm−1) (Maric et al. 1993). In our experiments, an intense Hartley band of unreacted ozone (λ max = 255 nm, ε255 = 3002.5 L mol−1 cm−1) (Sander et al. 2011b) is superimposed on the relatively weak chlorine band. For this reason, the gases were initially passed through the tube furnace. Its temperature was specially determined (see (Levanov et al. 2003b)) and took values 530–540 °C so that ozone was fully decomposed and Cl2 underwent no change. Then, the gases were directed into the optical cell of Agilent 8453 spectrophotometer, where the UV–vis spectra were recorded.
After the treatment, the experimental spectra showed the only peak—the signal of molecular chlorine with the maximum at 330 nm. Cl2 concentration, C (Cl2), mol/L, was calculated from the net absorbance (optical density) at 330 nm, A330, with the formula
where ℓ = 10.0 cm is the optical cell path length.
During the experiments, time dependencies C(Cl2)(t) were recorded, and from them, chlorine concentrations in the exit gases in the steady state C(Cl2)∞ were found. Chlorine emission rate (dnCl2/dt)/V liq, μmol L−1 min−1, was calculated by the formula
where C(Cl2)∞, μmol/L, is Cl2 concentration in the exit gases in the steady state, υ = 21 L/h = 0.35 L/min is gas mixture flow rate, and V liq = 0.4 L is the volume of reaction solution. The uncertainty of experimental determination of chlorine emission rate was estimated to be less than 4.2 μmol L−1 min−1.
Quantitative determination of chlorate ion in the reaction solution was performed using the indirect spectrophotometric method proposed in (Levanov et al. 2008). On completion of the treatment with ozone and UV light, molecular oxygen was bubbled through the solution during 6 min, which ensured withdrawal of ozone and molecular chlorine. Then, 2.5 ml of the solution sample was poured in a 25-ml graduated flask, 20 ml of the solution 10 M HCl + 1 mM KBr was added, and the rest of the flask was filled with distilled water. In thus prepared analytic solution, chlorate ion was largely transformed to stable complex ion BrCl2 − according to the reaction ClO3 − + 6H+ + 5Cl− + 3Br− → 3BrCl2 − + 3H2O (the equilibrium constants of BrCl2 − formation reactions are as follows: BrCl + Cl−⇄ BrCl2 −, K = 6 М−1 (Wang et al. 1994); Br− + Cl2 ⇄ BrCl2 −, K = 4.2×106 М−1 (Liu and Margerum 2001)). BrCl2 − ion possesses an intense and well-pronounced absorption line with the maximum at 232 nm, ε232 = 32700 L mol−1 cm−1 (Wang et al. 1994). Spectra of the analytic solution were recorded by Agilent 8453 spectrophotometer, blank sample being water. The maximum absorbance due to the presence of chlorate occurred at 237 nm. Chlorate ion concentration was determined from absorbance at 237 nm with the use of calibration curve. The uncertainty of chlorate determination in model solutions of 1 M NaCl and 10–300 μM ClO3 − was less than 5 μM. This corresponds to relative error less than 1.7 % at chlorate concentration 100–300 μM. The considerable advantage of this method compared to conventional iodometry lies in the fact that in acid media, bromide ion is not oxidized by oxygen present in the solution, as opposed to iodide ion.
The method is not specific for chlorate ion, and various oxidizers will interfere. However, formation of other nonvolatile oxychlorine oxidizing compounds (hypochlorite ion ClO−, hypochlorous acid HOCl, chlorite ion ClO2 −, and chlorine dioxide ClO2) as end products is excluded in our experiments. Indeed, in acid media, ClO− and HOCl undergoes fast protonation with the formation of Cl2; ClO−,ClO2 −, and ClO2 interact rapidly with ozone yielding chlorate ion ClO3 − in the end (Hoigné et al. 1985). As was noted previously, perchlorate ion ClO4 − was shown to be formed in small quantities as a byproduct of ozone interaction with aqueous chloride ion (Dasgupta et al. 2005; Kang et al. 2008; Rao et al. 2010). However, perchlorate cannot interfere with the chlorate determination if for no other reason than its great chemical inertness (Greenwood and Earnshaw 1997). Thus, the method described ensures exclusive determination of chlorate ion under our experimental conditions.Chlorate formation rate (dnClO3−/dt)/V liq, μmol L−1 min−1 was estimated by the formula
where Δt = 30 min is the time of treatment of reaction solution with ozone and UV light, Δ[ClO3 −], μmole/L, is the increase in chlorate ion concentration in reaction solution during the time Δt. The uncertainty of experimental determination of chlorate formation rate was 13 %. It should be noted that this uncertainty is significantly greater than that of chlorate ion determination in the model solutions.
With the purpose to explain the experimental kinetic regularities and to elucidate the chemical mechanism of photochemical interaction between ozone and chloride ion in aqueous solution, the mathematical modelling of the kinetics of this process has been performed. The reactor was represented in the model as a continuous stirring tank reactor with gas flow passing through. Chemical transformations were considered to proceed only in the liquid phase. Differential equations for the concentration of a substance A were written in the following way
where w Ais the rate of chemical reactions of the substance A in the liquid phase, mol L−1 s−1, C(A) and C(A)° are concentrations, mol/L, of A in the gas phase inside and at the entrance to the reactor (if A is a gaseous substance, O3 or Cl2), [A] is concentration of A in the liquid solution, mol/L, kLa is volumetric mass transfer coefficient, s−1, H A is dimensionless Henry’s law constant of A, equal to the ratio of molar concentration of A in the liquid solution to that in the gas phase in the equilibrium conditions, H A = [A]/C(A), υ = 21 L/h = 8.6 × 10−3 L/s is the volumetric rate of gas mixture flow through the reactor, V liq = 0.4 L is the volume of the liquid solution in the reactor, and V gas is the total volume of gas phase (gas bubbles) in the reactor, L, in our experiments V gas ≈ 0.1 V liq.
The set of chemical reactions included in the model was made up on the basis of general chemical knowledge about plausible chloride ion oxidation pathways, literature data on ozone photolysis and possible formation pathways of molecular chlorine and chlorate ion in aqueous solution, taking into account the values of rate constants of relevant reactions. Whenever possible, the rate and equilibrium constants of the model were taken for the temperature 23 °C; otherwise, the literature values related to the temperature 20–25 °C were used without recalculation. The influence of ionic strength on the rate constants of ion-ion reactions was neglected. The equilibrium acid dissociation constants was recomputed to the ion strength of 1 M.
Mass transfer between gaseous and liquid phases was considered for ozone O3 and molecular chlorine Cl2. Henry’s law constant for chlorine was taken from (Bartlett and Margerum 1999), H Cl2 = 2.4. The literature values of ozone Henry’s law constant in chloride solutions exhibit a large spread, see for example (Levanov et al. 2008); and hence, the two characteristic values H O3 = 0.24 (Levanov et al. 2003a) and H O3 = 0.16 (Levanov et al. 2008) were used in the model. It can be evaluated that under the conditions of our experiments, the Hatta number (Hatta 1932) for ozone is less than 10−3.Footnote 1 In this case, the rate of ozone dissolution is given by the term kLa (HO3C (O3) – [O3]) (Charpentier 1981; Sotelo et al. 1989). The volumetric mass transfer coefficient kLa was assumed to be the same for all gaseous substances; the rough estimate is that in our reactor kLa~0.2 s−1. The effect of kLa variation on the rates of chlorine emission and chlorate formation has been considered in the kinetic calculations.
The direct kinetics problem has been solved employing Kintecus computer program (Ianni 2006). The calculation results represented time dependencies of the concentrations of the reacting species. The experimental time dependencies C(Cl2)(t) showed that steady state was established in our reaction system. The steady state was due to the facts that the reactor was of continuous-flow type, the experimental parameters, in particular the gas mixture flow rate, ozone supply rate, and UV light intensity were kept constant and did not change with time, and decrease in chloride and hydrogen ion concentration during the experiments was negligible. In the steady state the calculated Cl2 concentration did not depend on time and was equal to the constant quantity C(Cl2)∞ calc, while ClO3 − concentration increased linearly as time passes. The calculated molecular chlorine emission rate was determined by substituting C(Cl2)∞ calc into relation (1), chlorate formation rate—according to relation (2), as a slope of the time dependence of ClO3 − concentration in the steady state.
Results and discussion
For the qualitative analysis, the exit gases were condensed on the cold “finger” at ~80 K, as described above. The condensate was non uniform along its height. Its upper part was a liquid of dark blue color, consisting mainly of unreacted ozone and carrier gas molecular oxygen. The lower part was a white solid. Its Raman spectra (Fig. 2) exhibit very strong and well-pronounced peaks with the maxima at 95.5, 112, 525, 532.5, and 540 cm−1. Comparison with the literature (Anderson and Sun 1970; Cahill and Leroi 1969; Suzuki et al. 1969) discloses that these signals constitute the spectrum of molecular chlorine in the crystalline state. The lines at 540, 532.5, and 525 cm−1 correspond to the fundamental vibrations 35Cl −35Cl, 37Cl −35Cl, and 37Cl −37Cl, while the peaks at 95.5 and 112 cm−1 are the most intense signals of lattice vibrations of molecular chlorine crystals. The fundamental lines intensities are consistent with the natural abundance of chlorine isotopes 35Cl and 37Cl. Also present in the condensate Raman spectrum are other signals of much lower intensity. The most prominent of them are the line at 1070 cm−1 of unknown origin, the peak of molecular oxygen at 1555 cm−1, and the broad band with a maximum at 3180 cm−1 corresponding to the stretching oscillations of OH groups of water bound with hydrogen bonds. Thus, molecular chlorine Cl2 is the only predominant gaseous product of the photochemical reaction between O3 and Cl−(aq).

Raman spectrum of exit gases condensed at the temperature of liquid nitrogen (ozone concentration in initial gases 60 g/m3, composition of reaction solution 0.2 М NaCl + 0.8 М HCl)
A typical infrared spectrum of the substrate obtained by vacuum evaporation of reaction solution is shown in Fig. 3. The spectrum shows intense signals of a nonvolatile reaction product with the absorption maxima at 483.5, 625, 937, 971, and 990 cm−1, while the initial substance sodium chloride does not possess infrared spectrum. When this spectrum is compared with the literature (Duverney 1962; Hollenberg and Dows 1960; Miller et al. 1960; Miller and Wilkins 1952), it is apparent that all the lines belong to crystalline sodium chlorate NaClO3. In the range 300–1200 cm−1, absorption maxima of this substance were observed at 484, 627 (Miller et al. 1960), 935, 965, and 990 cm−1 (Miller and Wilkins 1952). Thus, chlorate ion ClO3 − is the only detectable non-volatile product of photochemical oxidation of chloride ion by ozone in acid aqueous solutions. The formation of chlorate on photochemical (or thermal) interaction between ozone and chloride ion in acid media has not been reported in the literature so far.

Infrared spectrum of the substrate obtained by vacuum evaporation of the solution 1.5 M NaCl + 0.09 M HCl treated with ozone (initial concentration 53 g/m3) and UV light during 4 h
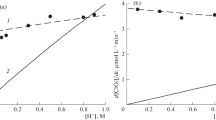
The rates of chlorine emission and chlorate formation are shown in Figs. 4 and 5 as functions of concentrations of ozone in initial gases and hydrogen ion in reaction solution. The influence of chloride ion on the rates has not been investigated in this work. Both the rates rise steadily as the O3 concentration increases and are quite slightly affected by the concentration of H+ ion. Chlorine emission rate rises moderately with increasing H+ concentration, while chlorate formation rate slightly falls (Fig. 5).

Effect of ozone concentration in initial gases on the rates of chlorine emission and chlorate formation. Points are the experimental data. Lines represent the results of model calculations: solid line—reactions (R1–R17), HO3 = 0.24, kLa = 0.3 s−1; dashed line—reactions (R1–R10, R12–R16, R18), HO3 = 0.24, kLa = 0.3 s−1, k 18 = 7.15 × 105 s−1, n 18 = 1.0725. Concentrations in reaction solution [H+] = 0.1 М, [Cl−] = 1 М, [Na+] = 0.9 М

Effect of H+ concentration in reaction solution on the rates of chlorine emission and chlorate formation. Points are the experimental data; rings
 show chlorine emission rate on addition of 0.008 M FeCl3 to reaction solution. Lines represent the results of model calculations: solid line—reactions (R1–R17), HO3 = 0.24, kLa = 0.3 s−1; dashed line—reactions (R1–R10, R12–R16, R18), HO3 = 0.24, kLa = 0.3 s−1, k
18 = 7.15 × 105 s−1, n
18 = 1.0725. Ozone concentration in initial gases 10.4 g/m3; concentrations in reaction solution [Cl−] = 1 М, [H+] + [Na+] = 1 М
show chlorine emission rate on addition of 0.008 M FeCl3 to reaction solution. Lines represent the results of model calculations: solid line—reactions (R1–R17), HO3 = 0.24, kLa = 0.3 s−1; dashed line—reactions (R1–R10, R12–R16, R18), HO3 = 0.24, kLa = 0.3 s−1, k
18 = 7.15 × 105 s−1, n
18 = 1.0725. Ozone concentration in initial gases 10.4 g/m3; concentrations in reaction solution [Cl−] = 1 М, [H+] + [Na+] = 1 М

Scheme of chloride ion oxidation in acid aqueous solution under the action of ozone and UV light
In all the experiments, chlorine emission rate is approximately an order of magnitude (7–14 times) greater than that of chlorate formation. On addition to the reaction solution of 0.008 M Fe(III), chlorine emission rate increases by approximately 20 % (Fig. 4). Thus, Fe(III) ions are catalysts of the photochemical reaction between O3 and Cl−(aq). Also, it is worth to notice that in the photochemical experiments of this work, Cl2 emission rate is appreciably greater than that in the thermal reaction in analogous conditions (compare Fig. 4 of this work with Fig. 5 of (Levanov et al. 2003a)Footnote 2 and Fig. 4 of (Levanov et al. 2005)), and the dependencies of (dnCl2/dt)/V liq on H+ concentration in the photochemical and thermal processes are different in character.
The succeeding sections of this work are devoted to the development of the scheme of chemical reactions and the corresponding quantitative kinetic model of Cl2 and ClO3 − formation during the oxidation of chloride ion under the action of ozone and UV radiation in acid medium. As is known, the main primary products of ozone photolysis by the light with λ < 300 nm are excited species singlet atomic oxygen O(1D) (quantum yield ~0.9) and singlet molecular oxygen O2(1∆g) (Bauer et al. 2000; Smith et al. 2000; Taniguchi et al. 2000; Wayne 1987).
Singlet molecular oxygen is rather unreactive; in aqueous solution, its main transformation route is fast quenching (τ1/2(O2(1∆g)) = 2.3 × 10−6 s) (Wilkinson et al. 1995) to form ground state triplet molecular oxygen O2(3Σg −). Singlet atomic oxygen O(1D) is extremely reactive. In particular, it interacts rapidly with solvent water with the formation of vibrationally excited hydrogen peroxide H2O2* (Biedenkapp et al. 1970). In aqueous media, the major portion of H2O2* undergoes thermalization with the formation of common ground state hydrogen peroxide H2O2, while the lesser part dissociates to give free hydroxyl radicals OH. According to (Reisz et al. 2003), on photolysis of ozone in aqueous solution by the light with the wavelength 254 nm (250–300 nm), the primary quantum yield of ozone photodecomposition takes the value of ~0.55, with one mole of photodecomposed O3 yielding 0.9 mole H2O2 and 0.1 mole OH.
In the kinetic model, the photolysis process was described by the chemical equationsFootnote 3
(Reisz et al. 2003).
(Reisz et al. 2003).Thus, the oxidizers in the photochemical reaction are not only ozone O3 but also very reactive hydroxyl radical OH. Besides, there exists an effective source of hydrogen peroxide H2O2, which is a reducing agent towards chlorine compounds in nonnegative oxidation states. Let us next consider the relevant reactions of O3, OH, and H2O2 with chloride ion and its oxidation products.
The mechanism of chloride ion interaction directly with ozone molecule is described in (Levanov et al. 2012b). Its primary stage is the oxidation of chloride via the mechanism of oxygen atom transfer from O3 to Cl−, and the formation of the compounds of chlorine ClO− and/or HOCl in the +1 oxidation state. The intermediates of this complex reaction has not been determined experimentally, and so it is described by the single equationFootnote 4
(Levanov et al. 2012b; Levanov et al. 2003a) with its rate constant being the function of temperature and H+ and Cl− concentrations according to (Levanov et al. 2012b; Levanov et al. 2003a). The subsequent rapid protonation reactions
(Adam et al. 1992)
(Wang and Margerum 1994), wherein the equilibria are almost entirely shifted to the left in acid media, lead to the end-product molecular chlorine Cl2.
The reactions of chloride ion oxidation by hydroxyl radical OH
(Yu 2004),
(Yu 2004), and
(Yu 2004) are well investigated (Jayson et al. 1973; Klaning and Wolff 1985; Yu 2004) and lead to the formation of labile anion-radical Cl2 −.
Hydrogen peroxide H2O2 is known to effectively interact with Cl2, Cl2 −, ClO−/HOCl, and other chlorine compounds in nonnegative oxidation states and reduce them to chloride ion (Connick 1947; Held et al. 1978; Makower and Bray 1933; Yu and Barker 2003; Yu 2004). The reactions of Cl2 and Cl2 − with Н2О2 (Connick 1947; Held et al. 1978; Makower and Bray 1933) and HO2 (the product of one-electron oxidation of H2O2)
(Connick 1947),
,(Yu 2004), and
(Bjergbakke et al. 1981), resulting in Cl− regeneration, should be significant under the conditions of our experiments.
The source of hydrogen peroxide due to ozone photolysis is rather strong. Therefore, if only known chemical reactions (R1–R12) are taken into consideration, then virtually no formation of chloride oxidation products should occur. This qualitative conclusion is supported by the kinetic calculations. If not only reactions (R1–R12), but a rather exhaustive set of known aqueous phase chemical reactions (union of sets of reactions between particles consisting of H, O, and Cl, given in (Bräuer et al. 2013; Knipping and Dabdub 2002; Sander et al. 2011a; Thomas et al. 2011)) is included in the model, then still the calculated steady-state chlorine emission rate is more than a thousand times less than the experimental one, and chlorate formation is not predicted.
A similar situation has been observed on modeling the kinetics of Cl2 formation on photochemical interaction of ozone with chloride ions of liquid aqueous aerosol, see, e.g., (Knipping and Dabdub 2002). The kinetic calculations with the use of detailed mechanism of established gas and liquid phase chemical reactions cannot explain the experimental Cl2 formation: the calculated Cl2 concentrations are about three orders of magnitude lower than the experimental ones (Knipping and Dabdub 2002). The agreement between experimental and theoretical Cl2 concentrations has been reached by invoking conjectural reaction
proceeding with anomalously high velocity on the aerosol/gas interface. In the conditions of our experiments, the contribution of interfacial reactions is negligible because of much lesser interface area. In our reactor, the relation of interface area to liquid phase volume is ~10 cm2/cm3, while in Knipping and Dabdub (2002), it equals to ~3 × 105 cm2/cm3.
By and large, the comprehensive atmospheric chemistry kinetic models of chemical and photochemical processes with the participation of chloride aerosols (e.g., (Bräuer et al. 2013; Knipping and Dabdub 2002; Sander et al. 2011a; Thomas et al. 2011)) do not predict formation of significant concentrations of Cl2 and other chloride ion oxidation products due to the chemical reactions in the bulk of aqueous solution. The main reason for this lies in the following. In these models, the significant primary process of Cl− oxidation is the complex reaction (R7-R9) of chloride ion with hydroxyl radical, which generates Cl and Cl2 − (the oxidation of Cl− with O3 is objectively insignificant in most instances). Molecular chlorine is formed for the most part through the recombination reactions
But in the system OH – Cl−(aq) – UV hydrogen peroxide H2O2 is always present in considerable concentrations, which makes impossible the formation of appreciable amounts of Cl2 and other chlorine compounds in positive oxidation states, because of H2O2 reducing power (reactions R10-R12 and others). Furthermore, the reactions of chlorine compounds in the oxidation states greater than +2 are not considered. Because of this, the models like Bräuer et al. (2013); Knipping and Dabdub (2002); Sander et al. (2011a); Thomas et al. (2011) are not able to describe such important experimental phenomena as formation of chlorate and perchlorate ions on interaction of O3 with Cl−(aq). The general conclusion is that the established chemical mechanisms with known chemical reactions cannot explain the formation of Cl2 and ClO3 − observed in the experiments of this work in acid media.
The chemistry of chlorine-oxygen compounds in aqueous solution is a wide and complex issue. On the whole, this subject is far from being explored completely, although many particular reactions have been studied in greater detail. The complex reaction between chlorate ClO3 − and chloride Cl− ions in acid solutions has been extensively investigated.Footnote 5 The mechanism of ClO3 − + Cl− reaction has been formulated in the final form in the review (Schmitz 2000) on the basis of careful and logically consistent analysis of almost all published works on the problem. This information will allow us to propose a justified hypothesis on chlorate formation pathways in the conditions of our experiments. The key stages of the mechanism are the reaction of an intermediate with the composition Cl2O2 (or its hydrated form H2Cl2O3). The intermediate was proposed for the first time in Taube and Dodgen (1949). It is considered to possess unsymmetrical structure ClClO2 or ClOClO (but not ClOOCl or OClClO) (Buxton and Subhani 1972a; Emmenegger and Gordon 1967; Gordon and Tachiyashiki 1991; Hong and Rapson 1968; Kieffer and Gordon 1968a, b; Taube and Dodgen 1949), and is formed on reversible interaction of chloric acid with chloride and hydrogen ions,
The reverse reaction of Cl2O2 hydrolysis gives rise to the formation of chloride and chlorate ions (Fabian and Gordon 1992; Jia et al. 2000; Mialocq et al. 1973; Peintler et al. 1990; Quiroga and Perissinotti 2005; Rabai and Orban 1993; Taube and Dodgen 1949),
(Quiroga and Perissinotti 2005).
The formation of Cl2O2 in aqueous solution can also take place in the fast dimerization reaction of chlorine monoxide radical (Buxton and Subhani 1972a, b; Klaning and Wolff 1985),
(Klaning and Wolff 1985).
We infer that the formation of chlorate ion under our experimental conditions results from proceeding of the sequence of reactions (R14) and (R13). It can be shown that other possible chlorate formation pathways (oxidation of hypochlorite ion ClO−⟶ ClO2 −⟶ ClO3 −, oxidation of chlorine monoxide ClO ⟶ ClO2 −⟶ ClO2 ⟶ ClO3 −, or ClO ⟶ ClO2 ⟶ ClO3, and subsequent hydrolysis of chlorine oxides) are insignificant. Indeed, hypochlorite ion is virtually absent in acid media as quickly protonates in reactions (R5–R6). The rate of oxidation of ClO with ozone and hydroxyl radical is low, due to the small value of ClO + O3 reaction rate constant (Atkinson et al. 2007) and low concentration of ClO and OH in the reaction solution.
Now, let us clarify the ways of chlorine monoxide formation in the reaction solution in our experiments. It is well known that in the gas phase, the reaction of free chlorine atom with ozone molecule (Beach et al. 2002)
is an efficient channel of ClO generation. Under our experiments in liquid aqueous solution, chlorine atoms are formed on oxidation of chloride ion with hydroxyl radical in reactions (R7–R8), and then promptly bind with excess chloride to give very stable anion-radical complex Cl2 − (reaction R9) (Buxton et al. 1998). It has been found that in aqueous solution, the particles Cl2 − and O3 interact rapidly with the rate constant 9 × 107 L mol−1 s−1 (Bielski 1993), but the interaction products have not been established. By analogy with the gas phase reaction, the reaction products should be ClO, O2, and Cl− (liberating from the complex Cl2 −), and the reaction equation takes the form
(Bielski 1993).
The formation of such products is thermodynamically favorable, since the standard Gibbs energy of reaction (R15) in aqueous solution Δ15G°298 = −113.5 kJ/mole is essentially negative (Δ15G° has been calculated from the reference data (Koppenol et al. 2010; Stanbury 1989; Wagman et al. 1982)). The quantum chemical calculations indicate the high exergonicity of the reaction and give the value Δ15G°298 = −133 kJ/mole (Naumov and von Sonntag 2011). Thus, the formation of chlorate ion under our experimental conditions apparently results from the sequence of transformations starting from the oxidation of Cl− by OH with the formation of Cl2 − (R7-R9), and goes on via reactions (R15), (R14), and (R13).
The set of reactions (R1–R15) can describe on the qualitative level the formation of both experimentally observed products molecular chlorine and chlorate ion. However, the kinetic calculations with the use of only reactions (R1-R15) were not consistent with the experiment. In particular, they gave too low concentrations of Cl2 and ClO3 −, and did not predict the steady state regime for these substances during the time of experiment. We believe that the principal reason of discrepancy between the calculations and the experiment is the proceeding in the real reaction system of new not yet investigated chemical reactions in the bulk of aqueous solution. These may be the reactions of chlorine monoxide ClO, which is effectively generated by the sequence of reactions (R7–R9, R15). Chlorine monoxide is a chemically active particle, but only few of its reactions were kinetically studied. Under the conditions of our experiment, the reactions of ClO with hydrogen peroxide and/or perhydroxyl free radical,
may be important. Their rate constants (k 16 < 3.05 × 108 L mol−1 s−1 (Su et al. 1979), k 17 = 4.2 × 109 L mol−1 s−1 (Atkinson et al. 2007)), have been determined in the gas phase, while in the liquid solution, these processes have not been explored. The inclusion in the model of reactions (R16–R17) with the rate constants
causes the increase in calculated Cl2 and ClO3 − concentrations and ensures establishment of the steady state concentration profiles. However, theoretical Cl2 emission and ClO3 − formation rates remain lower than the experimental values. Comparison of the experimental and theoretical rates is presented in Figs. 4 and 5, and also Figs. S1–S4 of the Supporting Information.
To attain the enhancement of both chlorine and chlorate theoretical formation rates, it is necessary to append to the model some additional effective processes of ClO and/or HOCl and/or Cl2 − and/or Cl and/or OH generation. In doing so, it is pertinent to note that hydrogen peroxide H2O2, generated during aqueous ozone photolysis (R1, R3), rapidly converts to perhydroxyl radical HO2 as a result of reaction (R16). Compared to hydroxyl radical OH, HO2 radical is rather nonreactive, cannot oxidize chloride ion, and possess reducing capabilities. As the rate of HO2 production is rather high, the addition of the processes to the reactions set (R1–R17) that bring about OH generation from HO2 would significantly improve the agreement between the calculations and the experiment.
We tried to add to the model a number of reactions, studied in the experiment or theoretically feasible, which should increase the calculated rates of ClO/HOCl/Cl2 −/Cl/OH generation. These reactions and the results of the calculations are presented in Table S2 of the Supporting Information. Also, the rate constants of reactions (R16–R17) were varied in the range from small values to the diffusion limit ~1 × 1010 L mol−1 s−1 (Caldin 2001). Besides, the effect of volumetric mass transfer coefficient kLa variation in the wide range was investigated (see Fig. S5 in the Supporting Information). However, it was impossible to get the theoretical rates of both ClO3 − and Cl2 formation to reach their experimental values. The situation did not change even if the kinetic calculations were performed on the basis of not only reactions (R1–R17) but also the exhaustive set of known relevant aqueous phase chemical reactions from (Bräuer et al. 2013; Knipping and Dabdub 2002; Sander et al. 2011a; Thomas et al. 2011).
The quantitative agreement between the calculated and experimental kinetic characteristics of generation of both chlorate ion and molecular chlorine can only be obtained if we assume that there exists a rapid process of transformation of rather unreactive HO2 free radicals to very active OH radicals
The chemical nature and mechanism of this process are unknown. The process (R18) should be considered as an empirical correction to the model, which enables us to adequately reproduce all the experimental data of this work. The variation of its apparent rate coefficient k 18 and stoichiometric coefficient n 18 (n 18 is the number of OH radicals produced from one HO2 radical) allows to raise the calculated rate of chloride oxidation and to reach the agreement between the calculated and experimental rates of both chlorine emission and chlorate formation. It is notable that separate processes of OH generation divorced from HO2 disappearance, or HO2 disappearance divorced from OH generation, cannot ensure an agreement between the experiment and calculations, see Tables S3–S4 of the Supporting Information.
If process (R18) is added to the reaction set (R1–R17), the rates of process (R18) and new reaction (R16) turn out to be quite high in the model, which results in kinetic insignificance of stages (R11) and (R17). On the other hand, all the other reactions (R1-R10, R12-R16) are indispensable. The optimized values of k 18 and n 18, which provide minimal difference between the theoretical and experimental values of the rates (dnClO3−/dt)/V liq and (dnCl2/dt)/V liq, depend on the parameters of the model—Henry’s law constant of ozone HO3 and volumetric mass transfer coefficient kLa, as demonstrated in Fig. S6 of the Supporting Information. It should be stressed that there is an uncertainty in the values of the parameters HO3 and kLa. However, the variation of these parameters does not cause fundamental changes in the calculated rates of Cl2 and ClO3 − formation (Figs. S1–S5 in the Supporting Information). In particular, it cannot alter the situation that the model with reactions (R1–R17) does not provide quantitative compliance with the experiment, while the incorporation of process (R18) and optimization of k 18 and n 18 permits quantitative agreement between calculations and experiment for kLa ≥ 0.2 s−1 at HO3 = 0.24 and kLa ≥ 0.3 s−1 at HO3 = 0.16.
The comparison between the formation rates of chlorine and chlorate on photochemical interaction between ozone and chloride ion in acid medium, obtained from the experiment and calculated according to the sets of reactions (R1–R17), and (R1–R10, R12–R16, R18) with optimized k 18 and n 18, is made in Figs. 4 and 5, and in Fig. S1–S4 of the Supporting Information. The model based on reactions (R1–R17) does not give strict agreement with the experiment, but reproduce important experimental regularities of chlorine emission and chlorate formation on the qualitative level, and the quantitative discrepancy is not very high. The theoretical and experimental kinetic curves of Cl2 and ClO3 − are the same in character, and the steady state regime is predicted. The model with reactions (R1–R10, R12–R16), and empirical correction term represented by process (R18), reproduces quantitatively the experimental dependencies of chlorine emission and chlorate formation.
According to the model, the rates of formation of chloride oxidation products are greatly influenced by the velocities of OH radicals and/or Cl atom generation. These velocities can be increased by addition of Fe(III) to the reaction solution. Trivalent iron ions are efficacious catalysts of OH production from H2O2. Their effect is explained by the reactions
which are stages of the radical mechanism of thermal Fenton reaction (Barb et al. 1949; Barb et al. 1951a, b; Pignatello et al. 2006). Besides OH radicals as well as Cl atoms are formed on UV photolysis of Fe (III) complexes (Kiwi et al. 2000; Langford and Carey 1975; Nadtochenko and Kiwi 1998)
Therefore, the addition of Fe(III) ions would raise chlorine emission rate, and this did take place: the presence of 0.008 M Fe(III) in the reaction solution caused the increase in the rate by ~20 %, see Fig. 4 (chlorate formation rate was not measured in these experiments). This fact is evidence in favor of significant role of free-radical particles OH and Cl in chloride oxidation in the course of our photochemical experiments.
The scheme of chloride ion oxidation under the conditions of our experiments is shown in Fig. 5.Footnote 6 The key oxidizer is hydroxyl radical OH, which is formed on photolysis of ozone by UV light in aqueous solution (reactions R1–R2). Oxidation of chloride ion proceeds via two routes.
The first, most important route constitutes the sequence of chemical transformations represented by reactions (R7–R9) and (R13–R16). It starts from the complex interaction (R7–R9) between Cl− and OH free radical and can be called “the radical mechanism”. The radical mechanism comprising of collection of reactions (R7–R9, R13–R16) has been first proposed in the present work. The kinetics and mechanism of complex reaction (R7–R9) and its role in the oxidation of Cl− have been thoroughly investigated, see (Jayson et al. 1973; Klaning and Wolff 1985; Yu 2004). On the contrary, reactions (R15) and (R16) have not yet been taken into consideration as kinetically significant stages of chloride ion oxidation. Meanwhile, these reactions are of primary significance in the reaction system O3 – OH – Cl−(aq) and in particular should play an important role in the course of oxidative chemical transformations in an atmospheric aerosol containing chloride ion.
The second, less significant route consists in interaction of Cl− directly with O3 molecule (“molecular mechanism”). In the model, it is represented as reaction (R4). Its detailed description has been given in (Levanov et al. 2012b).
An important kinetic role of reaction (R16) as well as process (R18) is that they ensure efficacious participation of hydrogen peroxide in the formation of end product molecular chlorine Cl2. Hydrogen peroxide is the main product of aqueous ozone photolysis by the light with the wavelength 254 nm. The majority of its reactions consist in reduction of Cl(0) and Cl(+1) to Cl− and thus retard Cl2 formation.
Conclusions
In the present work, the interaction of chloride ion with ozone in the bulk of acid aqueous solution (pH 0–2) under the action of ultraviolet radiation with the wavelength 254 nm (the reaction system O3 – OH – Cl−(aq)) has been investigated for the first time. Molecular chlorine Cl2 and chlorate ion ClO3 − have been discovered to be the end products of this complex interaction. The formation of chlorate under such conditions has not been reported previously. The influence of concentrations of ozone in initial gases and hydrogen ion in reaction solution on the rates of chlorine emission and chlorate formation has been investigated. The ions of trivalent iron have been found to be catalysts of the interaction. The chemical mechanism of chloride ion oxidation in the reaction system O3 – OH – Cl−(aq) has been proposed. The rates of Cl2 and ClO3 − formation calculated according to the mechanism agree with the experimental ones. In the mechanism, the key stages leading to chlorate and chlorine formation are the reactions
To the best of our knowledge, previously, these reactions have not been included in any kinetic models describing chloride ion oxidation. Reaction (R15) has been unknown in aqueous solution until the present time.
Notes
The Hatta number has been calculated with the formula Ha = \( \sqrt{D_{O3}{k}_{1,O3}/{k}_L} \) (Beltran 2004), where D O3 is ozone diffusion coefficient in aqueous solution, D O3 = 2 × 10−9 m2 s−1 (Beltran 2004), k L is the mass transfer coefficient, in the analogous reactors k L = 3 × 10−4 m s−1 (Benbelkacem et al. 2003; Khudoshin et al. 2008), k 1, O3 is the apparent pseudo-first-order rate constant of ozone reactions, in our experimental conditions k 1, O3 < 0.1 s−1. If Ha < 0.3, then ozone reactions are characterized as “slow”, and reactions in the interface film can be neglected compared with the reactions in the bulk of the liquid (Benbelkacem et al. 2003; Charpentier 1981; Sotelo et al. 1989).
Note that Fig. 5 in (Levanov et al. 2003a) presents the values of the apparent rate constant k app, L mol−1 min−1, of the thermal reaction between O3 and Cl− in aqueous solution, which is connected with the chlorine emission rate by the relation (dnCl2/dt)/V liq = k app∙[Cl−]∙HO3∙C(O3)∙1000/48, where [Cl−] = 1 M is chloride ion concentration in reaction solution, H O3 = 0.24 is dimensionless Henry’s constant of ozone (Levanov et al. 2003a), and C(O3), g/m3, is ozone concentration in the gas flow at the inlet to the reactor.
The photolysis primary stage (R1) was approximated as a reaction of first kinetic order with respect to ozone. Its rate constant was calculated with the formula k 1 = φ254 · N Φ · ε254 · ln10/(60 · NA), s−1, where φ254 = 0.55 (Reisz et al. 2003) is the primary quantum yield, N Φ = (4.34 ± 0.06) × 1020 photons L−1 min−1 is the rate of UV photons absorption by the reaction solution in the experiments of this work, and ε254 = 3000 L mol−1 cm−1 is the molar absorptivity of aqueous ozone solution, on the basis of (Bader and Hoigné 1982; Forni et al. 1982; Gilbert and Hoigne 1983; Hart et al. 1983; Hoigné 1998; Hoigne and Bader 1976; Kilpatrick et al. 1956; Taube 1957; von Sonntag and von Gunten 2012).
The rate constant k 4 was obtained with the use of ozone Henry’s law constant H O3 = 0.24 (Levanov et al. 2003a). If Henry’s law constant H O3 = 0.16 is used in the kinetic calculations, then the constant k 4 is recalculated correspondingly.
It is worth noting that the formation of chlorate ion on interaction of O3 with Cl−(aq) does not contradict the possibility of chlorate reduction with chloride in strong acid media (Gordon et al. 1972; Greenwood and Earnshaw 1997; Schmitz 2000),
ClO3 − + 5Cl− + 6H+ → 3Cl2 + 3H2O and ClO3 − + Cl− + 2H+ → ½Cl2 + ClO2 + H2O.
Quantitative estimates based on the kinetic data (Crisci and Lenzi 1971; Deshwal and Lee 2004; Hong et al. 1967; Sant'Anna et al. 2012) show that under the conditions of our experiments at pH 0–2, the rate of chlorate disappearance owing to these reactions is negligibly small.
Notice that the set of reactions (R1–R18) and the scheme of Fig. 6 account for the formation of only main products Cl2 and ClO3 −. The formation of an important minor product perchlorate ion is not described by the reactions and the scheme (and has not been investigated in this work), although it may well take place under the conditions of our experiments.
References
Adam LC, Fábián I, Suzuki K, Gordon G (1992) Hypochlorous acid decomposition in the pH 5–8 region. Inorg Chem 31:3534–3541
Alekseev VN (1969) Quantitative analysis. Mir Publishers, Moscow
Anderson A, Sun TS (1970) Raman spectra of molecular crystals I. Chlorine, bromine, and iodine. Chem Phys Lett 6:611–616. doi:10.1016/0009-2614(70)85239-3
Atkinson R et al (2007) Evaluated kinetic and photochemical data for atmospheric chemistry: volume III—gas phase reactions of inorganic halogens. Atmos Chem Phys 7:981–1191. doi:10.5194/acp-7-981-2007
Bader H, Hoigné J (1982) Colorimetric method for the measurement of aqueous ozone based on the decolorization of indigo derivatives. In: Masschelein WJ (ed) Ozonation manual for water and wastewater treatment. John Wiley and Sons, Chichester, UK, pp 169–172
Barb WG, Baxendale JH, George P, Hargrave KR (1949) Reactions of ferrous and ferric ions with hydrogen peroxide. Nature 163:692–694. doi:10.1038/163692a0
Barb WG, Baxendale JH, George P, Hargrave KR (1951a) Reactions of ferrous and ferric ions with hydrogen peroxide Part I.—The ferrous ion reaction. Trans Faraday Soc 47:462–500. doi:10.1039/TF9514700462
Barb WG, Baxendale JH, George P, Hargrave KR (1951b) Reactions of ferrous and ferric ions with hydrogen peroxide Part II.—The ferric ion reaction. Trans Faraday Soc 47:591–616. doi:10.1039/TF9514700591
Bartlett WP, Margerum DW (1999) Temperature dependencies of the Henry's Law constant and the aqueous phase dissociation constant of bromine chloride. Env Sci Technol 33:3410–3414. doi:10.1021/es990300k
Bauer D, D'Ottone L, Hynes AJ (2000) O 1D quantum yields from O3 photolysis in the near UV region between 305 and 375 nm. Phys Chem Chem Phys 2:1421–1424. doi:10.1039/B000159G
Baxendale JH, Wilson JA (1957) The photolysis of hydrogen peroxide at high light intensities. Trans Faraday Soc 53:344–356
Beach SD, Smith IWM, Tuckett RP (2002) Rate constants for the reaction of Cl atoms with O3 at temperatures from 298 to 184 K. In J Chem Kinet 34:104–109. doi:10.1002/kin.10033
Beltran FJ (2004) Ozone reaction kinetics for water and wastewater systems. . lewis publishers, CRC press LLC, boca raton, Florida
Benbelkacem H, Cano H, Mathe S, Debellefontaine H (2003) Maleic acid ozonation: reactor modeling and rate constants determination ozone. Sci Eng 25:13–24. doi:10.1080/713610647
Biedenkapp D, Hartshorn LG, Bair EJ (1970) The O (1D) + H2O reaction. Chem Phys Lett 5:379–381. doi:10.1016/0009-2614(70)85172-7
Bielski BHJ (1993) A pulse-radiolysis study of the reaction of ozone with cl2- radical-anion in aqueous-solutions. Radiat Phys Chem 41:527–530
Bjergbakke E, Navaratnam S, Parsons BJ, Swallow AJ (1981) Reaction between HO2 and chlorine IN aqueous solution. J Am Chem Soc 103:5926–5928. doi:10.1021/ja00409a059
Bräuer P, Tilgner A, Wolke R, Herrmann H (2013) Mechanism development and modelling of tropospheric multiphase halogen chemistry: the CAPRAM halogen module 2.0 (HM2). J Atmos Chem 70:19–52. doi:10.1007/s10874-013-9249-6
Broek VD (1862) Ueber die zersetzung der chlorwasserstoffsäure durch ozone (notizen). J Prakt Chem 86:317–318. doi:10.1002/prac.18620860142
Buxton GV, Bydder M, Salmon GA (1998) Reactivity of chlorine atoms in aqueous solution. Part 1. the equilibrium Cl• + Cl– = Cl2. J Chem Soc Faraday Trans 94:653–657. doi:10.1039/a707377a
Buxton GV, Subhani MS (1972a) Radiation chemistry and photochemistry of oxychlorine ions. Part 1. radiolysis of aqueous solutions of hypochlorite and chlorite ions. J Chem Soc Faraday Trans 1(68):947–957
Buxton GV, Subhani MS (1972b) Radiation chemistry and photochemistry of oxychlorine ions. Part 2. Photodecomposition of aqueous solutions of hypochlorite ions. J Chem Soc Faraday Trans 1(68):958–969
Cahill JE, Leroi GE (1969) Raman spectra of solid chlorine and bromine. J Chem Phys 51:4514–4519. doi:10.1063/1.1671821
Caldin EF (2001) The mechanisms of fast reactions in solution. Ios Press, Amsterdam
Charpentier J-C (1981) Mass-transfer rates in gas–liquid absorbers and reactors. In: Drew TB, Cokelet GR, Hoopes JW, Vermeulen T (eds) Advances in chemical engineering, vol.11, vol 11. Academic Pres, New York, pp 1–133. doi:10.1016/S0065-2377(08)60025-3
Connick RE (1947) The interaction of hydrogen peroxide and hypochlorous acid in acidic solutions containing chloride ion. J Am Chem Soc 69:1509–1514. doi:10.1021/ja01198a074
Crisci P, Lenzi F (1971) The kinetics and mechanism of the chloride–chlorate reaction in moderately concentrated solutions of HClO4 and H2SO4 at 25 °C. Can J Chem 49:2552–2562. doi:10.1139/v71-421
Dasgupta PK, Martinelango PK, Jackson WA, Anderson TA, Tian K, Tock RW, Rajagopalan S (2005) The origin of naturally occurring perchlorate: the role of atmospheric processes. Env Sci Tech 39:1569–1575
Deshwal BR, Lee HK (2004) Kinetics and mechanism of chloride based chlorine dioxide generation process from acidic sodium chlorate. J Hazard Mater 108:173–182. doi:10.1016/j.jhazmat.2003.12.006
Duverney MR (1962) Rayons restants infrarouges, de monocristaux de chlorate et de bromate de sodium. C R Acad Sci 254:1954–1956
Emmenegger F, Gordon G (1967) The rapid interaction between sodium chlorite and dissolved chlorine. Inorg Chem 6:633–635. doi:10.1021/ic50049a048
Fabian I, Gordon G (1992) Iron (III)-catalyzed decomposition of the chlorite ion: an inorganic application of the quenched stopped-flow method. Inorg Chem 31:2144–2150. doi:10.1021/ic00037a030
Forni L, Bahnemann D, Hart EJ (1982) Mechanism of the hydroxide ion-initiated decomposition of ozone in aqueous solution. J Phys Chem 86:255–259. doi:10.1021/j100391a025
Gilbert E, Hoigne J (1983) Messung von ozon in wasserwerken vergleich der dpd- und indigo-methode gas- und wasserfach. Wasser, Abwasser 124:527–531
Goldstein S, Aschengrau D, Diamant Y, Rabani J (2007) Photolysis of aqueous H2O2: quantum yield and applications for polychromatic UV actinometry in photoreactors env. Sci Technol 41:7486–7490. doi:10.1021/es071379t
Gordon G, Kieffer RG, Rosenblatt DH (1972) The chemistry of chlorine dioxide. In: Lippard SJ (ed) Progress in inorganic chemistry, vol 15, vol 15. Wiley, NewYork, pp 201–286. doi:10.1002/9780470166161.ch3
Gordon G, Tachiyashiki S (1991) Kinetics and mechanism of formation of chlorate ion from the hypochlorous acid/chlorite ion reaction at pH 6–10. Envirol Sci Technol 25:468–474. doi:10.1021/es00015a014
Greenwood NN, Earnshaw A (1997) Chemistry of the elements, Second Editionth edn. Butterworth-Heinemann, Oxford. doi:10.1016/B978-0-7506-3365-9.50003-1
Hart EJ, Sehested K, Holcman J (1983) Molar absorptivities of ultraviolet and visible bands of ozone in aqueous solutions. Anal Chem 55:46–49. doi:10.1021/ac00252a015
Hatta S (1932) Technological Reports of Tohoku University 10:613–662
Held AM, Halko DJ, Hurst JK (1978) Mechanisms of chlorine oxidation of hydrogen-peroxide. J Am Chem Soc 100:5732–5740. doi:10.1021/ja00486a025
Hoigné J (1998) Chemistry of Aqueous Ozone and Transformation of Pollutants by Ozonation and Advanced Oxidation Processes. In: Hrubec J (ed) The Handbook of Environmental Chemistry. Vol. 5, Part C. Quality and Treatment of Drinking Water II, vol 5 / 5C. The Handbook of Environmental Chemistry. Springer Verlag, Berlin - Heidelberg, pp 83–141. doi: 10.1007/978-3-540-68089-5_5
Hoigne J, Bader H (1976) Role of hydroxyl radical reactions in ozonation processes in aqueous solutions. Water Res 10:377–386
Hoigné J, Bader H, Haag WR, Staehelin J (1985) Rate constants of reactions of ozone with organic and inorganic compounds in water. III. Inorg Comp Rad Water Res 19:993–1004
Hollenberg JL, Dows DA, Spectrochim A (1960) Vibrational spectra of sodium chlorate. Spectrochim Acta 16:1155–1164. doi:10.1016/0371-1951(60)80220-2
Hong CC, Lenzi F, Rapson WH (1967) The kinetics and mechanism of the chloride-chlorate reaction. Can J Chem Eng 45:349–355. doi:10.1002/cjce.5450450605
Hong CC, Rapson WH (1968) Kinetics of disproportionation of chlorous acid. Can J Chem 46:2053–2060. doi:10.1139/v68-335
Hunt JP, Taube H (1952) The photochemical decomposition of hydrogen peroxide quantum yields, tracer and fractionation effects. J Am Chem Soc 74:5999–6002. doi:10.1021/ja01143a052
Ianni JC (2006) Kintecus , Windows Version 3.9, www.kintecus.com.
Jayson GG, Parsons BJ, Swallow AJ (1973) Some simple, highly reactive, inorganic chlorine derivatives in aqueous-solution. their formation using pulses of radiation and their role in mechanism of fricke dosimeter. J Chem Soc Faraday Trans I:1597–1607
Jia Z, Margerum DW, Francisco JS (2000) General-acid-catalyzed reactions of hypochlorous acid and acetyl hypochlorite with chlorite ion. Inorg Chem 39:2614–2620. doi:10.1021/ic991486r
Kang N, Jackson WA, Dasgupta PK, Anderson TA (2008) Perchlorate production by ozone oxidation of chloride in aqueous and dry systems. Sci Total Environ 405:301–309
Khudoshin AG, Mitrofanova AN, Lunin VV (2008) Kinetics and mechanism of the reactions of ozone with guaiacol, veratrol, and veratrol derivatives. Russ Chem Bull 57:283–288. doi:10.1007/s11172-008-0043-6
Kieffer RG, Gordon G (1968a) Disproportionation of chlorous acid. I. Stoichiometry Inorg Chem 7:235–239. doi:10.1021/ic50060a013
Kieffer RG, Gordon G (1968b) Disproportionation of chlorous acid. II. Kinet Inorg Chem 7:239–244. doi:10.1021/ic50060a014
Kilpatrick ML, Herrick CC, Kilpatrick M (1956) The decomposition of ozone in aqueous solution. J Am Chem Soc 78:1784–1789. doi:10.1021/ja01590a003
Kiwi J, Lopez A, Nadtochenko V (2000) Mechanism and kinetics of the oh-radical intervention during fenton oxidation in the presence of a significant amount of radical scavenger (cl-). Environ Sci Technol 34:2162–2168. doi:10.1021/es991406i
Klaning UK, Wolff T (1985) Laser flash photolysis of hclo, clo−, hbro, and bro− in aqueous solution reactions of cl- and br-atoms. Ber Bunsenges Phys Chem 89:243–245
Knipping EM, Dabdub D (2002) Modeling Cl2 formation from aqueous NaCl particles: evidence for interfacial reactions and importance of Cl2 decomposition in alkaline solution. J Geophys Res 107:4360. doi:10.1029/2001jd000867
Knipping EM et al (2000) Experiments and simulations of ion-enhanced interfacial chemistry on aqueous nacl aerosols. Science 288:301–306. doi:10.1126/science.288.5464.301
Koppenol WH, Stanbury DM, Bounds PL (2010) Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic Biol Med 49:317–322. doi:10.1016/j.freeradbiomed.2010.04.011
Langford CH, Carey JH (1975) The charge transfer photochemistry of the hexaaquoiron (iii) ion, the chloropentaaquoiron (iii) ion, and the μ-dihydroxo dimer explored with tert-butyl alcohol scavenging. Can J Chem 53:2430–2435. doi:10.1139/v75-344
Laskin A, Wang H, Robertson WH, Cowin JP, Ezell MJ, Finlayson-Pitts BJ (2006) A new approach to determining gas-particle reaction probabilities and application to the heterogeneous reaction of deliquesced sodium chloride particles with gas-phase hydroxyl radicals. J Phys Chem A 110:10619–10627. doi:10.1021/jp063263+
Levanov AV, Antipenko EE, Lunin VV (2012a) Primary stage of the reaction between ozone and chloride ions in aqueous solution: can chloride ion oxidation by ozone proceed via electron transfer mechanism? Russ J Phys Chem A 86:584–589. doi:10.1134/S0036024412040164
Levanov AV, Antipenko EE, Lunin VV (2012b) Primary stage of the reaction between ozone and chloride ions in aqueous solution: oxidation of chloride ions with ozone through the mechanism of oxygen atom transfer. Russ J Phys Chem A 86:519–522. doi:10.1134/s0036024412030193
Levanov AV, Kuskov IV, Antipenko EE, Lunin VV (2006a) The kinetics of reaction between permanganate and chlorine ions in acid solutions. Russ J Phys Chem 80:726–731. doi:10.1134/s0036024406050104
Levanov AV, Kuskov IV, Antipenko EE, Lunin VV (2006b) The oxidation of chlorine ions under the joint action of ozone and permanganate ions. Russ J Phys Chem 80:556–561. doi:10.1134/s0036024406040121
Levanov AV, Kuskov IV, Antipenko EE, Lunin VV (2008) The solubility of ozone and kinetics of its chemical reactions in aqueous solutions of sodium chloride. Russ J Phys Chem A 82:2045–2050. doi:10.1134/s0036024408120133
Levanov AV, Kuskov IV, Antipenko EE, Lunin VV (2012c) Stoichiometry and products of ozone reaction with chloride ion in an acidic medium. Russ J Phys Chem A 86:757–762. doi:10.1134/S0036024412050202
Levanov AV, Kuskov IV, Koiaidarova KB, Antipenko EI, Lunin VV (2006c) Interaction between ozone and the chloride ion in sulfuric acid solutions up to 6-M concentration. Kinet Catal 47:682–685. doi:10.1134/s0023158406050053
Levanov AV, Kuskov IV, Koiaidarova KB, Zosimov AV, Antipenko EE, Lunin VV (2005) Catalysis of the reaction of ozone with chloride ions by metal ions in an acidic medium. Kinet Catal 46:138–143. doi:10.1007/s10975-005-0021-z
Levanov AV, Kuskov IV, Zosimov AV, Antipenko EE, Lunin VV (2003a) Acid catalysis in reaction of ozone with chloride ions. Kinet Catal 44:740–746. doi:10.1023/B:KICA.0000009047.90252.2d
Levanov AV, Kuskov IV, Zosimov AV, Antipenko EE, Lunin VV (2003b) Photometric determination of chlorine in a gas flow in the presence of ozone. J Anal Chem 58:439–441. doi:10.1023/A:1024069912606
Levanov AV, Sakharov DV, Dashkova AV, Antipenko EE, Lunin VV (2011) Synthesis of hydrogen polyoxides H2O4 and H2O3 and their characterization by Raman spectroscopy. Eur J Inorg Chem 2011:5144–5150. doi:10.1002/ejic.201100767
Liu Q, Margerum DW (2001) Equilibrium and kinetics of bromine chloride hydrolysis. Environ Sci Technol 35:1127–1133
Makower B, Bray WC (1933) The rate of oxidation of hydrogen peroxide by chlorine in the presence of hydrochloric acid. J Am Chem Soc 55:4765–4776. doi:10.1021/ja01339a006
Maric D, Burrows JP, Meller R, Moortgat GK (1993) A study of the UV-visible absorption spectrum of molecular chlorine. J Photochem Photobiol A Chem 70:205–214
Mialocq JC, Barat F, Gilles L, Hickel B, Lesigne B (1973) Flash photolysis of chlorine dioxide in aqueous solution. J Phys Chem 77:742–749. doi:10.1021/j100625a003
Miller FA, Carlson GL, Bentley FF, Jones WH (1960) Infrared spectra of inorganic ions in the cesium bromide region (700–300 cm−1). Spectrochim Acta 16:135–235. doi:10.1016/0371-1951(60)80077-X
Miller FA, Wilkins CH (1952) Infrared spectra and characteristic frequencies of inorganic ions. Anal Chem 24:1253–1294. doi:10.1021/ac60068a007
Nadtochenko VA, Kiwi J (1998) Photolysis of feoh2+ and fecl2+ in aqueous solution. Photodissociation Kinetics and Quantum Yields Inorg Chem 37:5233–5238. doi:10.1021/ic9804723
Naumov S, von Sonntag C (2011) Standard gibbs free energies of reactions of ozone with free radicals in aqueous solution: quantum-chemical calculations. Environ Sci Technol 45:9195–9204. doi:10.1021/es2018658
Nissenson P, Thomas JL, Finlayson-Pitts BJ, Dabdub D (2008) Sensitivity and uncertainty analysis of the mechanism of gas-phase chlorine production from NaCl aerosols in the MAGIC model. Atmos Environ 42:6934–6941. doi:10.1016/j.atmosenv.2008.04.041
Oum KW, Lakin MJ, DeHaan DO, Brauers T, Finlayson-Pitts BJ (1998) Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 279:74–76. doi:10.1126/science.279.5347.74
Peintler G, Nagypal I, Epstein IR (1990) Systematic design of chemical oscillators. 60. kinetics and mechanism of the reaction between chlorite ion and hypochlorous acid. J Phys Chem 94:2954–2958. doi:10.1021/j100370a040
Pignatello JJ, Oliveros E, MacKay A (2006) Advanced Oxidation Processes for Organic Contaminant Destruction Based on the Fenton Reaction and Related Chemistry Critical Reviews in Environmental Science and Technology 36:1–84 doi:10.1080/10643380500326564
Quiroga SL, Perissinotti LJ (2005) Reduced mechanism for the 366 nm chlorine dioxide photodecomposition in N2-saturated aqueous solutions. J Photochem Photobiol A Chemistry 171:59–67. doi:10.1016/j.jphotochem.2004.09.006
Rabai G, Orban M (1993) General model for the chlorite ion based chemical oscillators. J Phys Chem 97:5935–5939. doi:10.1021/j100124a026
Rao B, Anderson TA, Redder A, Jackson WA (2010) Perchlorate formation by ozone oxidation of aqueous chlorine / oxy-chlorine species: role of clxoy radicals. Env Sci Technol 44:2961–2967
Reisz E, Schmidt W, Schuchmann HP, von Sonntag C (2003) Photolysis of ozone in aqueous solutions in the presence of tertiary butanol. Environ Sci Technol 37:1941–1948. doi:10.1021/es0113100
Sander R et al (2011a) The atmospheric chemistry box model CAABA/MECCA-3.0. Geosci Model Dev 4:373–380. doi:10.5194/gmd-4-373-201
Sander SP et al. (2011) Chemical kinetics and photochemical data for use in atmospheric studies, evaluation no. 17. Pasadena: JPL Publication 10–6, Jet Propulsion Laboratory, 2011.
Sant'Anna RTP, Santos CMP, Silva GP, Ferreira RJR, Oliveira AP, Côrtes CES, Faria RB (2012) Kinetics and mechanism of chlorate-chloride reaction. J Braz Chem Soc 23:1543–1550
Schmitz G (2000) Kinetics of the halates-halides-halogens reactions; apparent differences and fundamental similarities. In: Ribnikar S, Anic S (eds) Proceedings of the 5th International conference on fundamental and applied aspects of physical chemistry. Society of Physical Chemists, Belgrade, pp 129–140
Smith GD, Molina LT, Molina MJ (2000) Temperature dependence of O (1D) quantum yields from the photolysis of ozone between 295 and 338 nm. J Phys Chem A 104:8916–8921. doi:10.1021/jp001006d
Sotelo JL, Beltrán FJ, Benitez FJ, Beltrán-Heredia J (1989) Henry's law constant for the ozone-water system. Water Res 23:1239–1246. doi:10.1016/0043-1354(89)90186-3
Stanbury DM (1989) Reduction potentials involving inorganic free radicals in aqueous solution. in advances in inorganic chemistry, vol 33. pp 69–138
Su F, Calvert JG, Lindley CR, Uselman WM, Shaw JH (1979) Fourier transform infrared kinetic study of hypochlorous acid and its absolute integrated infrared band intensities. J Phys Chem 83:912–920. doi:10.1021/j100471a006
Suzuki M, Yokoyama T, Ito M (1969) Raman spectrum and intermolecular forces of the chlorine crystal. J Chem Phys 50:3392–3398
Taniguchi N, Takahashi K, Matsumi Y (2000) Photodissociation of O3 around 309 nm. J Phys Chem A 104:8936–8944. doi:10.1021/jp001706i
Taube H (1957) Photochemical reactions of ozone in solution. Trans Farad Soc 53:656–665. doi:10.1039/TF9575300656
Taube H, Dodgen H (1949) Applications of radioactive chlorine to the study of the mechanisms of reactions involving changes in the oxidation state of chlorine. J Am Chem Soc 71:3330–3336. doi:10.1021/ja01178a016
Thomas JL, Jimenez-Aranda A, Finlayson-Pitts BJ, Dabdub D (2006) Gas-phase molecular halogen formation from nacl and nabr aerosols: when are interface reactions important? J Phys Chem A 110:1859–1867. doi:10.1021/jp054911c
Thomas JL, Stutz J, Lefer B, Huey LG, Toyota K, Dibb JE, von Glasow R (2011) Modeling chemistry in and above snow at Summit, Greenland—part 1: model description and results. Atmos Chem Phys 11:4899–4914. doi:10.5194/acp-11-4899-2011
von Sonntag C, von Gunten U (2012) Chemistry of ozone in water and wastewater treatment. From basic principles to applications. IWA Publishing, London
Wagman DD et al. (1982) The NBS tables of chemical thermodynamic properties. J. Phys. Chem. Ref. Data, 1982, 11, Supplement №2. National Bureau of Standards, Washington D. C.
Wang TX, Kelley MD, Cooper JN, Beckwith RC, Margerum DW (1994) Equilibrium, kinetic, and UV-spectral characteristics of aqueous bromine chloride, bromine, and chlorine species. Inorg Chem 33:5872–5878
Wang TX, Margerum DW (1994) Kinetics of reversible chlorine hydrolysis: temperature dependence and general-acid/base-assisted mechanisms. Inorg Chem 33:1050–1055. doi:10.1021/ic00084a014
Wayne RP (1987) The photochemistry of ozone. Atmos Environ 21:1683–1694. doi:10.1016/0004-6981(87)90107-7
Wilkinson F, Helman WP, Ross AB (1995) Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. An expanded and revised compilation. J Phys Chem Ref Data 24:663–677. doi:10.1063/1.555965
Yeatts LRB, Taube H (1949) The kinetics of the reaction of ozone and chloride ion in acid aqueous solution. J Am Chem Soc 71:4100–4105
Yu X-Y, Barker JR (2003) Hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions I. Chem Mech J Phys Chem A 107:1313–1324. doi:10.1021/jp0266648
Yu XY (2004) Critical evaluation of rate constants and equilibrium constants of hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions. J Phys Chem Ref Data 33:747–763. doi:10.1063/1.1695414
Acknowledgments
The authors acknowledge partial support from M.V. Lomonosov Moscow State University Program of Development.
Conflict of Interest
The authors declare no competing interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Electronic supplementary material
Electronic Supporting Information available:
The experimental and calculated dependencies of chlorine emission and chlorate formation rates on concentrations of H+ in reaction solution and O3 in initial gases (Figs. S1-S4).
The effect of variation of volumetric mass transfer coefficient kLa on calculated rates of chlorine emission and chlorate formation (Fig. S5).
Optimized values of coefficients k18 and n18 as functions of Henry’s law constant of ozone HO3 and volumetric mass transfer coefficient kLa (Fig. S6).
Summary of the reactions included in the mechanism of photochemical chloride oxidation with ozone (Table S1).
Effect of addition to the reaction set (R1 – R17) of various reactions, and also processes of HO2 disappearance and OH generation, on the calculated rates of chlorine emission and chlorate formation. (Tables S2-S4).
ESM 1
(DOCX 234 kb)
Rights and permissions
About this article

Cite this article
Levanov, A.V., Isaykina, O.Y., Amirova, N.K. et al. Photochemical oxidation of chloride ion by ozone in acid aqueous solution. Environ Sci Pollut Res 22, 16554–16569 (2015). https://doi.org/10.1007/s11356-015-4832-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-4832-9