
Abstract
Purpose
p-Chloroamphetamine (PCA) is a neurotoxin that selectively degenerates the serotonin (5-HT) axon terminals. In order to study the brain metabolic consequences induced by serotonergic denervation, a single dose of PCA (2.5 or 10 mg/kg i.p.) was administered to male adult rats.
Procedures
In vivo regional brain metabolism was evaluated 3 and 21 days after PCA (2.5 or 10 mg/kg; i.p.) injection by 2-deoxy-2-[18F] fluoro-d-glucose ([18F] FDG) positron emission tomography (PET). At day 22, the following markers of neurotoxicity were determined: (a) 5-HT axon terminal lesion by 5-HT transporter (SERT) autoradiography, (b) reactive gliosis by glial fibrillary acidic protein immunohistochemistry, and (c) eventual neurodegeneration by DAPI/Fluoro-Jade C labeling.
Results
An average of 20 % reduction of [18F] FDG uptake in most brain areas was observed at day 21 under 10 mg/kg PCA treatment. Instead, 2.5 mg/kg PCA only reduced metabolic activity in neocortex. Likewise, the high dose of PCA exerted a strong decrease (>30 %) in SERT density in several 5-HT innervated regions, but no effect was found in midbrain raphe nuclei, the main source of serotonergic neurons. Although PCA induced astroglial activation both in hippocampus and cortex in response to axotomy, no signs of neuronal death in these areas were detected.
Conclusions
Overall, [18F] FDG PET revealed that the reduction of the brain metabolic activity induced by PCA is related to 5-HT axon terminal lesion, with no apparent affectation of neuronal viability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
p-Chloroamphetamine (PCA) is an amphetamine derivative which shows a high degree of selectivity to lesion serotonergic terminals [1, 2]. This compound exerts its actions by getting into the axon terminal through the serotonin (5-hydroxytryptamine; 5-HT) transporter (SERT) which is located on presynaptic serotonergic neurons.
Systemic administration of PCA has shown to induce a wide range of physiological and behavioral changes in rodents. Shortly after the PCA injection, a 5-HT syndrome with a high mortality rate has been observed [3–5]. Other effects attributed to PCA include memory impairment [6, 7], nociception augmented [8], and aggressive behavior [9].
Regarding the serotonergic neural system, at short term, PCA induces a sharp release of 5-HT at the synaptic cleft [2, 3, 5, 10], whereas at long term, it has shown to deplete the 5-HT concentration and its metabolite 5-hydroxyindolacetic acid (5-HIAA) in several brain regions [11]. In addition, a reduction of the expression and density of SERT has been found when PCA is administrated at a high dose [4, 12]. Although a neuroadaptative mechanism for the fenfluramine-mediated SERT reduction has been proposed [13], either by its down-regulation or internalization, further evidence of a common neurotoxic action, shared by PCA and other substituted amphetamines such as 3,4-methylenedioxy-N-methylamphetamine (MDMA), has been proved [14–18]. Due to the selective presence of SERT in serotonergic neurons, it is regarded as a specific marker for serotonergic neuron integrity [19]. Thus, the lasting loss in expression and density of SERT is considered an index of the neurotoxic action mediated by the chemical PCA. In several cases, different signs of neuronal death after administration of several amphetamine derivatives have been observed [17, 20, 21].
In vivo evaluation of brain metabolism by positron emission tomography (PET) neuroimaging may provide a screening platform for identifying drugs that are potentially protective against amphetamine derivative-induced serotonergic neurotoxicity in animal models. Thus, in order to complement assays on histochemically stained brain slices which do not fully reflect conditions in the living brain, it is possible that 2-deoxy-2-[18F] fluoro-d-glucose ([18F] FDG) uptake quantification can be used reliably for directly evaluating the neurotoxicity of PCA in the living brain and for monitoring the response to eventually neuroprotective drugs.
With such a view, the main aim of this study was to evaluate whether an eventual disturbance in brain metabolism upon PCA treatment might occur and, if so, to determine its potential correlation with several neurochemical markers. Furthermore, whether those metabolic disturbances are due to selective serotonergic denervation and/or neuronal death will be assessed.
Materials and Methods
Animal Treatment
Male adult Sprague–Dawley rats weighing approximately 250 g at the beginning of the experiment were used (Harlan Interfauna Iberica, Sant Feliu de Codines, Spain). The animals (6–8 rats/group) were individually housed and maintained under a 12 h dark/12 h light cycle. Standard rodent food and tap water were available ad libitum. A single dose of the serotonergic neurotoxin PCA (Sigma-Aldrich, St. Louis, MO) dissolved in saline was administered (2.5 or 10 mg/kg i.p.). Instead, control rats only received saline. [18F] FDG PET scanning was performed at day 3 and day 21 after PCA injection. At the end of the experiment, at day 22, rats were sacrificed by decapitation and their brains were quickly removed. The samples were immediately frozen in dry-ice chilled isopentane and stored at −80 °C until further processing.
The use of animals in this study conformed to the guidelines of The European Communities Council Directive 2010/63/UE and was approved by the Animal Research Ethical Committee of the Universidad Complutense de Madrid. All efforts were made to minimize the number of animals used and their suffering.
[18F]FDG PET Imaging
At days 3 and 21, longitudinal [18F] FDG PET studies were performed for measuring the effect of PCA on the brain metabolic activity (Albira ARS dual scanner; Oncovision, Valencia, Spain). The acquisition protocol is described in detail in [22] with some modifications. Briefly, fasted rats were injected i.v. into the tail vein with [18F] FDG (approximately 18.5 MBq in 0.2 ml of 0.9 % NaCl; Instituto Tecnológico PET, Madrid, Spain). After an uptake period of 30 min, the animals were anesthetized by inhalation of a mixture of isoflurane/oxygen (5 % for induction and 2 % for maintenance) and placed on the bed of the tomograph. The duration of the PET acquisition was 20 min, and it was immediately followed by a CT (computed tomography) scanning. For the metabolic activity quantification, the procedure used was as follows: first, the CT image of the skull from each animal was co-registered to a magnetic resonance image (MRI) rat brain template in which the regions of interest (ROIs) were previously delineated. Then, the spatial mathematic transformation was applied to its own fused PET image, allowing the correct matching between the PET image and the MRI template as described by Jupp and O’Brien [23]. For these tasks, PMOD 3.0 software (PMOD Technologies Ltd., Zurich, Switzerland) was used. As index of regional metabolic activity, the standardized uptake value (SUV) was obtained. SUV is a widely used PET quantifier, calculated as a ratio of a ROI radioactivity concentration (kBq/ml) measured by the PET scanner and the actual injected dose (kBq), decay-corrected at the time of the injection, divided by the body weight of the animal (g).
SERT Autoradiography
Thirty-micrometer hemibrain coronal sections from Bregma −2.80 to −7.80 mm [24] were obtained by using a cryostat (Leica CM1850, Germany). The slices (four slices/glass slide) were thaw-mounted onto Superfrost Plus slides (Thermo Scientific, Germany) and stored at −80 ° C until assayed. The autoradiography was performed using [3H] paroxetine (Perkin Elmer, USA; 847.3 GBq/mmol) as SERT ligand as previously described [12, 25]. After exposing and developing the autoradiographic films (Kodak BioMax MR, Carestream, NY), the images were digitally acquired (DFC425 camera, Leica, Germany). For quantification purposes, commercial tritium microscale strips (Amersham Biosciences, GE Healthcare, Spain) were co-exposed with the brain slices. The quantification procedure is briefly described as follows: First, a background-subtracted image of each slice was obtained by running a rolling ball algorithm. Afterwards, the optical densities (OD) of the different brain areas and microscales were measured. Then, a calibration curve was fitted, allowing the conversion from OD values to radioactivity units (nCi/mg). Finally, the SERT density values for each structure were obtained. All these steps were carried out with the built-in functions of ImageJ software (freely available on the Internet at http://rsb.info.nih.gov/ij/download.html). To calculate each individual value, the average of four slices was used. The results were expressed as mean ± SEM.
GFAP Immunohistochemistry
GFAP was visualized by a one-step technique using fluorescent labeled anti-GFAP antibody conjugated with the cyanine dye Cy3. First, the slices were fixed into ice-cold acetone for 5 min. After drying, they were rinsed in Tris-buffered saline (TBS) for 30 min and then blocked (3 % BSA, 0.1 % triton X-100 in TBS for 45–60 min). Then, the slices were incubated with the fluorescent cyanine anti-GFAP-Cy3 antibody (dilution 1:500; Sigma-Aldrich, St. Louis, MO) at 4 °C overnight. The next day, the slices were washed in 0.1 % Tween 20 (3 × 5 min) to remove the excess of unbound antibody. Finally, the slices were coverslipped with Flouromount aqueous mounting medium (Sigma-Aldrich, St. Louis, MO). Tissue sections were examined under a fluorescence microscope (Olympus IX51; Olympus Europa Holding, Germany) using TRITC (tetramethylrhodamine isothiocyanate) filter. Digital captures were obtained and further processed with ImageJ software.
DAPI/Fluoro-Jade C Labeling
The protocol originally described by Schmued et al. [26] was followed with minor modifications. First, the slices were fixed with 4 % formaldehyde in phosphate buffer pH 7.5 for 10 min, followed by two washes in phosphate buffer. They were then sequentially rinsed for 5 min in basic alcohol, 2 min in ethanol, 2 min in distilled water, and 10 min in potassium permanganate solution. The slides were transferred for 10 min to a 0.01 % acetic acid solution containing 0.0001 % Fluoro-Jade C (Millipore) and 0.0001 % DAPI (Sigma-Aldrich, St. Louis, MO). Then, the glass slides were subsequently rinsed three times in distilled water for 1 min. Once the sections were air-dried, they were rinsed in xylene for 2 min and afterwards they were coverslipped with DPX (Fluka). Finally, tissue sections were examined under a fluorescence microscope (Olympus IX51). Fluorescein isothiocyanate and blue/cyan filters were used to visualize Fluoro-Jade C and DAPI labeling, respectively. After capturing the fluorescence images for DAPI and Fluoro-Jade C, they were processed and merged by using ImageJ software.
Statistical Analyses of Data
Data are presented as means ± SEM. Data were analyzed using one-way analysis of variance (ANOVA) followed by the post hoc Student–Newman–Keuls test for multiple comparisons. Differences at p <0.05 were considered statistically significant (SigmaPlot 11.0, Systat Software Inc., Chicago, IL).
Results
Brain Glucose Metabolism
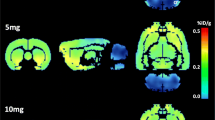
Three days after the administration of the high dose of PCA (10 mg/kg), an overall brain glucose hypometabolism was detected, whereas no significant changes were observed with the low dose (2.5 mg/kg) compared with [18F] FDG uptake of the saline group (Fig. 1a). Furthermore, brain glucose metabolism reduction seems to be time dependent as after 3 weeks from the neurotoxin administration, the [18F] FDG uptake was reduced even a bit more. Thus, 10 mg/kg PCA caused a significant reduction (p < 0.05) of 15.95 and 14.43 % in hippocampus and cortex, respectively, after 3 days (Fig. 1a), and this decrement reached 20.61 and 19.42 % after 3 weeks (Fig. 1a) when compared to control animals. Moreover, when analyzed at day 21, the metabolic activity of the 10 mg/kg PCA group was significantly lower (p < 0.05) than that of the animals which received 2.5 mg/kg PCA. At this time point, the cortex was the only brain structure which, upon 2.5 mg/kg PCA dose, significantly underwent a decrease in glucose metabolic activity when compared to saline group (p < 0.05; Fig. 1b). Representative PET images showing the different brain glucose metabolism of the three experimental groups (saline, 2.5 mg/kg PCA, and 10 mg/kg PCA) at the two time points are displayed in Fig. 2.

Changes in regional brain glucose metabolism in control (Saline, n = 6), 2.5 mg/kg PCA (n = 6), and 10 mg/kg PCA (n = 4) groups at a day 3 and at b day 21 after the injection. Data are expressed as means ± SEM (*p < 0.05, significantly different from saline group, #p < 0.05 significantly different from 2.5 mg/kg PCA group)

Representative [18F] FDG PET images showing the rat brain metabolic activity from saline, 2.5 mg/kg PCA, and 10 mg/kg PCA groups after 3 and 21 days from the neurotoxin (or saline) injection in coronal, sagittal, and trans-axial views. The upper rows show the CT images, the middle rows show the PET images (in SUV scale), and the bottom rows represent both tomographic images merged
SERT Density
As depicted in the left panel of Fig. 4, brain serotonergic innervation shows a differential regional distribution in naive male Sprague–Dawley rats. Thus, the ventral hippocampus was the brain area analyzed with the highest SERT density (1.29 ± 0.04 nCi/mg), followed by the occipital cortex (1.21 ± 0.07 nCi/mg). Finally, the dorsal hippocampus was found to be the area with the lowest SERT expression (0.72 ± 0.04 nCi/mg).
A dose-dependent reduction of [3H] paroxetine binding was observed in ventral hippocampus when measured after 22 days from the neurotoxin administration (Fig. 3). Thus, in this region, the low PCA dose administered (2.5 mg/kg) produced an 18.46 % decrease (p < 0.05), whereas a 55.15 % reduction was detected when the high dose (10 mg/kg) was injected (p < 0.01). In the rest of the structures analyzed, the low dose of PCA did not produce any significant change in the expression of the transporter.

Effect of PCA on SERT density in occipital cortex, ventral hippocampus, and dorsal hippocampus from saline (n = 4), 2.5 mg/kg PCA (n = 5), and 10 mg/kg PCA (n = 7) groups, 22 days after the injection. Data are expressed as mean of percentage of saline-control ± SEM (*p < 0.05 vs. saline group; **p < 0.01 vs. saline group; #p < 0.05 vs. 2.5 mg/kg PCA group; ##p < 0.01 vs. 2.5 mg/kg PCA group)
Instead, the high PCA dose (10 mg/kg) significantly reduced the SERT density in all the regions analyzed: occipital cortex (31.54 %, p < 0.01) and dorsal hippocampus (30.64 %, p < 0.01). Furthermore, the reduction of SERT densities observed by the administration of 10 mg/kg PCA was statistically higher (Fig. 3, p < 0.05 or p < 0.01 regarding the brain region) than the displayed at the dose of 2.5 mg/kg. These results support a dose-dependent reduction of SERT density mediated by PCA.
As the raphe nuclei are the main source of serotonergic innervation of forebrain and limbic regions [27], we also measured SERT density in this area for evaluating an eventual lesion exerted by PCA at the somatodendritic level. In accordance with the effects induced by other amphetamine derivatives [15], our results support a sole and specific action of PCA on the axonal transporters, as SERT density in the raphe nuclei was not modified (% change from control group; saline group = 100.00 ± 3.77; 10 mg/kg PCA group = 104.52 ± 6.93).
Representative coronal autoradiographic images of [3H] paroxetine binding from the three experimental groups that show a PCA-mediated SERT density reduction at the level of Bregma −4.30 mm are depicted in Fig. 4.

Representative autoradiograms of coronal hemibrain sections at the level of Bregma −4.30 mm, showing the gradual reduction of SERT expression, 22 days after the PCA injection. OC occipital cortex, DH dorsal hippocampus, VH ventral hippocampus. Scale bar: 1 mm
GFAP Immunohistochemistry
In order to evidence astroglial activation, non-quantitative GFAP immunohistochemistry was performed. As shown in the fluorescence microphotographs (Fig. 5), PCA administration produced a noticeable long-term increase in GFAP labeling both in occipital cortex and dorsal hippocampus compared to the same brain areas from non-treated animals.

GFAP immunohistochemistry in coronal brain slices upon long-term effect of a single PCA administration. GFAP labeling is shown at the level of occipital cortex (upper row) and dorsal hippocampus (bottom row), 22 days after injection of saline (left column), 2.5 mg/kg PCA (middle column), and 10 mg/kg PCA (right column). A strong gliosis is observed when the neurotoxin PCA was injected compared to the saline group. oc occipital cortex, cg cingulum, hil hilus, dg dentate gyrus. Scale bar: 250 μm
Assessment of Eventual Neurodegeneration by DAPI/Fluoro-Jade C Double Labeling
As shown in Fig. 6, Fluoro-Jade C labeling showed no signs of post- and presynaptic neurodegeneration at the level of hippocampus and cortex at any of the two doses of PCA administered. In addition, DAPI labeling was fairly similar in PCA-treated and non-treated animals.

Double DAPI/Fluoro-Jade C fluorescence micrographs at the level of the occipital cortex (upper row) and the dentate gyrus of the dorsal hippocampus (bottom row). The images show no signs of neurodegeneration 22 days after PCA injection. Left panels, saline group; middle panels, 2.5 mg/kg PCA; right panels, 10 mg/kg PCA. oc occipital cortex, cg cingulum, hil hilus, dg dentate gyrus. Scale bar: 250 μm
A parallel assay was performed on rats receiving intrahippocampal kainic acid, a well-known neuroexcitotoxic agent, as a positive control of Fluoro-Jade C labeling. In accordance with Schmued et al. [26], these animals showed an intense Fluoro-Jade C signal (data not shown).
Discussion
In this study, we first used longitudinal in vivo [18F] FDG PET to monitor both short- and long-term effects (3 and 21 days, respectively) of a single dose (2.5 or 10 mg/kg i.p.) of PCA on brain glucose metabolism in rats. Secondly, at day 22, we performed an autoradiographic study describing the pattern change on the SERT density in several 5-HT innervated brain regions in order to evaluate any putative axon damage. SERT density was also measured in raphe nuclei for evaluating a putative reduction in somatodendritic SERT. Additionally, the degree of reactive astrogliosis was evaluated by GFAP immunohistochemistry. Finally, double DAPI/Fluoro-Jade C staining was carried out to detect any eventual sign of neurodegeneration.
For the present study, we selected two different PCA doses at seeking both a severe and a mild lesion on the serotonergic tracks [4, 12, 28, 29]. Rat exposure to 10 mg/kg PCA produced a significant reduction of [18F] FDG uptake (in SUV units) in all brain regions studied at the two time points. Instead, the low dose (2.5 mg/kg) only reduced the metabolic activity in cortex after 21 days from the neurotoxin exposure (Fig. 1b). Currently, the most widely used quantification index in small-animal [18F] FDG PET studies is the semi-quantitative SUV measure [30]. It is important to note that the differences found in the SUV values at day 3 and 21 of the control (saline) group could be attributed to inter-scan interval as recently reported by Deleye et al. [30]. For this reason, the comparisons of [18F] FDG SUV values of PCA-treated animals were performed not against basal acquisitions made before the neurotoxin administration but against the corresponding control (saline) group. The control group of rats was scanned at the same two time points as that of the PCA-injected rats, aiming at avoiding such eventual SUV inter-scan variation.
The data from the [3H] paroxetine autoradiography showed a dose-related reduction of the SERT density in cortex and hippocampus after PCA injection and paralleled the findings of the [18F] FDG PET study. Although a neuroadaptative mechanism for the SERT change has been proposed [13, 31], a presynaptic neurotoxic action for PCA and other amphetamine derivatives is generally accepted [14–18, 32]. Specifically, these kinds of compounds have the potential to produce a distal axotomy of serotonergic neurons [15, 18]. In this context, the degrees of denervation revealed at both doses were in agreement with previous reports [4, 12]. Moreover, the sustained fall in SERT density observed 22 days after a single 10 mg/kg PCA injection (Fig. 3) points out a non-recoverable neurotoxic-like effect.
Indeed, the PCA-induced decrease of SERT density in cortex and hippocampus at day 22 is concomitant to a reduction of [18F] FDG uptake in the same brain areas. Thereby, it can be inferred that the lowered synaptic activity, as a consequence of the PCA-induced 5-HT denervation, contributes to the reduction of the brain glucose metabolism.
A single PCA injection is shown here to cause extensive astrogliosis in 5-HT innervated regions such as cortex and hippocampus (Fig. 5). Astrogliosis is considered to be one of the markers for neuronal injury [33]. It should be noted that only amphetamine derivatives with neurotoxic properties have shown to generate astrogliosis following microglial activation [17, 21, 34]. This reactive gliosis seems to be an essential event for the early neuronal damage by increasing the generation of reactive oxygen species [17, 35]. In turn, after a brain insult, it has been proposed that a slow astrocytic response can maintain microglia activation and eventually lead to a chronic brain lesion [36].
Fluoro-Jade staining is widely recognized to specifically localizing not only degenerating nerve cell bodies but also distal dendrites, axons, and terminals, regardless of being the result of brain trauma, stroke, or neurotoxic insult [26]. This makes it ideal for localizing degenerating neuronal structures. Our results indicate that a single dose of PCA is related solely to distal axotomy in agreement with previous observations [1, 2, 18], but not to neurodegeneration as no labeled neuron structures were detected at hippocampal and cortical areas by using double DAPI/Fluoro-Jade C staining, 22 days after the PCA injection (Fig. 6).
Furthermore, the serotonergic neuronal bodies appeared not to be affected by PCA, as SERT density in the midbrain raphe nuclei (the main area that contains the majority of the serotonergic neurons) was not significantly modified 22 days after PCA administration. This observation is in agreement with the MDMA-induced long-term neurotoxicity observed in monkeys by Hatzidimitriou et al. [15]. These authors reported persistent regional deficits in axons with abnormal brain 5-HT innervation patterns in hippocampus and cortex, being still evident even 7 years after MDMA exposure. Furthermore, no loss of 5-HT nerve cell bodies in the rostral raphe nuclei was found, suggesting that abnormal innervation patterns in MDMA-treated monkeys are not necessarily associated with loss of a particular 5-HT nerve cell group.
A regional brain hypometabolism found non-invasively by in vivo [18F] FDG PET as a result of a single dose of PCA is reported here for the first time. At long term, the cortex seems to be more affected by this neurotoxin than the rest of the brain regions analyzed, as the radiotracer uptake into this brain area, upon 2.5 mg/kg PCA, was significantly reduced (Fig. 1b). The dissimilar metabolic affectation observed at long term can be explained by the different constitutive SERT expression on 5-HT axon terminals, as this kind of compounds uses the transporter to enter the neuron [1]. Thus, the thin 5-HT axons that innervate the cortex are more vulnerable than axons that innervate the hippocampal formation [1, 37]. In this regard, supporting our finding, Riezzo et al. [38] reported that the frontal cortex was the only brain structure in which enzymatic and non-enzymatic cellular antioxidant defense systems were acutely reduced after MDMA administration.
It has been reported that 5-HT is involved in neurogenesis [39], promotes the survival of cortical neurons in vitro [40], and can display neuroprotective properties through a mechanism that involves up-regulation of the bcl-xl protein thereby diminishing the risk of mitochondrial-dependent activation of caspases [41]. Thus, it cannot be completely ruled out a secondary mechanism of postsynaptic damage involved in the PCA-induced hypometabolism due to the reported long-lasting 5-HT depletion.
Finally, the present study shows that the cortical and hippocampal hypometabolism induced by PCA is associated with its axotomic properties since the metabolic disturbance is accompanied by a long-term SERT density reduction in those brain areas. Furthermore, following axonal denervation, the reactive gliosis observed might also account for the damage caused by PCA. Further studies assessing the molecular events coupled to the reported long-term metabolic impairment will give us a deeper insight of the neurotoxicity triggered by PCA.
References
Brown P, Molliver ME (2000) Dual serotonin (5-HT) projections to the nucleus accumbens core and shell: relation of the 5-HT transporter to amphetamine-induced neurotoxicity. J Neurosci 20:1952–1963
Fuller RW (1992) Effects of p-chloroamphetamine on brain serotonin neurons. Neurochem Res 17:449–456
Colado MI, Murray TK, Green AR (1993) 5-HT loss in rat brain following 3,4-methylenedioxymethamphetamine (MDMA), p-chloroamphetamine and fenfluramine administration and effects of chlormethiazole and dizocilpine. Br J Pharmacol 108:583–589
Fernandez F, Aguerre S, Mormede P, Chaouloff F (2001) Differential sensitivities to the lethal, but not the neurotoxic, effects of p-chloroamphetamine in inbred rat strains. Neurosci Lett 297:53–57
Sugimoto Y, Ohkura M, Inoue K, Yamada J (2001) Involvement of serotonergic and dopaminergic mechanisms in hyperthermia induced by a serotonin-releasing drug, p-chloroamphetamine in mice. Eur J Pharmacol 430:265–268
Hsieh MT, Kuo LH, Tsai FH et al (2002) Effects of puerarin on scopolamine-, mecamylamine-, p-chloroamphetamine- and dizocilpine-induced inhibitory avoidance performance impairment in rats. Planta Med 68:901–905
Solana-Figueroa R, Salado-Castillo R, Quirarte GL et al (2002) Enhanced inhibitory avoidance training protects against the amnesic effect of p-chloroamphetamine. Life Sci 71:391–399
Maneepak M, le Grand S, Srikiatkhachorn A (2009) Serotonin depletion increases nociception-evoked trigeminal NMDA receptor phosphorylation. Headache 49:375–382
Kanno H, Sekiguchi K, Yamaguchi T et al (2009) Effect of yokukansan, a traditional Japanese medicine, on social and aggressive behaviour of para-chloroamphetamine-injected rats. J Pharm Pharmacol 61:1249–1256
Trulson ME, Jacobs BL (1976) Behavioral evidence for the rapid release of CNS serotonin by PCA and fenfluramine. Eur J Pharmacol 36:149–154
Sanders-Bush E, Sulser F (1970) P-chloroamphetamine: in vivo investigations on the mechanism of action of the selective depletion of cerebral serotonin. J Pharmacol Exp Ther 175:419–426
Lauzurica N, Garcia-Garcia L, Pinto S et al (2010) Changes in NPY and POMC, but not serotonin transporter, following a restricted feeding/repletion protocol in rats. Brain Res 1313:103–112
Rothman RB, Jayanthi S, Wang X et al (2003) High-dose fenfluramine administration decreases serotonin transporter binding, but not serotonin transporter protein levels, in rat forebrain. Synapse 50:233–239
Green AR, Mechan AO, Elliott JM et al (2003) The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharmacol Rev 55:463–508
Hatzidimitriou G, McCann UD, Ricaurte GA (1999) Altered serotonin innervation patterns in the forebrain of monkeys treated with (+/−) 3,4-methylenedioxymethamphetamine seven years previously: factors influencing abnormal recovery. J Neurosci 19:5096–5107
McLane MW, McCann U, Ricaurte G (2011) Identifying the serotonin transporter signal in Western blot studies of the neurotoxic potential of MDMA and related drugs. Synapse 65:1368–1372
Thomas DM, Dowgiert J, Geddes TJ et al (2004) Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett 367:349–354
Xie T, Tong L, McLane MW et al (2006) Loss of serotonin transporter protein after MDMA and other ring-substituted amphetamines. Neuropsychopharmacology 31:2639–2651
Nielsen K, Brask D, Knudsen GM, Aznar S (2006) Immunodetection of the serotonin transporter protein is a more valid marker for serotonergic fibers than serotonin. Synapse 59:270–276
Capela JP, Araujo SD, Costa VM et al (2013) The neurotoxicity of hallucinogenic amphetamines in primary cultures of hippocampal neurons. Neurotoxicology 34:254–263
Fukumura M, Cappon GD, Pu C, Broening HW, Vorhees CV (1998) A single dose model of methamphetamine-induced neurotoxicity in rats: effects on neostriatal monoamines and glial fibrillary acidic protein. Brain Res 806:1–7
de Cristóbal J, García-García L, Delgado M et al (2014) A longitudinal FDG-PET study of transgenic mice overexpressing GSK-3β in the brain. Curr Alzheim Res 11:175–181
Jupp B, O’Brien TJ (2007) Application of coregistration for imaging of animal models of epilepsy. Epilepsia 48(Suppl 4):82–89
Paxinos G, Watson C (2005) The rat brain in stereotaxic coordinates. Elsevier Academic, San Diego
Rehavi M, Roz N, Weizman A (2002) Chronic clozapine, but not haloperidol, treatment affects rat brain vesicular monoamine transporter 2. Eur Neuropsychopharmacol 12:261–268
Schmued LC, Stowers CC, Scallet AC, Xu L (2005) Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31
Jacobs BL, Azmitia EC (1992) Structure and function of the brain serotonin system. Physiol Rev 72:165–229
Kanarik M, Matrov D, Kõiv K et al (2008) Changes in regional long-term oxidative metabolism induced by partial serotonergic denervation and chronic variable stress in rat brain. Neurochem Int 52:432–437
Tõnissaar M, Mällo T, Eller M et al (2008) Rat behavior after chronic variable stress and partial lesioning of 5-HT-ergic neurotransmission: effects of citalopram. Prog Neuropsychopharmacol Biol Psychiatry 32:164–177
Deleye S, Verhaeghe J, Wyffels L et al (2014) Towards a reproducible protocol for repetitive and semi-quantitative rat brain imaging with (18) F-FDG: exemplified in a memantine pharmacological challenge. Neuroimage 96:276–287
Biezonski DK, Meyer JS (2011) The nature of 3, 4-methylenedioxymethamphetamine (MDMA)-induced serotonergic dysfunction: evidence for and against the neurodegeneration hypothesis. Curr Neuropharmacol 9:84–90
Freo U, Larson DM, Tolliver T et al (1991) Parachloroamphetamine selectively alters regional cerebral metabolic responses to the serotonergic agonist metachlorophenylpiperazine in rats. Brain Res 544:17–25
Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 23:570–577
Martínez-Clemente J, López-Arnau R, Abad S et al (2014) Dose and time-dependent selective neurotoxicity induced by mephedrone in mice. PLoS One 9:e99002. doi:10.1371/journal.pone.0099002
Thomas DM, Kuhn DM (2005) Cyclooxygenase-2 is an obligatory factor in methamphetamine-induced neurotoxicity. J Pharmacol Exp Ther 313:870–876
Gao Z, Zhu Q, Zhang Y et al (2013) Reciprocal modulation between microglia and astrocyte in reactive gliosis following the CNS injury. Mol Neurobiol 48:690–701
Mamounas LA, Mullen CA, O’Hearn E, Molliver ME (1991) Dual serotoninergic projections to forebrain in the rat: morphologically distinct 5-HT axon terminals exhibit differential vulnerability to neurotoxic amphetamine derivatives. J Comp Neurol 314:558–586
Riezzo I, Cerretani D, Fiore C et al (2010) Enzymatic–nonenzymatic cellular antioxidant defense systems response and immunohistochemical detection of MDMA, VMAT2, HSP70, and apoptosis as biomarkers for MDMA (Ecstasy) neurotoxicity. J Neurosci Res 88:905–916
Mahar I, Bambico FR, Mechawar N, Nobrega JN (2014) Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci Biobehav Rev 38:173–192
Dooley AE, Pappas IS, Parnavelas JG (1997) Serotonin promotes the survival of cortical glutamatergic neurons in vitro. Exp Neurol 148:205–214
Stankovski L, Alvarez C, Ouimet T et al (2007) Developmental cell death is enhanced in the cerebral cortex of mice lacking the brain vesicular monoamine transporter. J Neurosci 27:1315–1324
Acknowledgments
We thank Dr. José Luis López-Lacomba, director of the Instituto de Estudios Biofuncionales UCM, and Dr. Enrique Martínez-Campos for their assistance in the use of the fluorescence microscope.
This work was financially supported by grants from the Spanish Ministerio de Ciencia e Innovación (SAF2009-09020) and Comunidad de Madrid (I2M2, S2010/BMD-2349).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
García-García, L., Delgado, M., Al-Sayed, A.A. et al. In Vivo [18F] FDG PET Imaging Reveals that p-Chloroamphetamine Neurotoxicity is Associated with Long-Term Cortical and Hippocampal Hypometabolism. Mol Imaging Biol 17, 239–247 (2015). https://doi.org/10.1007/s11307-014-0794-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-014-0794-4