
Abstract
Background
Until recently, plant metabolomics have provided a deep understanding on the metabolic regulation in individual plants as experimental units. The application of these techniques to agricultural systems subjected to more complex interactions is a step towards the implementation of translational metabolomics in crop breeding.
Aim of Review
We present here a review paper discussing advances in the knowledge reached in the last years derived from the application of metabolomic techniques that evolved from biomarker discovery to improve crop yield and quality.
Key Scientific Concepts of Review
Translational metabolomics applied to crop breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
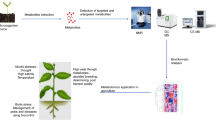
The ideal conditions to maximize crop yield is the adjustment of all plant cell metabolic events with the environmental conditions. Although optimization of plant architecture has been suggested as a focus to maximize impact on yield (Jiang et al. 2013; Schauer et al. 2006, 2008; Cai et al. 2016), the capacity of a crop to produce and redistribute biomass is a direct consequence of the relationship between the nutrient availability with the source-sink metabolic activity. The ability to manipulate this complex signalling network is limited by our knowledge of the metabolic pathways involved in these processes (reviewed by Rossi et al. 2015 and; Sonnewald and Fernie 2018). The integration of different metabolomic platforms allowing the identification and quantification of known and unknown metabolites remains a non-trivial step in deciphering complete metabolomes (Alseekh and Fernie 2018). However, knowing and understanding the control of plant metabolism is not only critical for maximizing crop yield but more importantly for attending human nutrition aspects which have, to date, been disregarded in crop plant breeding programs. In this review we discuss knowledge advances reached in the last years that tackle the above aspects of crop yield and quality derived from the application of metabolomic techniques, based on mass spectrometry (MS) and nuclear magnetic resonance (1H NMR). Furthermore, we discuss how the obtained data when analyzed with bioinformatic tools is facilitating the development of metabolomics beyond applications in biomarker discovery towards meaningful contributions to the improvement of crops. The illustrative figure shown in panel 1 summarizes all topics covered in this minireview and is intended to guide the reader throughout the paper (Fig. 1).

Overview of the boarded topics in plant metabolomics advances toward crop yield and quality improvement. Each box refers to a specific section addressed in the text
2 Elucidation and discovery of metabolic pathways
The dynamism of plant metabolism across plant phenological stages led to put efforts in developing sophisticated integration methods of plant metabolomics data with metabolic networks (Topfer et al. 2015). Without requiring a priori knowledge of biochemical reactions, correlation based network analyses represent an attractive approach to study the mode of interaction of known metabolites, to propose unknown candidates for pathways elucidation (Sonawane et al. 2016) and to identify association between metabolites and yield related traits (Schauer et al. 2006; Lisec et al. 2011).
The biosynthesis and endogenous turnover of dhurrin in developing sorghum grains was recently studied by metabolite profiling (LC-MS/MS) and time-resolved transcriptome analyses (Nielsen et al. 2016). The results presented in this article reveal the existence of two endogenous dhurrin turnover pathways in sorghum, identify genes putatively involved in these transformations and show that dhurrin, in addition to its insect deterrent properties, may serve as a storage form of reduced nitrogen (N).
GC-MS-based sterol profiling recently led to discover the entire route of cholesterol biosynthesis in plants, specifically in tomato where the pathway is a key precursor for thousands of bioactive plant metabolites, including the well-known Solanum steroidal glycoalkaloids (Sonawane et al. 2016). Despite the importance of this work for understanding plant metabolism evolution; the finding brings a new dimension for the economic exploitation of the plant secondary metabolism derived compounds. More recently, combining metabolomics with multi-omics tools Zhu et al. (2018) have identified alleles of genes associated with reduction of anti-nutritional steroidal glycoalkaloids during the processes of tomato domestication and improvement. The widespread adoption of tomato for plant metabolomics (Tohge and Fernie 2015) coupled to the diversity of the Solanum genus and the availability of hundreds of genome sequences from cultivars, landraces and wild species (TGSC 2012, Bolger et al. 2014; Lin et al. 2014; Aflitos et al. 2014) embrace a largely untapped resource that harbor a tremendous potential impact to improve this species. The collection of similar scales of data for rice and maize are already available (Kusano et al. 2015; Venkatesh et al. 2016) and it seems reasonable to anticipate many more will shortly be released, rendering this an approach which will soon be available for many of our crops.
Returning to tomato, Tieman et al. (2017) combining tasting panels with the identification of a large list of volatile organic compounds in a diverse set of modern and ancient tomato varieties identified flavorful components that have been lost along tomato breeding, paving the way for their recovery. In this study they were able to classify tomato heirloom varieties by their contents of nutritionally important molecules and based on their genetic background. Such variability is suitable to harness association mapping for metabolic quality traits using this germplasm as an experimental population and facilitate breeding for quality-related compounds in tomato fruits (Baldina et al. 2016). The application of metabolomic techniques to identify and reintroduce the lost diversity into elite varieties provides an opportunity to revert the genetic bottleneck enforced by domestication and breeding in this species (López et al. 2015; Cortina et al. 2018; D’Angelo et al. 2018).
3 Primary metabolite profiling in crop breeding and management
Carbon/nitrogen (C/N) status regulates, through a complex signalling network, the source-sink relationship that end-up in biomass partitioning, ultimately impacting yield (see reviews from Fukushima and Kusano 2014 and; Rossi et al. 2015). The consequences of primary metabolism manipulation on biomass partitioning have been reported decades ago (Stitt and Schulze 1994). Furthermore, metabolic markers diagnostic of the plant N status are commonly used for the sustainable fertilization in wheat, barley and maize crops (Justes et al. 1997; Uddling et al. 2008). However, this practice is not straightforward since metabolite traits are associated with the genotype, the environment, the interactions between them and other traits. These properties, however, render metabolic markers ideal tools for identifying metabolite quantitative trait loci (mQTLs) and finding the underlying causal genes (Schauer et al. 2006, 2008; Gong et al. 2013, see the recent review of; Kumar et al. 2017).
Optimizing N-application in agriculture is especially relevant both from an economic and environmental perspective. Application of post-genomic tools in combination with network analysis studies, have been largely used to elucidate the regulation of N metabolism and its coordination with C metabolism in crop species, such as rice (Li et al. 2016a), tomato (Benard et al. 2015), maize (Wen et al. 2015, 2018), legumes (Ramalingam et al. 2015), soybean (Yang et al. 2017) and forest trees (Gargallo-Garriga et al. 2017). Beyond this, Beatty et al. (2016) applied the metabolic profiling and introduced a theoretical framework for a whole plant model of N use efficiency, describing the complex flow of N in a real world agricultural setting using metabolomic data, such as isotope labeling rates and analyses of nutrient uptake, to refine the proposed model.
N-use efficiency becomes especially relevant for those crops requiring large amount of fertilizer to obtain maximum yield (Raun et al. 2002). Basic knowledge concerning the response of the plants to various growth and environmental conditions are needed. Amiour et al. (2012) conducted a metabolomic analysis in maize leaves subjected to long-term N deprivation. Their results showed that N availability plays an important role by controlling both primary and secondary leaf C metabolism during vegetative development. A similar response has been reported for several other abiotic stresses (Stoop et al. 1996) such as high and low temperature (Kaplan et al. 2004). Furthermore, the N limitation showed a direct effect on plant cell wall biosynthesis intermediates (arabinose and galactose) and the amount of starch were severely affected in leaves. Thus, these results have direct applications when deciding crop cultural practices such as production destiny: grain or forage (Amiour et al. 2012). On the other hand, the excess apply of N fertilizer in attempts to boost yield by farmers led to superfluous N lost from the plant-soil system, causing environmental damage, and the inhibition of K+ uptake. Kong et al. (2017) used metabolic data to demonstrate that excessive N application in wheat caused less accumulation of many non-enzymatic antioxidants, which associated with less N remobilization, lower resistance to pathogen attacks, decreased resistance to heat stress led to reduced grain yield.
Partitioning to sink organs during developmental leaf senesce was studied from transcriptomics and metabolomics angles. Mitochondrial pathways were revealed as essential in providing the C backbone for N remobilization, a process of utmost importance not only to ensure the quality of grain in crop species, but also representing a valuable target for minimizing the negative ecological impact of the current fertilization practices (Chrobok et al. 2016).
The glutamine/glutamate synthases (GS/GOGAT) cycle is the almost exclusive pathway by which ammonium is assimilated by plants (Lancien et al. 2000). However, the regulatory role of this cycle in the C/N balance under crop cultivation conditions is not well understood. Yang et al. (2016), by combining mutant analyses with metabolic profiling, reported a rice ferredoxin-dependent-GOGAT essential for plant growth and development by modulating N assimilation and C/N balance, pointing this enzyme as a breeding target. A similar study also conducted in rice observed that the levels of glutamate and aspartate are the best indicator of stresses related to N limitation and high plant density conditions (Misyura et al. 2014). The shuttling of 2-oxoglutarate into the production of these amino acids is one of the key indicators of biomass accumulation as opposed to ATP production (Sweetlove et al. 2010).
A comparison between glycine and nitrate as N supply in lettuce showed that glycine treatment decreased the growth and final biomass of the plants. Metabolite profile analyses showed that pyruvic acid content was lower in glycine-treated lettuce than in the control. However, glycine supply increased the accumulation of glycosylated flavonoids, ascorbate and most amino acids but reduced the levels of phenolic acids and tricarboxylic acid (TCA) cycle intermediates (Yang et al. 2018). Thus, this fertilization practice can be used as a potential strategy to obtain vegetable products of higher nutraceutical value.
In maize, Rao et al. (2014) generated a comprehensive metabolic map by integrating transcriptomic, proteomic and metabolomic data providing a unique tool for maize breeding towards improved kernel quality and yield. Interestingly, the detection of two anti-nutrient compounds, inositol hexaphosphate (IP6) and sorbitol in high quantities were reported. These finding could be used to improve kernel quality and yield either by screening for mutants or by engineering these pathways. Another recent metabolomics approach determined a clear relationship between certain primary metabolites (such as sucrose, arabitol and histidine) and maize yield by quantifying the phloem sap composition of a diverse panel of maize inbred lines (Yesbergenova-Cuny et al. 2016). In a similar vein, the role of primary metabolites in stomatal movement has been hypothesized for many years. A metabolomic study (Misra et al. 2015) has shown that sugars such as sucrose, glucose and fructose, do not exert an apoplastic osmotic effect on these cells, but rather are sensed by hexokinase to stimulate stomatal closure, thus coordinating photosynthesis and sugar levels with transpiration (Kelly et al. 2013). These studies highlighted the importance of selecting genotypes with cool leaf temperature which in some species is associated with longer stomatal opening and higher yield.
Light regimes (quality and photoperiod) are inductive for production of desirable metabolites, allowing plausible manipulation targets to enhance nutritional capacity of crop plants. Lakshmanan et al. (2015) combining transcript and metabolite profiles with constraint-based modeling showed the feasibility of metabolism modulation by increasing the accumulation of molecules of high nutritional value, such as terpenoids and phenolic compounds, under different light conditions.
Many attempts have been made to link primary metabolites abundance to the mechanistic relationships that regulate fruit quality (see for examples; Chambers et al. 2014; Desnoues et al. 2014; Tohge et al. 2015; Vimolmangkang et al. 2016). Li et al. (2016b) have recently reported proteomic and metabolomics analyses in apple showing that both sorbitol and sucrose are synthesized in leaves and then metabolized in fruits where at least 80% of the total C flux goes through fructose resulting in higher organoleptic quality.
Lipid profiles of plant crop species has been delayed mainly for technical reasons. MS and MS/MS spectral information obtained as lipidomic data is being of primary importance to elucidate biosynthetic mechanisms of lipid production and metabolism. Examples of this are the reports of Okazaki et al. (2013) and Shimojima et al. (2015). The first one applied lipidomic analyses to a series of mutants subjected to phosphorous (P) starvation. This allowed the elucidation of a new plastidial biosynthetic pathway which results in the accumulation of a novel class of plant lipid: glucuronosyldiacylglycerol. The second paper also showed that P-starvation induced oil accumulation in vegetative tissues without activating transcription of triacylglyceride (TAG) biosynthetic genes.
Lipid metabolism has been reported to be affected by N deficiency. In tea plants (Camellia sinensis L.), as in many other volatile-producing species, lipids are precursors of flavor/aroma compounds. A recent lipidomic report by Liu et al. (2017) shows that under low N levels tea plants can remobilize the C stored in TAG to maintain C/N balance, with the consequent impact on tea quality. Conclusions from this report highlight the importance of appropriate application of N fertilizer to balance lipid metabolism and the formation of flavor/aroma origin compounds. Another link between lipid metabolism and quality has been recently reported in tomato. A combination of QTL mapping, metabolic and transcriptomic data led to the identification and cloning of novel lipid–genes with major effects on the production of multiple fatty-acid-derived flavor volatiles. These compounds are positively correlated with consumer liking and crucial for key agronomical traits to improve the quality of this crop (Garbowicz et al. 2018).
4 Metabolomics and stress responses in crop plant species
A very prolific field for plant metabolomic applications is stress response-related studies; a plethora of works have been published in the last years boarding both plant biotic and abiotic stresses. However, most of them are focused in the discovery of new biomarkers and have been reviewed recently by Nakabayashi and Saito (2015), Abdelrahman et al. (2017) and Ahmad et al. (2017). Even more, a comprehensive compendium of abiotic stress responses, at least for primary metabolism, was published years ago by Obata and Fernie (2012).
Anyhow, here below we briefly discuss the latest advances that link metabolomics with stress responses which may point out important pathways to be boarded in crop breeding.
Kim et al. (2017) reported an essential drought-responsive network in which plants trigger a dynamic metabolic flux conversion from glycolysis into acetate synthesis to stimulate the jasmonate (JA) signalling pathway conferring drought tolerance. This novel acetate function is evolutionarily conserved across different plant species as a survival strategy against environmental changes. The findings highlight the role of acetate as orchestrator connecting primary metabolism, epigenetic regulation and hormone signalling by which plants adapt to drought. Another integrative approach was taken by Moschen et al. (2017) in sunflower to reveals hub transcription factors involved in drought stress response. Vital et al. (2017) described a novel phospho and redox proteomic analyses, combined with metabolomics data that revealed the importance of posttranslational regulation mechanisms in sugarcane under drought stress. The shift to soluble sugar, secondary metabolite production and activation of ROS eliminating processes in response to drought were the most noticeable results of this work. In the non-model plant, Phoenix dactylifera (date palm), heat and drought stresses induced carbohydrate metabolism and cell wall biogenesis (Safronov et al. 2017). Drought stress response and photorespiration was also studied by metabolomic approaches in maize (Obata et al. 2015) and in sorghum (Ogbaga et al. 2016). Variations in soluble sugars and amino acids in roots of salt-tolerant barley plants under stress was reported by Shelden et al. (2016). In rice, flavone-glycosides profiles were surveyed for protectants of abiotic stress and herbivores (Matsuda et al. 2012; Peng et al. 2017).
Wild species of Tibetan barley were screened for salt tolerance by applying ionic, metabolomics and proteomics approaches (Shen et al. 2016). Accessions differed greatly in salt tolerance and the stress caused significant reduction in glycolysis metabolites and elevated levels of TCA cycle intermediates. More recently, Quan et al. (2017) by using an ionomic strategy reported that microelement contents in shoots of Tibetan barley species decreased under low N stress being Fe and Cu the most dramatically affected. In soybean seedlings were also found metabolic differences when comparing wild and cultivated genotypes under salt stress conditions (Zhang et al. 2016). Results suggested that the energy generation from β-oxidation, glycolysis and the TCA cycle plays important roles under alkali-salt stress. Also in soybean, metabolic profiles were applied to flooding-tolerant mutants and found that accumulated fructose might play a role in initial flooding tolerance through regulation of hexokinase and phosphofructokinase (Wang et al. 2017).
Biotic stresses triggers metabolic responses that vary depending on the kind of interactions established between the plant cell and the pathogen. Results are extremely difficult to interpret mainly due to technical limitations identifying the source of the metabolites being measured. Nevertheless, the detection of altered host metabolic factors is contributing to highlight those pathways suitable for the design of engineered disease resistance. Bellow, we discuss few recent papers where metabolic processes associated to the plant pathogen responses were described.
Metabolic variations in citrus rootstock cultivars associated with different responses to Huanglongbing (HLB) showed that tolerance did not appear to be associated with accumulation of higher amounts of protective metabolites in response to infection (Albrecht et al. 2016). However, the study revealed that lower availability of specific sugars necessary for survival of the pathogen may be a contributing factor in the decreased disease severity observed in the tolerant cultivars. A comprehensive metabolomic investigation combining GC- and UHPLC-HR-MS profiles was applied to leaf rust infection in barley, rice and wheat to understand durable anti-fungal resistance in cereals provided by Lr34 (a leaf rust resistance gene) (Bucher et al. 2017). An integrated RNAseq 1H-NMR approach identified candidate genes and metabolites that can be used in soybean breeding for resistance to Rizoctonia solani AG1-IA infection. Additionally, it was found that alcohol and its associated gene product alcohol dehydrogenase (ADH) may have important roles in plant resistance to this pathogen (Copley et al. 2017). Another metabolomics approach was used to study the impacts of Rhizobium infection on seed productivity and protection of Pisum sativum upon disease caused by Didymella pinodes. This work showed increased redox, cell wall associated-metabolites and secondary metabolites such as the seed triterpenoid soyasapogenol (Ranjbar Sistani et al. 2017).
5 Advances in implementing translational metabolomics in crop yield and quality
Metabolomic techniques were proven to be useful tools for revealing mechanisms associated to biomass accumulation, photosynthesis and yield performance in many agricultural important plants (Hu et al. 2014). There are two ways to meet the projected increased demand for food production: allocate more land for crops and/or increase the yield of existing planted areas (Pretty and Bharucha 2014). The latter is considered a more sustainable option to feed the growing population (FAO 2009, 2017). However, the required yield improvements and timeframe can no longer be achieved using traditional means because of changing weather patterns (Powell and Reinhard 2016). Under this section we highlight recent papers showing direct links between basic metabolism knowledge with the understanding of two very complex traits such as yield and quality.
Recent studies combined metabolite profiling and genetic analyses in crop species identified dozens of genomic regions associated with variations in the contents of Brassica seeds glucosinolates (Feng et al. 2012), rice seed lipids (Ying et al. 2012), maize grain carotenoids (Owens et al. 2014) and tocochromanols (Lipka et al. 2013; Diepenbrock et al. 2017; Wang et al. 2018) and hundreds of not linked loci for variations in the contents of intermediate compounds from primary and secondary metabolic pathways.
Metabolomic studies have also afforded greater insights in fruit biology specially related to ripening and quality in tomato (Tohge and Fernie 2015; Upadhyaya et al. 2017; Perez-Fons et al. 2014; Quadrana et al. 2014; Di Paola; Naranjo et al. 2016), apple (Cebulj et al. 2017; Hatoum et al. 2014, 2016), kiwifruit (Nardozza et al. 2013; Ainalidou et al. 2016), orange (Pan et al. 2012), grape (Domingos et al. 2016; Son et al. 2009; Cuadros-Inostroza et al. 2010; Flamini et al. 2015), pear (Oikawa et al. 2015) and strawberry (Mikulic-Petkovsek et al. 2013; Nagpala et al. 2016). Metabolite profiling coupled with Genome-wide association studies (GWASs) allowed the identification of 44 SNP loci associated to 19 metabolic traits and provided candidate genes involved in the genetic architecture of tomato fruit metabolic traits (Sauvage et al. 2014). A combination of 1H-NMR and GC–MS methods allowed the selection of tomato elite recombinant lines by linking metabolites and agronomic traits and by calculating their heritability values (López et al. 2015). A broader metabolome-based GWAS (mGWAS) have recently identified those molecules which were negatively selected during the history of breeding (Zhu et al. 2018), providing major insights into how this process changed the tomato metabolome, a knowledge base for fruit quality improvement.
In legume, metabolomics studies remain limited to model species. The influence of rhizobial node factor in Medicago was investigated and a significant effect on the level of oxylipins was reported, suggesting that this pathway facilitates Nod factor signalling during the early stages of symbiosis (Zhang et al. 2012).
In cereals, various authors reported on natural metabolic variations in rice (Kusano et al. 2007, 2015; Redestig et al. 2011; Gong et al. 2013; Hu et al. 2014, 2016; Chen et al. 2014; Okazaki and Saito 2016) and in maize (Matsuda et al. 2012; Wen et al. 2015, 2018; Venkatesh et al. 2016). Chen et al. (2014) have presented a comparative mQTL fine mapping between rice and maize. Moreover, this work provides evidences on metabotype-phenotype linkage in rice grain and discovers novel relationships between pathways, structural genes and agronomically important phenotypes. A non-targeted metabolomic analysis of Sorghum bicolor leaf tissue showed that 956 of 1,181 metabolites varied among different inbred lines. Both univariate and multivariate analyses determined relationships between metabolites and morpho-physiological traits, and 384 metabolites correlated with at least one trait, including many secondary metabolites such as glycosylated flavonoids and chlorogenate. The later and shikimate showed to be associated with photosynthesis, early plant growth and final biomass accumulation (Turner et al. 2016). Chlorogenate contents were also found to be associated with larger kernels in a study with maize lines by Cañas et al. (2017). This study combines metabolomic, biochemical, fluxomic and metabolic modelling approaches to identify maize ideotypes with high grain yield potential.
Combining genomic data (SNPs) with leaf metabolomic data, Riedelsheimer et al. (2012a, b) showed that metabolic profiles of diverse maize inbred lines allow prediction of their testcross performance in multi-location field trials, which provides a reliable screening of large collections of diverse inbred lines for their potential to create superior hybrids. Moreover, mGWAS explained up to 32.0% of the observed genetic variance. Remarkably, the use of metabolites as yield predictor variables showed only 6.7% lower accuracy than SNPs. By contrast, in wheat, Zhao et al. (2015) failed to improve prediction of hybrid performance by using metabolome data. The authors explain this failure due to the sampling of flag leafs under field conditions. Thus, further research is needed to investigate the option of improving prediction accuracy through metabolite profiling under strictly controlled conditions. In rice, hybrid yield prediction was assessed by means of a LC/UFLC-MS/MS metabolomic platform and the identification of 76 significant metabolites with high predictive values (Xu et al. 2016). Authors hypothesize that each metabolite represents a biologically built-in genetic network for yield. The fact that metabolomic prediction is better, in this case, than genomic prediction for yield of hybrid rice is surprising, considering: (i) the small number of metabolites relative to the large number of SNPs; and (ii) the metabolic profile being a snapshot at a specific development stage.
Determine whether the rate of biomass accumulation correlates with metabolic composition was a matter of significant efforts at the onset of metabolomics applied to plant crop species, especially in grasses as possible biomass energy crops (Meyer et al. 2007). More recently, Ghaffari et al. (2016) pointed the metabolites and enzymes involved in central metabolism that determine biomass accumulation at the vegetative and reproductive stages.
This body of knowledge encouraged and allowed the application of metabolomic techniques to less known plant crop species. An example is the paper from Tuttle et al. (2015) in cotton fiber; where 206 compounds were detected by applying ion-optimized GC-MS. The glutathione pathway related to amino acids and ROS management, as well as galactose pathway were remarked as critical points for quality of the fibers. It is also shown that enhanced ascorbate biosynthesis and recycling contributes to extend the fiber elongation period and thus, results in longer final length. Another example of metabolomic applications to even more exotic crop species is given in the paper published by Price et al. (2017). Yam (Dioscorea spp.) production has doubled in the last years, particularly in low income food deficit countries. Combined analysis on leaf and tuber tissues identified a subset of metabolites which allow accurate species classification and highlighted the potential of predicting tuber composition from leaf profiles.
6 The importance of metabolomics for plant engineering
C4 crops have higher photosynthetic efficiency, greater biomass, and better tolerance to abiotic stress than C3 plants. Great efforts are being made in transferring C4 metabolic pathways to C3 plants. Qi et al. (2017) used transgenic wheat lines (C3) containing the maize (C4) phosphoenolpyruvate carboxylase (PEPC) gene to study the effects of high temperature on their photosynthetic physiological characteristics and related metabolic responses. Transgenic plants showed lower rate of superoxide anion production, H2O2 and malondialdehyde content and higher photosynthetic rate under high temperatures. Overall, the results of this report indicate that the maize PEPC gene enhances photochemical and antioxidant enzyme activity, upregulates the expression of photosynthesis-related genes, delays degradation of chlorophyll, changes contents of proline and other metabolites and ultimately improves wheat heat tolerance.
Metabolomic analyses have been recognized as a valuable tool in risk assessment of genetically modified plants (Harrigan et al. 2016). Recently, Benevenuto et al. (2017) challenged the presumption of molecular stability of commercially approved GM maize cultivars through a proteomic and metabolomic approach under different environmental conditions.
Plant-based vaccines is a technology that involves the introduction of genes encoding antigen proteins into the plant genome. Recombinant antigen protein expressed in transgenic plants is expected to induce specific immune responses when the plant material is consumed. A broad profiling analysis indicated that hydrophilic primary metabolite and lipid contents are able to separate transgenic rice lines expressing a modified cholera toxin B (CTB) from their corresponding wild type control, suggesting that the expression of modified CTB affected rice seed metabolic activity (Ogawa et al. 2017). This result is particularly important in cases were safety assessment are needed in substantial equivalence analyses (Llorente et al. 2011).
Translational metabolomics in crop plants has successfully rendered to the production of multifarious nutritional metabolites that have important role in human diet (Tatsis and O’Connor 2016). Metabolic engineering of plant pathways for improved human health and diet is a focus of many current plant biotechnology programmes, and there are some progress made in to engineering secondary metabolites for human health (reviewed by Davies and Espley 2013). A well-known and successful example is the Golden Rice that accumulates higher levels of pro-vitamin A, in which three carotenoid biosynthetic enzymes were engineered for simultaneous expression in rice endosperm (Moghissi et al. 2016; Ye and Beyer 2000). Following the Gold Rice success, nowadays several crop species have been targeted for vitamin biofortification such as cassava (Sayre et al. 2011), maize and potato (Bai et al. 2011). Crop engineering allowed increasing the levels of antioxidant phenylpropanoids to promote health properties in fruit and vegetable crops (Korkina 2007). Phenylpropanoid production was substantially accumulated in tomato fruits by introducing fruit-specific expression of the transcription factor AtMYB12 (Zhang et al. 2015). In addition, the accumulation of endogenous sugars and simple sugar derivatives have been successfully increased in sugar-storing sink organs such as fruits, sugarcane culms and sugarbeet tubers, via plant metabolic engineering (Patrick et al. 2013). Furthermore, metabolic engineering approaches resulted in increments if the levels of lysine in corn, folate in tomatoes and ferritin in lettuce (Yonekura-Sakakibara and Saito 2006). Despite the controversy associated with genetically modified plants, these examples prove that the nutrient content of crop plants can be improved by metabolic engineering. Thus, the application of translational metabolomics to crop species is a current challenge for the scientific community, which will impact on human health and food security.
7 Perspectives
In this paper, we reviewed recent reports demonstrating the applicability of metabolomic techniques on crop species, with trending advances in implementing translational metabolomics in breeding.
Overall, it is estimated that there are about 200,000 different metabolites in the plant kingdom (Bino et al. 2004) but, in each experiment, only a few hundred of them can be measured. It is important to remark that the few works discussed in this mini-review exemplify that, in some cases, this small proportion already predicts the yield and allow assess quality better than genomic data. If we can increase the number of metabolites to a few thousands, the predictability may be further improved.
New emerging technologies like metabolome-scale labelling and single-cell metabolomics will surely improve metabolite annotation, metabolic pathway elucidation and MS-relative quantification at the very basic level of biological organization and overcome limitations associated to the averaging effect of tissues with mixed cell types (Freund and Hegeman 2017; Misra et al. 2014). Another technology which merits special attention is the single-probe MS technique that has the potential to allow for near in situ metabolomic analyses, opening the door to targeted analyses minimizing cell manipulation and sampling artifacts, while preserving metabolic variability at the cellular level (Sun et al. 2018).
References
Abdelrahman, M., Burritt, D. J., & Tran, L. P. (2017) The use of metabolomic quantitative trait locus mapping and osmotic adjustment traits for the improvement of crop yields under environmental stresses. In Seminars in Cell & Developmental Biology. Cambridge: Academic Press
Aflitos, S., Schijlen, E., de Jong, H., de Ridder, D., Smit, S., Finkers, R., Wang, J., Zhang, G., Li, N., Mao, L., Bakker, F., Dirks, R., Breit, T., Gravendeel, B., Huits, H., Struss, D., Swanson-Wagner, R., van Leeuwen, H., van Ham, R. C., Fito, L., Guignier, L., Sevilla, M., Ellul, P., Ganko, E., Kapur, A., Reclus, E., de Geus, B., van de Geest, H., Hekkert, T. L., van Haarst, B., Smits, J., Koops, L., Sanchez-Perez, A., van Heusden, G., Visser, A. W., Quan, R., Min, Z., Liao, J., Wang, L., Wang, X., Yue, G., Yang, Z., Xu, X., Schranz, N., Smets, E., Vos, E., Rauwerda, R., Ursem, J., Schuit, R., Kerns, C., van den Berg, M., Vriezen, J., Janssen, W., Datema, A., Jahrman, E., Moquet, T., Bonnet, F., J. and Peters, S. (2014) Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. The Plant Journal: For Cell and Molecular Biology, 80, 136–148.
Ahmad, R., Jamil, S., Shahzad, M., Zörb, C., Irshad, U., Khan, N., et al. (2017). Metabolic profiling to elucidate genetic elements due to salt stress. Clean - Soil, Air, Water. https://doi.org/10.1002/clen.201600574.
Ainalidou, A., Tanou, G., Belghazi, M., Samiotaki, M., Diamantidis, G., Molassiotis, A., & Karamanoli, K. (2016). Integrated analysis of metabolites and proteins reveal aspects of the tissue-specific function of synthetic cytokinin in kiwifruit development and ripening. Journal of Proteomics, 143, 318–333.
Albrecht, U., Fiehn, O., & Bowman, K. (2016). Metabolic variations in different citrus rootstock cultivars associated with different responses to Huanglongbing. Plant Physiol Biochem, 107, 33–44.
Alseekh, S., & Fernie, A. R. (2018). Metabolomics 20 years on: What have we learned and what hurdles remain? The Plant Journal: For Cell and Molecular Biology, 94, 933–942.
Amiour, N., Imbaud, S., Clement, G., Agier, N., Zivy, M., Valot, B., Balliau, T., Armengaud, P., Quillere, I., Canas, R., Tercet-Laforgue, T., & Hirel, B. (2012). The use of metabolomics integrated with transcriptomic and proteomic studies for identifying key steps involved in the control of nitrogen metabolism in crops such as maize. Journal of Experimental Botany, 63, 5017–5033.
Bai, C., Twyman, R. M., Farré, G., Sanahuja, G., Christou, P., Capell, T., & Zhu, C. (2011). A golden era-pro-vitamin A enhancement in diverse crops. In Vitro Cellular and Developmental Biology—Plant. https://doi.org/10.1007/s11627-011-9363-6.
Baldina, S., Picarella, M. E., Troise, A. D., Pucci, A., Ruggieri, V., Ferracane, R., Barone, A., Fogliano, V., & Mazzucato, A. (2016). Metabolite profiling of italian tomato landraces with different fruit types. Frontiers in Plant Science, 7, 664.
Beatty, P., Klein, M., Fischer, J., Lewis, I., Muench, D., & Good, A. (2016). Understanding plant nitrogen metabolism through metabolomics and computational approaches. Plants, 5(4), 39. https://doi.org/10.3390/plants5040039.
Benard, C., Bernillon, S., Biais, B., Osorio, S., Maucourt, M., Ballias, P., Deborde, C., Colombie, S., Cabasson, C., Jacob, D., Vercambre, G., Gautier, H., Rolin, D., Genard, M., Fernie, A. R., Gibon, Y., & Moing, A. (2015). Metabolomic profiling in tomato reveals diel compositional changes in fruit affected by source-sink relationships. Journal of Experimental Botany, 66, 3391–3404.
Benevenuto, R. F., Agapito-Tenfen, S. Z., Vilperte, V., Wikmark, O. G., van Rensburg, P. J., & Nodari, R. O. (2017). Molecular responses of genetically modified maize to abiotic stresses as determined through proteomic and metabolomic analyses. PLoS ONE, 12, e0173069.
Bino, R. J., Hall, R. D., Fiehn, O., Kopka, J., Saito, K., Draper, J., et al. (2004). Potential of metabolomics as a functional genomics tool. Trends in Plant Science, 9(9), 418–425. https://doi.org/10.1016/j.tplants.2004.07.004.
Bolger, A., Scossa, F., Bolger, M. E., Lanz, C., Maumus, F., Tohge, T., Quesneville, H., Alseekh, S., Sorensen, I., Lichtenstein, G., Fich, E. A., Conte, M., Keller, H., Schneeberger, K., Schwacke, R., Ofner, I., Vrebalov, J., Xu, Y., Osorio, S., Aflitos, S. A., Schijlen, E., Jimenez-Gomez, J. M., Ryngajllo, M., Kimura, S., Kumar, R., Koenig, D., Headland, L. R., Maloof, J. N., Sinha, N., van Ham, R. C., Lankhorst, R. K., Mao, L., Vogel, A., Arsova, B., Panstruga, R., Fei, Z., Rose, J. K., Zamir, D., Carrari, F., Giovannoni, J. J., Weigel, D., Usadel, B., & Fernie, A. R. (2014). The genome of the stress-tolerant wild tomato species Solanum pennellii. Nature Genetics, 46, 1034–1038.
Bucher, R., Veyel, D., Willmitzer, L., Krattinger, S., Keller, B., & Bigler, L. (2017). Combined GC- and UHPLC-HR-MS based metabolomics to analyze durable anti-fungal resistance processes in cereals. CHIMIA International Journal for Chemistry, 71(4), 156–159. https://doi.org/10.2533/chimia.2017.156.
Cai, G., Yang, Q., Chen, H., Yang, Q., Zhang, C., Fan, C., & Zhou, Y. (2016). Genetic dissection of plant architecture and yield-related traits in Brassica napus. Scientific Reports, 6, 21625. https://doi.org/10.1038/srep21625.
Cañas, R. A., Yesbergenova-Cuny, Z., Simons, M., Chardon, F., Armengaud, P., Quillere, I., Cukier, C., Gibon, Y., Limami, A. M., Nicolas, S., Brule, L., Lea, P. J., Maranas, C. D., & Hirel, B. (2017). Exploiting the genetic diversity of maize using a combined metabolomic, enzyme activity profiling, and metabolic modeling approach to link leaf physiology to kernel yield. The Plant Cell, 29, 919–943.
Cebulj, A., Cunja, V., Mikulic-Petkovsek, M., & Veberic, R. (2017). Importance of metabolite distribution in apple fruit. Scientia Horticulturae, 214, 214–220.
Chambers, A. H., Pillet, J., Plotto, A., Bai, J., Whitaker, V. M., & Folta, K. M. (2014). Identification of a strawberry flavor gene candidate using an integrated genetic-genomic-analytical chemistry approach. BMC Genomics, 15, 217. https://doi.org/10.1186/1471-2164-15-217.
Chen, W., Gao, Y., Xie, W., Gong, L., Lu, K., Wang, W., Li, Y., Liu, X., Zhang, H., Dong, H., Zhang, W., Zhang, L., Yu, S., Wang, G., Lian, X., & Luo, J. (2014). Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nature Genetics, 46, 714–721.
Chrobok, D., Law, S. R., Brouwer, B., Linden, P., Ziolkowska, A., Liebsch, D., Narsai, R., Szal, B., Moritz, T., Rouhier, N., Whelan, J., Gardestrom, P., & Keech, O. (2016). Dissecting the metabolic role of mitochondria during developmental leaf senescence. Plant Physiology, 172, 2132–2153.
Copley, T. R., Duggavathi, R., & Jabaji, S. (2017). The transcriptional landscape of Rhizoctonia solani AG1-IA during infection of soybean as defined by RNA-sEq. PLoS ONE, 12, e0184095.
Cortina, P. R., Santiago, A. N., Sance, M. M., Peralta, I. E., Carrari, F., & Asis, R. (2018). Neuronal network analyses reveal novel associations between volatile organic compounds and sensory properties of tomato fruits. Metabolomics, 14, 57.
Cuadros-Inostroza, A., Giavalisco, P., Hummel, J., Eckardt, A., Willmitzer, L., & Pena-Cortes, H. (2010). Discrimination of wine attributes by metabolome analysis. Analytical Chemistry, 82, 3573–3580.
D’Angelo, M., Zanor, M. I., Sance, M., Cortina, P. R., Boggio, S. B., Asprelli, P., Carrari, F., Santiago, A. N., Asis, R., Peralta, I. E., & Valle, E. M. (2018). Contrasting metabolic profiles of tasty Andean varieties of tomato fruit in comparison with commercial ones. Journal of the Science of Food and Agriculture, 98, 4128–4134.
Davies, K. M., & Espley, R. V. (2013). Opportunities and challenges for metabolic engineering of secondary metabolite pathways for improved human health characters in fruit and vegetable crops. New Zealand Journal of Crop and Horticultural Science, 41(3), 154–177. https://doi.org/10.1080/01140671.2013.793730.
Desnoues, E., Gibon, Y., Baldazzi, V., Signoret, V., Génard, M., & Quilot-Turion, B. (2014). Profiling sugar metabolism during fruit development in a peach progeny with different fructose-to-glucose ratios. BMC Plant Biology, 14(1), 336. https://doi.org/10.1186/s12870-014-0336-x-.
Di Paola Naranjo, R. D., Otaiza, R. D., Saragusti, S., Baroni, A. C., Carranza, V., Peralta, AdelV., I. E., et al (2016). Hydrophilic antioxidants from Andean tomato landraces assessed by their bioactivities in vitro and in vivo. Food Chemistry, 206, 146–155. https://doi.org/10.1016/j.foodchem.2016.03.027.
Diepenbrock, C. H., Kandianis, C. B., Lipka, A. E., Magallanes-Lundback, M., Vaillancourt, B., Gongora-Castillo, E., Wallace, J. G., Cepela, J., Mesberg, A., Bradbury, P. J., Ilut, D. C., Mateos-Hernandez, M., Hamilton, J., Owens, B. F., Tiede, T., Buckler, E. S., Rocheford, T., Buell, C. R., Gore, M. A., & DellaPenna, D. (2017). Novel loci underlie natural variation in vitamin E levels in maize grain. The Plant Cell, 29, 2374–2392.
Domingos, S., Fino, J., Cardoso, V., Sanchez, C., Ramalho, J. C., Larcher, R., Paulo, O. S., Oliveira, C. M., & Goulao, L. F. (2016). Shared and divergent pathways for flower abscission are triggered by gibberellic acid and carbon starvation in seedless Vitis vinifera L. BMC Plant Biology, 16, 38.
FAO. Food and Agriculture Organisation. (2009). How to feed the world in 2050. Insights from an Expert Meeting at FAO, 2050(1), 1–35. https://doi.org/10.1111/j.1728-4457.2009.00312.x.
FAO. Food and Agriculture Organisation. (2017). The future of food and agriculture: Trends and challenges. http://www.fao.org/3/a-i6583e.pdf.
Feng, J., Long, Y., Shi, L., Shi, J., Barker, G., & Meng, J. (2012). Characterization of metabolite quantitative trait loci and metabolic networks that control glucosinolate concentration in the seeds and leaves of Brassica napus. The New Phytologist, 193, 96–108.
Flamini, R., De Rosso, M., & Bavaresco, L. (2015) Study of grape polyphenols by liquid chromatography-high-resolution mass spectrometry (UHPLC/QTOF) and suspect screening analysis. Journal of Analytical Methods in Chemistry, 2015, 350259.
Freund, D. M., & Hegeman, A. D. (2017). Recent advances in stable isotope-enabled mass spectrometry-based plant metabolomics. Current Opinion in Biotechnology, 43, 41–48.
Fukushima, A., & Kusano, M. (2014). A network perspective on nitrogen metabolism from model to crop plants using integrated “omics” approaches. Journal of Experimental Botany, 65(19), 5619–5630. https://doi.org/10.1093/jxb/eru322.
Garbowicz, K., Liu, Z., Alseekh, S., Tieman, D., Taylor, M., Kuhalskaya, A., Ofner, I., Zamir, D., Klee, H. J., Fernie, A. R., & Brotman, Y. (2018) Quantitative trait loci analysis identifies a prominent gene involved in the production of fatty-acid-derived flavor volatiles in tomato. Molecular Plant. S1674-2052(18)30190-4.
Gargallo-Garriga, A., Ayala-Roque, M., Sardans, J., Bartrons, M., Granda, V., Sigurdsson, B. D., Leblans, N. I. W., Oravec, M., Urban, O., Janssens, I. A., & Penuelas, J. (2017) Impact of soil warming on the plant metabolome of icelandic grasslands. Metabolites, 7.
Ghaffari, M. R., Shahinnia, F., Usadel, B., Junker, B., Schreiber, F., Sreenivasulu, N., & Hajirezaei, M. R. (2016). The metabolic signature of biomass formation in barley. Plant & Cell Physiology, 57, 1943–1960.
Gong, L., Chen, W., Gao, Y., Liu, X., Zhang, H., Xu, C., Yu, S., Zhang, Q., & Luo, J. (2013). Genetic analysis of the metabolome exemplified using a rice population. Proceedings of the National Academy of Sciences of the United States of America, 110, 20320–20325.
Harrigan, G. G., Venkatesh, T. V., Leibman, M., Blankenship, J., Perez, T., Halls, S., Chassy, A. W., Fiehn, O., Xu, Y., & Goodacre, R. (2016). Evaluation of metabolomics profiles of grain from maize hybrids derived from near-isogenic GM positive and negative segregant inbreds demonstrates that observed differences cannot be attributed unequivocally to the GM trait. Metabolomics, 12, 82.
Hatoum, D., Annaratone, C., Hertog, M.L.A.T.M., Geeraerd, A. H., & Nicolai, B. M. (2014). Targeted metabolomics study of ‘Braeburn’ apples during long-term storage. Postharvest Biology and Technology, 96, 33–41.
Hatoum, D., Hertog, M. L. A. T. M., Geeraerd, A. H., & Nicolai, B. M. (2016). Effect of browning related pre- and postharvest factors on the ‘Braeburn’ apple metabolome during CA storage. Postharvest Biology and Technology, 111, 106–116. https://doi.org/10.1016/j.postharvbio.2015.08.004.
Hu, C., Shi, J., Quan, S., Cui, B., Kleessen, S., Nikoloski, Z., Tohge, T., Alexander, D., Guo, L., Lin, H., Wang, J., Cui, X., Rao, J., Luo, Q., Zhao, X., Fernie, A. R., & Zhang, D. (2014). Metabolic variation between japonica and indica rice cultivars as revealed by non-targeted metabolomics. Scientific Reports, 4, 5067.
Hu, C., Tohge, T., Chan, S.-A., Song, Y., Rao, J., Cui, B., et al. (2016). Identification of conserved and diverse metabolic shifts during rice grain development. Scientific Reports, 6, 20942. https://doi.org/10.1038/srep20942.
Jiang, K., Liberatore, K. L., Park, S. J., Alvarez, J. P., & Lippman, Z. B. (2013). Tomato yield heterosis is triggered by a dosage sensitivity of the florigen pathway that fine-tunes shoot architecture. PLoS Genetics, 9, e1004043. https://doi.org/10.1371/journal.pgen.1004043.
Justes, E., Mary, B., & Meynard, J. M. (1997). Evaluation of a nitrate test indicator to improve the nitrogen fertilisation of winter wheat crops, diagnostic procedures for crop N management. Proceedings of a workshop held in Poitiers, France, 22–23 November, 1995 Paris, France. Institut National de la Recherche Agronomique (INRA) (pp. 93–110).
Kaplan, F., Kopka, J., Haskell, D. W., Zhao, W., Schiller, K. C., Gatzke, N., Sung, D. Y., & Guy, C. L. (2004). Exploring the temperature-stress metabolome of Arabidopsis. Plant Physiology, 136, 4159–4168.
Kelly, G., Moshelion, M., David-Schwartz, R., Halperin, O., Wallach, R., Attia, Z., Belausov, E., & Granot, D. (2013). Hexokinase mediates stomatal closure. The Plant Journal: For Cell and Molecular Biology, 75, 977–988.
Kim, J. M., To, T. K., Matsui, A., Tanoi, K., Kobayashi, N. I., Matsuda, F., Habu, Y., Ogawa, D., Sakamoto, T., Matsunaga, S., Bashir, K., Rasheed, S., Ando, M., Takeda, H., Kawaura, K., Kusano, M., Fukushima, A., Endo, T. A., Kuromori, T., Ishida, J., Morosawa, T., Tanaka, M., Torii, C., Takebayashi, Y., Sakakibara, H., Ogihara, Y., Saito, K., Shinozaki, K., Devoto, A., & Seki, M. (2017). Acetate-mediated novel survival strategy against drought in plants. Nature Plants, 3, 17097.
Kong, L., Xie, Y., Hu, L., Si, J., & Wang, Z. (2017). Excessive nitrogen application dampens antioxidant capacity and grain filling in wheat as revealed by metabolic and physiological analyses. Scientific Reports, 7, 43363.
Korkina, L. G. (2007). Phenylpropanoids as naturally occurring antioxidants: From plant defense to human health. Cellular and Molecular Biology (Noisy-le-Grand, France), 53(1), 15–25.
Kumar, R., Bohra, A., Pandey, A. K., Pandey, M. K., & Kumar, A. (2017). Metabolomics for plant improvement: Status and prospects. Frontiers in Plant Science, 8, 1302.
Kusano, M., Fukushima, A., Kobayashi, M., Hayashi, N., Jonsson, P., Moritz, T., Ebana, K., & Saito, K. (2007). Application of a metabolomic method combining one-dimensional and two-dimensional gas chromatography-time-of-flight/mass spectrometry to metabolic phenotyping of natural variants in rice. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 855, 71–79.
Kusano, M., Yang, Z., Okazaki, Y., Nakabayashi, R., Fukushima, A., & Saito, K. (2015). Using metabolomic approaches to explore chemical diversity in rice. Molecular Plant, 8, 58–67.
Lakshmanan, M., Lim, S.-H., Mohanty, B., Kim, J. K., Ha, S.-H., & Lee, D.-Y. (2015). Unraveling the light-specific metabolic and regulatory signatures of rice through combined in silico modeling and multi-omics analysis. Plant Physiology, 169, 01379. https://doi.org/10.1104/pp.15.01379. 2015.
Lancien, M., Gadal, P., & Hodges, M. (2000). Enzyme redundancy and the importance of 2-oxoglutarate in higher plant ammonium assimilation. Plant Physiology, 123(3), 817–824. https://doi.org/10.1104/pp.123.3.817.
Li, B., Zhang, Y., Mohammadi, S. A., Huai, D., Zhou, Y., & Kliebenstein, D. J. (2016a). An integrative genetic study of rice metabolism, growth and stochastic variation reveals potential C/N partitioning loci. Scientific Reports, 6, 30143.
Li, M., Li, D., Feng, F., Zhang, S., Ma, F., & Cheng, L. (2016b). Proteomic analysis reveals dynamic regulation of fruit development and sugar and acid accumulation in apple. Journal of Experimental Botany, 67, 5145–5157.
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., Zhang, Z., Lun, Y., Li, S., Wang, X., Huang, Z., Li, J., Zhang, C., Wang, T., Zhang, Y., Wang, A., Zhang, Y., Lin, K., Li, C., Xiong, G., Xue, Y., Mazzucato, A., Causse, M., Fei, Z., Giovannoni, J. J., Chetelat, R. T., Zamir, D., Stadler, T., Li, J., Ye, Z., Du, Y., & Huang, S. (2014). Genomic analyses provide insights into the history of tomato breeding. Nature Genetics, 46, 1220–1226.
Lipka, A. E., Gore, M. A., Magallanes-Lundback, M., Mesberg, A., Lin, H., Tiede, T., Chen, C., Buell, C. R., Buckler, E. S., Rocheford, T., & DellaPenna, D. (2013) Genome-wide association study and pathway-level analysis of tocochromanol levels in maize grain. G3 (Bethesda, Md.), 3, 1287–1299.
Lisec, J., Romisch-Margl, L., Nikoloski, Z., Piepho, H. P., Giavalisco, P., Selbig, J., Gierl, A., & Willmitzer, L. (2011). Corn hybrids display lower metabolite variability and complex metabolite inheritance patterns. The Plant Journal: For Cell and Molecular Biology, 68, 326–336.
Liu, M. Y., Burgos, A., Ma, L., Zhang, Q., Tang, D., & Ruan, J. (2017). Lipidomics analysis unravels the effect of nitrogen fertilization on lipid metabolism in tea plant (Camellia sinensis L.). BMC Plant Biology, 17, 165.
Llorente, B., Alonso, G. D., Bravo-Almonacid, F., Rodriguez, V., Lopez, M. G., Carrari, F., Torres, H. N., & Flawia, M. M. (2011). Safety assessment of nonbrowning potatoes: Opening the discussion about the relevance of substantial equivalence on next generation biotech crops. Plant Biotechnology Journal, 9, 136–150.
López, M. G., Zanor, M. I., Pratta, G. R., Stegmayer, G., Boggio, S. B., Conte, M., Bermúdez, L., Leskow, C., Rodríguez, C., Picardi, G. R., Zorzoli, L. A., Fernie, R., Milone, A. R., Asís, D., Valle, R., E.M. and Carrari, F. (2015). Metabolic analyses of interspecific tomato recombinant inbred lines for fruit quality improvement. Metabolomics, 11, 1416–1431.
Matsuda, F., Okazaki, Y., Oikawa, A., Kusano, M., Nakabayashi, R., Kikuchi, J., Yonemaru, J., Ebana, K., Yano, M., & Saito, K. (2012). Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. The Plant Journal: For Cell and Molecular Biology, 70, 624–636.
Meyer, R. C., Steinfath, M., Lisec, J., Becher, M., Witucka-Wall, H., Torjek, O., Fiehn, O., Eckardt, A., Willmitzer, L., Selbig, J., & Altmann, T. (2007). The metabolic signature related to high plant growth rate in Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the United States of America, 104, 4759–4764.
Mikulic-Petkovsek, M., Schmitzer, V., Slatnar, A., Weber, N., Veberic, R., Stampar, F., Munda, A., & Koron, D. (2013). Alteration of the content of primary and secondary metabolites in strawberry fruit by Colletotrichum nymphaeae infection. Journal of Agricultural and Food Chemistry, 61, 5987–5995.
Misra, B. B., Acharya, B. R., Granot, D., Assmann, S. M., & Chen, S. (2015). The guard cell metabolome: Functions in stomatal movement and global food security. Frontiers in Plant Science, 6, 334.
Misra, B. B., Assmann, S. M., & Chen, S. (2014). Plant single-cell and single-cell-type metabolomics. Trends in Plant Science, 19, 637–646.
Misyura, M., Guevara, D., Subedi, S., Hudson, D., McNicholas, P. D., Colasanti, J., & Rothstein, S. J. (2014). Nitrogen limitation and high density responses in rice suggest a role for ethylene under high density stress. BMC Genomics, 15, 681.
Moghissi, A. A., Pei, S., & Liu, Y. (2016). Golden rice: Scientific, regulatory and public information processes of a genetically modified organism. Critical Reviews in Biotechnology. https://doi.org/10.3109/07388551.2014.993586.
Moschen, S., Di Rienzo, J. A., Higgins, J., Tohge, T., Watanabe, M., Gonzalez, S., Rivarola, M., Garcia-Garcia, F., Dopazo, J., Hopp, H. E., Hoefgen, R., Fernie, A. R., Paniego, N., Fernandez, P., & Heinz, R. A. (2017). Integration of transcriptomic and metabolic data reveals hub transcription factors involved in drought stress response in sunflower (Helianthus annuus L.). Plant Molecular Biology, 94, 549–564.
Nagpala, E. G., Guidarelli, M., Gasperotti, M., Masuero, D., Bertolini, P., Vrhovsek, U., & Baraldi, E. (2016). Polyphenols variation in fruits of the susceptible strawberry cultivar alba during ripening and upon fungal pathogen interaction and possible involvement in unripe fruit tolerance. Journal of Agricultural and Food Chemistry, 64(9), 1869–1878. https://doi.org/10.1021/acs.jafc.5b06005.
Nakabayashi, R., & Saito, K. (2015). Integrated metabolomics for abiotic stress responses in plants. Current Opinion in Plant Biology, 24, 10–16.
Nardozza, S., Boldingh, H. L., Osorio, S., Hohne, M., Wohlers, M., Gleave, A. P., MacRae, E. A., Richardson, A. C., Atkinson, R. G., Sulpice, R., Fernie, A. R., & Clearwater, M. J. (2013). Metabolic analysis of kiwifruit (Actinidia deliciosa) berries from extreme genotypes reveals hallmarks for fruit starch metabolism. Journal of Experimental Botany, 64, 5049–5063.
Nielsen, L. J., Stuart, P., Picmanova, M., Rasmussen, S., Olsen, C. E., Harholt, J., Moller, B. L., & Bjarnholt, N. (2016). Dhurrin metabolism in the developing grain of Sorghum bicolor (L.) Moench investigated by metabolite profiling and novel clustering analyses of time-resolved transcriptomic data. BMC Genomics, 17, 1021.
Obata, T., & Fernie, A. R. (2012). The use of metabolomics to dissect plant responses to abiotic stresses. Cellular and Molecular Life Sciences: CMLS, 69, 3225–3243.
Obata, T., Witt, S., Lisec, J., Palacios-Rojas, N., Florez-Sarasa, I., Yousfi, S., Araus, J. L., Cairns, J. E., & Fernie, A. R. (2015). Metabolite profiles of maize leaves in drought, heat, and combined stress field trials reveal the relationship between metabolism and grain yield. Plant Physiology, 169, 2665–2683.
Ogawa, T., Kashima, K., Yuki, Y., Mejima, M., Kurokawa, S., Kuroda, M., Okazawa, A., Kiyono, H., & Ohta, D. (2017). Seed metabolome analysis of a transgenic rice line expressing cholera toxin B-subunit. Scientific Reports, 7, 5196.
Ogbaga, C. C., Stepien, P., Dyson, B. C., Rattray, N. J., Ellis, D. I., Goodacre, R., & Johnson, G. N. (2016). Biochemical analyses of sorghum varieties reveal differential responses to drought. PLoS ONE, 11, e0154423.
Oikawa, A., Otsuka, T., Nakabayashi, R., Jikumaru, Y., Isuzugawa, K., Murayama, H., Saito, K., & Shiratake, K. (2015). Metabolic profiling of developing pear fruits reveals dynamic variation in primary and secondary metabolites, including plant hormones. PLoS ONE, 10, e0131408.
Okazaki, Y., Otsuki, H., Narisawa, T., Kobayashi, M., Sawai, S., Kamide, Y., Kusano, M., Aoki, T., Hirai, M. Y., & Saito, K. (2013). A new class of plant lipid is essential for protection against phosphorus depletion. Nature Communications, 4, 1510.
Okazaki, Y., & Saito, K. (2016). Integrated metabolomics and phytochemical genomics approaches for studies on rice. GigaScience, 5, 11.
Owens, B. F., Lipka, A. E., Magallanes-Lundback, M., Tiede, T., Diepenbrock, C. H., Kandianis, C. B., Kim, E., Cepela, J., Mateos-Hernandez, M., Buell, C. R., Buckler, E. S., DellaPenna, D., Gore, M. A., & Rocheford, T. (2014). A foundation for provitamin A biofortification of maize: genome-wide association and genomic prediction models of carotenoid levels. Genetics, 198, 1699–1716.
Pan, Z., Zeng, Y., An, J., Ye, J., Xu, Q., & Deng, X. (2012). An integrative analysis of transcriptome and proteome provides new insights into carotenoid biosynthesis and regulation in sweet orange fruits. Journal of Proteomics, 75, 2670–2684.
Patrick, J. W., Botha, F. C., & Birch, R. G. (2013). Metabolic engineering of sugars and simple sugar derivatives in plants. Plant Biotechnology Journal. https://doi.org/10.1111/pbi.12002.
Peng, M., Ying, P., Liu, X., Li, C., Xia, R., Li, J., & Zhao, M. (2017). Genome-wide identification of histone modifiers and their expression patterns during fruit abscission in litchi. Frontiers in Plant Science, 8, 639.
Perez-Fons, L., Wells, T., Corol, D. I., Ward, J. L., Gerrish, C., Beale, M. H., Seymour, G. B., Bramley, P. M., & Fraser, P. D. (2014). A genome-wide metabolomic resource for tomato fruit from Solanum pennellii. Scientific Reports, 4, 3859.
Powell, J. P., & Reinhard, S. (2016). Measuring the effects of extreme weather events on yields. Weather and Climate Extremes, 12, 69–79. https://doi.org/10.1016/j.wace.2016.02.003.
Pretty, J., & Bharucha, Z. P. (2014). Sustainable intensification in agricultural systems. Annals of Botany, 114(8), 1571–1596. https://doi.org/10.1093/aob/mcu205.
Price, E. J., Bhattacharjee, R., Lopez-Montes, A., & Fraser, P. D. (2017). Metabolite profiling of yam (Dioscorea spp.) accessions for use in crop improvement programmes. Metabolomics, 13, 144.
Qi, X., Xu, W., Zhang, J., Guo, R., Zhao, M., Hu, L., Wang, H., Dong, H., & Li, Y. (2017). Physiological characteristics and metabolomics of transgenic wheat containing the maize C4 phosphoenolpyruvate carboxylase (PEPC) gene under high temperature stress. Protoplasma, 254, 1017–1030.
Quadrana, L., Almeida, J., Asis, R., Duffy, T., Dominguez, P. G., Bermudez, L., Conti, G., Correa da Silva, J. V., Peralta, I. E., Colot, V., Asurmendi, S., Fernie, A. R., Rossi, M., & Carrari, F. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nature Communications, 5, 3027.
Quan, X., Zeng, J., Han, Z., & Zhang, G. (2017). Ionomic and physiological responses to low nitrogen stress in Tibetan wild and cultivated barley. Plant Physiology and Biochemistry, 111, 257–265. https://doi.org/10.1016/j.plaphy.2016.12.008.
Ramalingam, A., Kudapa, H., Pazhamala, L. T., Weckwerth, W., & Varshney, R. K. (2015). Proteomics and metabolomics: Two emerging areas for legume improvement. Frontiers in Plant Science, 6, 1116.
Ranjbar Sistani, N., Kaul, H. P., Desalegn, G., & Wienkoop, S. (2017) Rhizobium impacts on seed productivity, quality, and protection of Pisum sativum upon disease stress caused by Didymella pinodes: Phenotypic, proteomic, and metabolomic traits. Frontiers in Plant Science, 8, 1961.
Rao, J., Cheng, F., Hu, C., Quan, S., Lin, H., Wang, J., Chen, G., Zhao, X., Alexander, D., Guo, L., Wang, G., Lai, J., Zhang, D., & Shi, J. (2014). Metabolic map of mature maize kernels. Metabolomics, 10, 775–787.
Raun, W., Solie, J. B., Johnson, G. V., Stone, M., Mullen, R. W., Freeman, K. W., Thomason, W., & Lukina, E. V. (2002). Improving nitrogen use efficiency in cereal grain production with optical sensing and variable rate application. Agronomy Journal, 94, 815–820.
Redestig, H., Kusano, M., Ebana, K., Kobayashi, M., Oikawa, A., Okazaki, Y., Matsuda, F., Arita, M., Fujita, N., & Saito, K. (2011). Exploring molecular backgrounds of quality traits in rice by predictive models based on high-coverage metabolomics. BMC Systems Biology, 5, 176.
Riedelsheimer, C., Czedik-Eysenberg, A., Grieder, C., Lisec, J., Technow, F., Sulpice, R., et al. (2012a). Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nature Genetics, 44(2), 217–220. https://doi.org/10.1038/ng.1033.
Riedelsheimer, C., Lisec, J., Czedik-Eysenberg, A., Sulpice, R., Flis, A., Grieder, C., Altmann, T., Stitt, M., Willmitzer, L., & Melchinger, A. E. (2012b). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proceedings of the National Academy of Sciences of the United States of America, 109, 8872–8877.
Rossi, M., Bermudez, L., & Carrari, F. (2015). Crop yield: Challenges from a metabolic perspective. Current Opinion in Plant Biology, 25, 79–89.
Safronov, O., Kreuzwieser, J., Haberer, G., Alyousif, M. S., Schulze, W., Al-Harbi, N., Arab, L., Ache, P., Stempfl, T., Kruse, J., Mayer, K. X., Hedrich, R., Rennenberg, H., Salojarvi, J., & Kangasjarvi, J. (2017). Detecting early signs of heat and drought stress in Phoenix dactylifera (date palm). PLoS ONE, 12, e0177883.
Sauvage, C., Segura, V., Bauchet, G., Stevens, R., Do, P. T., Nikoloski, Z., Fernie, A. R., & Causse, M. (2014). Genome-wide association in tomato reveals 44 candidate loci for fruit metabolic traits. Plant Physiology, 165, 1120–1132.
Sayre, R., Beeching, J. R., Cahoon, E. B., Egesi, C., Fauquet, C., Fellman, J., et al. (2011). The BioCassava plus program: Biofortification of cassava for sub-saharan Africa. Annual Review of Plant Biology. https://doi.org/10.1146/annurev-arplant-042110-103751.
Schauer, N., Semel, Y., Balbo, I., Steinfath, M., Repsilber, D., Selbig, J., Pleban, T., Zamir, D., & Fernie, A. R. (2008). Mode of inheritance of primary metabolic traits in tomato. The Plant Cell, 20, 509–523.
Schauer, N., Semel, Y., Roessner, U., Gur, A., Balbo, I., Carrari, F., Pleban, T., Perez-Melis, A., Bruedigam, C., Kopka, J., Willmitzer, L., Zamir, D., & Fernie, A. R. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nature Biotechnology, 24, 447–454.
Shelden, M. C., Dias, D. A., Jayasinghe, N. S., Bacic, A., & Roessner, U. (2016). Root spatial metabolite profiling of two genotypes of barley (Hordeum vulgare L.) reveals differences in response to short-term salt stress. Journal of Experimental Botany, 67, 3731–3745.
Shen, Q., Fu, L., Dai, F., Jiang, L., Zhang, G., & Wu, D. (2016). Multi-omics analysis reveals molecular mechanisms of shoot adaption to salt stress in Tibetan wild barley. BMC Genomics, 17, 889.
Shimojima, M., Madoka, Y., Fujiwara, R., Murakawa, M., Yoshitake, Y., Ikeda, K., Koizumi, R., Endo, K., Ozaki, K., & Ohta, H. (2015). An engineered lipid remodeling system using a galactolipid synthase promoter during phosphate starvation enhances oil accumulation in plants. Frontiers in Plant Science, 6, 664.
Son, H.-S., Hwang, G.-S., Kim, K. M., Ahn, H.-J., Park, W.-M., Van Den Berg, F., et al. (2009). Metabolomic studies on geographical grapes and their wines using 1H NMR analysis coupled with multivariate statistics. Journal of Agricultural and Food Chemistry, 57(4), 1481–1490. https://doi.org/10.1021/jf803388w.
Sonawane, P. D., Pollier, J., Panda, S., Szymanski, J., Massalha, H., Yona, M., Unger, T., Malitsky, S., Arendt, P., Pauwels, L., Almekias-Siegl, E., Rogachev, I., Meir, S., Cardenas, P. D., Masri, A., Petrikov, M., Schaller, H., Schaffer, A. A., Kamble, A., Giri, A. P., Goossens, A., & Aharoni, A. (2016). Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nature Plants, 3, 16205.
Sonnewald, U., & Fernie, A. R. (2018). Next-generation strategies for understanding and influencing source-sink relations in crop plants. Current Opinion in Plant Biology, 43, 63–70.
Stitt, M., & Schulze, D. (1994). Does Rubisco control the rate of photosynthesis and plant growth? An exercise in molecular ecophysiology. Plant, Cell & Environment, 17, 465–487. https://doi.org/10.1111/j.1365-3040.1994.tb00144.x.
Stoop, J. M. H., Williamson, J. D., & Mason Pharr, D. (1996). Mannitol metabolism in plants: A method for coping with stress. Trends in Plant Science, 1, 139–144.
Sun, M., Yang, Z., & Wawrik, B. (2018). Metabolomic fingerprints of individual algal cells using the single-probe mass spectrometry technique. Frontiers in Plant Science, 9, 571.
Sweetlove, L. J., Beard, K. F., Nunes-Nesi, A., Fernie, A. R., & Ratcliffe, R. G. (2010). Not just a circle: Flux modes in the plant TCA cycle. Trends in Plant Science, 15, 462–470.
Tatsis, E. C., & O’Connor, S. E. (2016). New developments in engineering plant metabolic pathways. Current Opinion in Biotechnology. https://doi.org/10.1016/j.copbio.2016.04.012.
The Tomato Genome Consortium. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature, 485, 635–641.
Tieman, D., Zhu, G., Resende, M. F. Jr., Lin, T., Nguyen, C., Bies, D., Rambla, J. L., Beltran, K. S., Taylor, M., Zhang, B., Ikeda, H., Liu, Z., Fisher, J., Zemach, I., Monforte, A., Zamir, D., Granell, A., Kirst, M., Huang, S., & Klee, H. (2017). A chemical genetic roadmap to improved tomato flavor. Science, 355, 391–394.
Tohge, T., & Fernie, A. R. (2015). Metabolomics-inspired insight into developmental, environmental and genetic aspects of tomato fruit chemical composition and quality. Plant & Cell Physiology, 56, 1681–1696.
Tohge, T., Scossa, F., & Fernie, A. R. (2015). Integrative approaches to enhance understanding of plant metabolic pathway structure and regulation. Plant Physiology, 169(3), 1499–1511.
Topfer, N., Kleessen, S., & Nikoloski, Z. (2015). Integration of metabolomics data into metabolic networks. Frontiers in Plant Science, 6, 49.
Turner, M., Heuberger, A., Kirkwood, J., Collins, C., Wolfrum, C., Broeckling, E., Prenni, C., J. and Jahn, C. (2016). Non-targeted metabolomics in diverse sorghum breeding lines indicates primary and secondary metabolite profiles are associated with plant biomass accumulation and photosynthesis. Frontiers in Plant Science, 7, 953.
Tuttle, J. R., Nah, G., Duke, M. V., Alexander, D. C., Guan, X., Song, Q., et al. (2015). Metabolomic and transcriptomic insights into how cotton fiber transitions to secondary wall synthesis, represses lignification, and prolongs elongation. BMC Genomics, 16(1), 1–28. https://doi.org/10.1186/s12864-015-1708-9.
Uddling, J., Gelang-Alfredsson, J., Karlsson, P. E., Selldén, G., & Pleijel, H. (2008). Source–sink balance of wheat determines responsiveness of grain production to increased [CO2] and water supply. Agriculture, Ecosystems and Environment, 127, 215–222.
Upadhyaya, P., Tyagi, K., Sarma, S., Tamboli, V., Sreelakshmi, Y., & Sharma, R. (2017). Natural variation in folate levels among tomato (Solanum lycopersicum) accessions. Food Chemistry, 217, 610–619. https://doi.org/10.1016/j.foodchem.2016.09.031.
Venkatesh, T. V., Chassy, A. W., Fiehn, O., Flint-Garcia, S., Zeng, Q., Skogerson, K., & Harrigan, G. G. (2016). Metabolomic assessment of key maize resources: GC-MS and NMR profiling of grain from B73 hybrids of the nested association mapping (NAM) founders and of geographically diverse landraces. Journal of Agricultural and Food Chemistry, 64, 2162–2172.
Vimolmangkang, S., Zheng, H., Peng, Q., Jiang, Q., Wang, H., Fang, T., et al. (2016). Assessment of sugar components and genes involved in the regulation of sucrose accumulation in peach fruit. Journal of Agricultural and Food Chemistry, 64(35), 6723–6729. https://doi.org/10.1021/acs.jafc.6b02159.
Vital, C. E., Giordano, A., de Almeida Soares, E., Williams, R., Mesquita, T. C., Vidigal, R. O., de Santana Lopes, P. M. P., Pacheco, A., Rogalski, T. G., M., de O. Ramos, H.J. and Loureiro, M. E. (2017). An integrative overview of the molecular and physiological responses of sugarcane under drought conditions. Plant Molecular Biology, 94, 577–594.
Wang, H., Xu, S., Fan, Y., Liu, N., Zhan, W., Liu, H., Xiao, Y., Li, K., Pan, Q., Li, W., Deng, M., Liu, J., Jin, M., Yang, X., Li, J., Li, Q., & Yan, J. (2018). Beyond pathways: Genetic dissection of tocopherol content in maize kernels by combining linkage and association analyses. Plant Biotechnology Journal, 16, 1464–1475.
Wang, X., Zhu, W., Hashiguchi, A., Nishimura, M., Tian, J., & Komatsu, S. (2017). Metabolic profiles of flooding-tolerant mechanism in early-stage soybean responding to initial stress. Plant Molecular Biology, 94, 669–685.
Wen, W., Jin, M., Li, K., Liu, H., Xiao, Y., Zhao, M., Alseekh, S., Li, W., de Abreu, E. L. F., Brotman, Y., Willmitzer, L., Fernie, A. R., & Yan, J. (2018). An integrated multi-layered analysis of the metabolic networks of different tissues uncovers key genetic components of primary metabolism in maize. The Plant Journal: For Cell and Molecular Biology, 93, 1116–1128.
Wen, W., Li, K., Alseekh, S., Omranian, N., Zhao, L., Zhou, Y., Xiao, Y., Jin, M., Yang, N., Liu, H., Florian, A., Li, W., Pan, Q., Nikoloski, Z., Yan, J., & Fernie, A. R. (2015). Genetic determinants of the network of primary metabolism and their relationships to plant performance in a maize recombinant inbred line population. The Plant Cell, 27, 1839–1856.
Xu, S., Xu, Y., Gong, L., & Zhang, Q. (2016). Metabolomic prediction of yield in hybrid rice. The Plant Journal: For Cell and Molecular Biology, 88, 219–227.
Yang, F., Xu, X., Wang, W., Ma, J., Wei, D., He, P., Pampolino, M. F., & Johnston, A. M. (2017). Estimating nutrient uptake requirements for soybean using QUEFTS model in China. PLoS ONE, 12, e0177509.
Yang, X., Feng, L., Zhao, L., Liu, X., Hassani, D., & Huang, D. (2018). Effect of glycine nitrogen on lettuce growth under soilless culture: A metabolomics approach to identify the main changes occurred in plant primary and secondary metabolism. Journal of the Science of Food and Agriculture, 98, 467–477.
Yang, X., Nian, J., Xie, Q., Feng, J., Zhang, F., Jing, H., Zhang, J., Dong, G., Liang, Y., Peng, J., Wang, G., Qian, Q., & Zuo, J. (2016). Rice ferredoxin-dependent glutamate synthase regulates nitrogen-carbon metabolomes and is genetically differentiated between japonica and indica subspecies. Molecular Plant, 9, 1520–1534.
Ye, X., & Beyer, P. (2000). Engineering the provitamin A (β-carotene) biosynthetic pathway into (carotenoid-free) rice endosperm. Science. https://doi.org/10.1126/science.287.5451.303.
Yesbergenova-Cuny, Z., Dinant, S., Martin-Magniette, M. L., Quillere, I., Armengaud, P., Monfalet, P., Lea, P. J., & Hirel, B. (2016). Genetic variability of the phloem sap metabolite content of maize (Zea mays L.) during the kernel-filling period. Plant Science: An International Journal of Experimental Plant Biology, 252, 347–357.
Ying, J. Z., Shan, J. X., Gao, J. P., Zhu, M. Z., Shi, M., & Lin, H. X. (2012). Identification of quantitative trait Loci for lipid metabolism in rice seeds. Molecular Plant, 5, 865–875.
Yonekura-Sakakibara, K., & Saito, K. (2006). Review: Genetically modified plants for the promotion of human health. Biotechnology Letters. https://doi.org/10.1007/s10529-006-9194-4.
Zhang, J., Luo, W., Zhao, Y., Xu, Y., Song, S., & Chong, K. (2016). Comparative metabolomic analysis reveals a reactive oxygen species-dominated dynamic model underlying chilling environment adaptation and tolerance in rice. The New Phytologist, 211, 1295–1310.
Zhang, N., Venkateshwaran, M., Boersma, M., Harms, A., Howes-Podoll, M., den Os, D., Ane, J. M., & Sussman, M. R. (2012). Metabolomic profiling reveals suppression of oxylipin biosynthesis during the early stages of legume-rhizobia symbiosis. FEBS Letters, 586, 3150–3158.
Zhang, Y., Butelli, E., Alseekh, S., Tohge, T., Rallapalli, G., Luo, J., et al. (2015). Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nature Communications. https://doi.org/10.1038/ncomms9635.
Zhao, Y., Li, Z., Liu, G., Jiang, Y., Maurer, H. P., Wurschum, T., Mock, H. P., Matros, A., Ebmeyer, E., Schachschneider, R., Kazman, E., Schacht, J., Gowda, M., Longin, C. F., & Reif, J. C. (2015). Genome-based establishment of a high-yielding heterotic pattern for hybrid wheat breeding. Proceedings of the National Academy of Sciences of the United States of America, 112, 15624–15629.
Zhu, G., Wang, S., Huang, Z., Zhang, S., Liao, Q., Zhang, C., Lin, T., Qin, M., Peng, M., Yang, C., Cao, X., Han, X., Wang, X., van der Knaap, E., Zhang, Z., Cui, X., Klee, H., Fernie, A. R., Luo, J., & Huang, S. (2018). Rewiring of the fruit metabolome in tomato breeding. Cell, 172, 249–261.e212.
Acknowledgements
Work in our laboratories is funded in part by ANPCyT, CONICET, INTA, UBA (Argentina); CAPES and USP (Brazil); Max Planck Society (Germany) and the European Union Horizon 2020 Research and Innovation Programme (Grant Agreement Number 679796).
Author information
Authors and Affiliations
Contributions
SA, LAdH and LB surveyed and discussed scientific literature and participated in writing the manuscript. LAdH and LB designed and drawn the illustrative figure. ARF wrote and edited the manuscript and FC wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Alseekh, S., Bermudez, L., de Haro, L.A. et al. Crop metabolomics: from diagnostics to assisted breeding. Metabolomics 14, 148 (2018). https://doi.org/10.1007/s11306-018-1446-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-018-1446-5