
Abstract
Introduction
Advances in high-resolution mass spectrometry have created renewed interest for studying global lipid biochemistry in disease and biological systems.
Objectives
Here, we present an untargeted 30 min. LC-MS/MS platform that utilizes positive/negative polarity switching to perform unbiased data dependent acquisitions (DDA) via higher energy collisional dissociation (HCD) fragmentation to profile more than 1000–1500 lipid ions mainly from methyl-tert-butyl ether (MTBE) or chloroform:methanol extractions.
Methods
The platform uses C18 reversed-phase chromatography coupled to a hybrid QExactive Plus/HF Orbitrap mass spectrometer and the entire procedure takes ~10 h from lipid extraction to identification/quantification for a data set containing 12 samples (~4 h for a single sample). Lipids are identified by both accurate precursor ion mass and fragmentation features and quantified using LipidSearch and Elements software.
Results
Using this approach, we are able to profile intact lipid ions from up to 18 different main lipid classes and 66 subclasses. We show several studies from different biological sources, including cultured cancer cells, resected tissues from mice such as lung and breast tumors and biological fluids such as plasma and urine.
Conclusions
Using mouse embryonic fibroblasts, we showed that TSC2−/− KD significantly abrogates lipid biosynthesis and that rapamycin can rescue triglyceride (TG) lipids and we show that SREBP−/− shuts down lipid biosynthesis significantly via mTORC1 signaling pathways. We show that in mouse EGFR driven lung tumors, a large number of TGs and phosphatidylmethanol (PMe) lipids are elevated while some phospholipids (PLs) show some of the largest decrease in lipid levels from ~ 2000 identified lipid ions. In addition, we identified more than 1500 unique lipid species from human blood plasma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Lipids play many important biological roles that include being the structural and functional component of membranes, a key form of energy storage within lipid droplets, and critical intracellular and extracellular signaling molecules (Rolim et al. 2015). This protocol addresses the need to quantify and identify lipid molecules extracted from cell lines or tissue samples. A surge in interest in lipidomics profiling over the last few years due to the emergence of modern high-resolution mass spectrometers has created a demand for protocols for high-throughput lipidomics profiling. Orbitrap and QTOF mass spectrometers were largely introduced to serve the proteomics community over the last two decades, aimed at increasing specificity, sensitivity and throughput for very complex proteomes (Coombs 2011; Cox and Mann 2011; Sajic et al. 2015). However, in recent years, small molecule metabolomics and lipidomics have become increasing popular, especially in the study of cancer metabolism (Yuan et al. 2012; Junot et al. 2014; Ma and Chowdhury 2013; Theodoridis et al. 2012; Xiao et al. 2012; Cajka and Fiehn 2014). High-resolution mass spectrometry (MS) is needed for stable isotope-labeling experiments from 13C and 15N for both proteomics (e.g., stable isotope labeling of amino acids in cell culture (SILAC) and tandem mass tags (TMT) and metabolomics (i.e., metabolic flux analysis) (Ben-Sahra et al. 2013; Ying et al. 2012; Nicolay et al. 2013). Similarly, lipidomics requires high-resolution MS to obtain the high mass accuracy required to accurately identify head groups and fatty acid chains, including mass shifts due to differences in saturation of fatty acid chains (Cajka and Fiehn 2014; Junot et al. 2014; Schwudke et al. 2011; Vaz et al. 2015). Many mass spectrometry based lipidomics studies have been completed using either direct infusion (previously termed shotgun lipidomics) or with chromatographic separation (mostly reversed phase) followed by MALDI-MS (Han et al. 2012; Kofeler et al. 2012; Li et al. 2014; Wang et al. 2015). If specific molecules or lipid classes are known, product and/or neutral loss scans can be performed using triple quadrupole mass spectrometers (Hou et al. 2011; Lam and Shui 2013; Yang et al. 2015). Lipidomics can be performed in either positive ion or negative ion mode depending upon the lipid class. Lipid identifications with mass spectrometry can be difficult due to the abundance of structural isomers and the various adducts that can generate lipid ions and confuse the fragmentation pattern (Godzien et al. 2015). Several LC-MS based lipidomic studies have been reported whose identification rely upon LC retention time and high mass accuracy MS1 data, some of which focus on specific lipid classes (van der Kloet et al. 2013; Tyurina et al. 2014; Hartler et al. 2011; Hein et al. 2010; Collins et al. 2016). LC-MS/MS based studies have also been shown that utilize high resolution all-ion or MSE fragmentation, several of which are vendor specific (Bilgin et al. 2016; Ahmed et al. 2015; Fauland et al. 2011). Bird et al. utilized full scan MS and HCD all ion fragmentation on an orbitrap mass spectrometer to characterize triacylglycerides (Bird et al. 2011). Here, we chose to incorporate positive/negative polarity switching so that we can profile a broad range of lipids during a single acquisition. Here, we present a high-resolution lipidomics platform that uses positive/negative ion switching with C18 reversed-phase chromatography in data dependent analysis (DDA) mode on a QExactive Plus Orbitrap mass spectrometer in a similar way that bottom-up proteomics is performed. Individual intact lipid molecules are subsequently identified using LipidSearch 4.1 software (Yamada et al. 2013a, b). The speed, sensitivity and specificity of the platform allows for more than 1000 lipid identifications from a single 30 min LC-MS/MS run (Asara et al. 2016; Breitkopf et al. 2015, 2016; Narvaez-Rivas and Zhang 2016). The platform can identify most lipid families including phospholipids (P-choline, P-ethanolamine, P-serine, P-glycerol, P-inositol, P-ethanol, P-methanol, P-acid), monoacylglycerol, diacylglycerol, triacylglycerol, neutral glycerolipids, fatty acids, cardiolipins, sphingolipids, sphingoid bases, glycosphingolipids, neutral glycosphingolipids, glycoglycerolipids, sterols, etc. A total of 66 different lipid subclasses (Table S1) can be identified using this platform independent of sample source.
While cells from most adult tissues rely primarily on the uptake of circulating lipids, de novo lipid synthesis occurs abundantly in certain normal cell types, such as adipocytes, hepatocytes, and activated immune cells, as well as in certain pathologies, such as cancer. In de novo fatty acid synthesis, the enzyme fatty acid synthase (FASN) uses cytosolic acetyl-coA derived from acetate or carbon from the citric acid cycle to produce fatty acids such as palmitate (C16:0) and stearate (C18:0) (Menendez and Lupu 2007). Both palmitate and stearate can be desaturated by stearoyl-coA desaturase 1 (SCD1) to form palmitoleate (C16:1) and oleate (C18:1), respectively. FASN and members of the elongase family can further extend the length of saturated and unsaturated fatty acids, with elongases producing very long-chain fatty acids from those species derived from either de novo synthesis or the diet. Through these reactions, many different lengths and degrees of unsaturation of fatty acids can be produced and incorporated, via acyltransferases, resulting in thousands of distinct lipid species existing within a given cell. Lipid synthesis pathways are frequently altered in diseases such as cancer and diabetes and can potentially be targeted with drugs (Hopperton et al. 2014; Linden et al. 2015; Menendez and Lupu 2007; Ricoult et al. 2015). However, how the global population of lipid species in cells and tissues are altered under different disease states is poorly understood and requires precision lipidomic approaches for comparative analyses.
Extracting lipids from cells and tissues is performed using liquid–liquid extraction methods designed for phase separation of non-polar compounds. The Folch method is the most popular and uses 2:1 chloroform:methanol (Folch et al. 1957), although newer less harmful solvents, such as methyl-tert-butyl ether (MTBE), can be used for extraction with similar results (Matyash et al. 2008). These methods contrast with the purification of polar metabolites, which typically use methanol for extraction (Yuan et al. 2012). Many lipids have been categorized in databases such as LIPID MAPS (Fahy et al. 2007), the Human Metabolite Database (HMDB) (Wishart et al. 2009) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Chou et al. 2009) that can be interrogated for identification and classification of lipids and metabolites. The software used in the platform described here is able to distinguish between individual lipid ions without any derivatization based on their intact mass and fragmentation features, since different head groups and fatty acid chains give rise to distinct mass signatures. It is also unique that we use positive/negative switching to capture lipids using high resolution MS, similarly to our approach for small molecule metabolomics (Yuan et al. 2012). The platform can detect lipids from simple fatty acids to glycerophospholipids to complex triglycerides and cardiolipins in an untargeted manner. The important features of our platform are that (1) a single reversed-phase (C18) liquid chromatographic run is required, (2) the MS acquisition time per sample is relatively short (~28 min), and (3) more than 1000 species are routinely identified and quantified.
We use relative quantification across biological conditions where one or more of the profiled biological samples serve as a reference control. Relative quantification relies on a reproducible platform and it is recommended that samples be run with at least three biological replicates so that statistics can be applied for quality control and to meet the requirements for use of informatics software packages such as LipidSearch, MetaboAnalyst (http://www.MetaboAnalyst.ca) (Xia and Wishart) or Elements (http://www.proteomesoftware.com).
Biological triplicates rather than technical triplicates are suggested since the platform routinely shows R 2 values >0.90 for technical replicate analyses and average CV values of <0.15. If one chooses to use a simple ratio or fold change analysis for quantifying between samples, it is best to first normalize the MS1 peak areas of all samples across conditions, including triplicates, according to the median peak area of all identified species in each sample followed by taking an average for each species within the biological triplicates for each sample condition. One can then calculate fold changes between biological conditions. Alternatively, we recommend using the free online MetaboAnalyst software, which is used for statistical analysis of the data as well as for creating heat maps, clustergrams, principal component analysis (PCA) and pathway analysis(Xia and Wishart, Xia et al. 2009). Absolute quantification (not covered here), can be achieved using a concentration curve for each lipid molecule of interest or by spiking the samples using an internal standard (Rodamer et al. 2011).
Mass spectrometry with positive/negative switching allows for the acquisition of Q1/Q3 MRM transition mass spectra in both ionization modes from a single LC-MS/MS analysis (Koyama et al. 2011; Zhang et al. 2011; Hara et al. 2004; Yuan et al. 2012). While hybrid triple quadrupole style instruments such as the 5500 QTRAP mass spectrometer (AB/SCIEX) has the ability to switch polarities in as little as 50 msec, the high resolution QExactive Plus Orbitrap has the ability to switch polarities in several hundred msec, allowing for fast duty cycling. When coupled to a data dependent acquisition (DDA) platform with dynamic exclusion whereby the same molecules are only targeted one time for MS/MS and the MS scan is used for quantification, the switching time is sufficient for obtaining ~1000–1500 identified and quantified lipid molecules per sample in 28 min from a single run in both positive and negative ion mode. This represents a comprehensive lipid dataset that can be obtained without the need to prepare two sets of samples and two different single polarity LC-MS/MS runs, thus preserving precious samples. Similar platforms have been implemented for untargeted analysis of polar metabolites (Kluger et al. 2014). It is important to note that the current number of identified lipids can likely be increased by expanding the chromatographic gradient and MS acquisition time, however, to maximize throughput, a half hour or less is recommended. While this protocol focuses on cell lines, blood plasma and mouse tissue, the platform could be used to analyze non-polar lipids from any biological source regardless of organism or tissue of origin such as urine, liver, mammary gland, lung and tumor tissues as well as serum and primary cultured cells. While the lipid class profiles vary amongst sample type, the overall number and range of identified lipids is similar across various biological materials.
2 Materials
2.1 Reagents
-
LC/MS grade water (Fisher Scientific, WG-4))
-
LC/MS grade acetonitrile (Acros, AC61514-0025)
-
HPLC grade methanol (Fisher Scientific, AC61009-0040)
-
LC/MS grade isopropanol (Fisher Scientific, A461-4)
-
Ammonium formate crystals, >99.0% (Fluka, 09735)
-
Formic acid, ~98% (Fluka, 94318)
-
Methyl tert-butyl ether (MTBE), anhydr. 99.8% (Sigma Aldrich, 306975-1L)
-
HPLC grade chloroform (Acros, 268320010)
2.2 Reagent setup
2.2.1 Tissue
-
For soft tissue samples, including tumor tissue, start with ~5–10 mg of solid tissue or the equivalent of at least 1–2 million cells, place in 1.5 mL microcentrifuge tube and snap freeze tissue sample in liquid nitrogen (−196°C) as close to the time of resection as possible.
2.2.2 Fluid
-
For biological fluid samples, start with 50–200 µL of serum, plasma, urine or cerebrospinal fluid in a 1.5 mL microcentrifuge tube. If less fluid is available, add 1× PBS to a total volume of 200 µL.
2.2.3 Cells
-
For cultured cells, use the equivalent of ≥1–2 million cells, or one 10 cm plate at ~80% confluence.
2.2.4 Methyl tert-butyl ether (MTBE)
-
MTBE is used directly at room temperature without preparation. Be sure to prepare aliquots of MTBE in glass vials to avoid sample loss due to adsorption of lipids to the plastic surface and possible release of contaminants.
-
Note: Due to MTBE’s distinctive odor, it is recommended to work in a chemical fume hood and close vial lids immediately after usage as it evaporates quickly.
-
Note: 2:1 chloroform:methanol can be used for extraction in place of MTBE according to the Folch method. MTBE is preferred since it is less hazardous and extracts a broad range of lipids.
2.2.5 HPLC buffer A
(pH = 3.5: 39.9% (v/v) water, 60% (v/v) acetonitrile, 10 mM ammonium formate, 0.1% formic acid)
-
To prepare HPLC A buffer, add 0.63055 g of ammonium formate to 1 L HPLC bottle and add 399 mL of LC/MS grade water and gently stir by rotating bottle. Next, add 1.0 mL of formic acid followed by 600 mL of HPLC grade acetonitrile. Cap bottle and mix by gently shaking. Check pH to assure it is ~ 3.5.
2.2.6 HPLC B buffer B
(89.9% (v/v) isopropanol, 10.0% (v/v) acetonitrile, 10 mM ammonium formate, 0.1% formic acid)
-
To prepare HPLC B buffer, add 0.63055 g of ammonium formate to 1 L HPLC bottle and add 899 mL of LC/MS grade isopropanol and stir by rotating bottle. Next, add 1.0 mL of formic acid followed by 100 mL of HPLC grade acetonitrile. Cap bottle and mix by shaking until fully dissolved.
2.2.7 Equipment
-
Cadenza CD-C18 HPLC column (3.0 µm) 2.0 mm i.d. x 150 mm length (Imtakt, CD025)
Note: The Imtakt Cadenza columns are robust, reproducible and have a long life for lipidomics analyses (≥600 analyses with high reproducibility). Alternatively, most other C18 HPLC columns of similar dimensions can be used in place of the Imtakt column.
-
Peek Pre-Column Filter (IDEX Health & Science, A-356)
-
Peek Frit for pre-column filter (IDEX Health & Science, A-701)
-
QExactive Plus Orbitrap (QE+) mass spectrometer (Thermo Fisher Scientific) capable of polarity switching times <500 msec with HCD fragmentation or other orbitrap of time-of flight (TOF) based high resolution mass spectrometer with high mass accuracy (<3 ppm)
-
1100 HPLC (Agilent) or generic HPLC instrument capable of high run-to-run reproducibility and capable of flow rates in the several hundred microliters per minute range (up to ~1 mL/min). The HPLC does not need a chilled autosampler attached to the unit capable of holding all of the lipid samples in your sample set (~24–96). Note that equipment from many HPLC vendors can be used for this purpose and room temperature sampling is preferred since long and multi-branched lipids such as triglycerides can stiffen and precipitate at low temperatures.
-
SpeedVac concentrator or lyophilizer for drying metabolite supernatants to a pellet (Thermo Electron or other vendor)
-
12 × 32 mm glass autosampler tubes with caps (National Scientific, MSCERT400-36LVW)
-
12 mL glass vial (Fisher Scientific, 03-340-47A)
-
22 mL glass vial (Fisher Scientific, B7990-5)
-
Small pestle or tissue grinder for use in 1.5–2.0 mL glass tubes
2.3 Equipment setup
2.3.1 Mass spectrometer
Use an Orbitrap or quadrupole-TOF mass spectrometer capable of positive/negative polarity switching time of 500 msec or below such as the QExactive Plus Orbitrap (Thermo Fisher Scientific) This allows for data-dependent acquisition (DDA) scans of 1 MS1 scan followed by 8 MS2 scans per cycle resulting in a sufficient number of MS1 scans (~ 8–11) per lipid peak using a standard HPLC. The QExactive Plus is capable of 12 scans/sec (Hz) at 17,500 resolution. In our platform, the resolution of the orbitrap mass analyzer was set to 70,000 for MS1 scans and 35,000 for MS2 scans.
2.3.2 HPLC
For the procedure described, a C18 or other reversed-phase HPLC column is needed (see equipment). Ultra-high pressure HPLC (UHPLC) is not suitable for this application since polarity switching DDA mode described here benefits from peak widths at least 8–10 s at FWHH and base peak widths of 16–20 s. UHPLC would limit the number of lipid MS1 scans per lipid.
2.3.3 Software
-
Software such as XCalibur 2.2 (Thermo Fisher Scientific) is needed for data acquisition
-
LipidSearch software is needed for the identification, alignment and quantification of lipids across a sample set. LipidSearch can identify 18 classes of lipids including more than 66 subclasses as well as align lipid peaks across sample sets for relative quantification. In addition, one can chart the relative abundance of lipid classes and fatty acid chain distributions within each lipid class.
-
As alternative software, Elements for Metabolomics from Proteome Software (http://www.proteomesoftware.com/products/elements/metabolomics/) can be used with a search set up against NIST (http://www.proteomesoftware.com/) or HMDB databases (http://www.hmdb.ca/), or LipidMaps (http://www.lipidmaps.org), although the lipid identifications are limited to molecule and spectral entries in the databases selected.
-
Software for computational biology calculations and statistics is needed such as free online Metaboanalyst 3.0 software at http://www.MetaboAnalyst.ca that includes normalization, clustering tools, heat maps and pathway analysis.
-
Additional software and customized informatics tools include R, a freeware biostatistical suite (http://www.r-project.org/), dChip visualization and expression software (https://sites.google.com/site/dchipsoft/), MetATT for two-factor and time-series data (http://metatt.metabolomics.ca/) and pathway analysis from KEGG IDs in TICL (http://mips.helmholtz-muenchen.de/proj/cmp/), a freeware online tool. Microsoft Excel can also be used to manually calculate fold changes, plot lipids, bar graphs, etc.
-
The LipidMaps consortium (http://www.lipidmaps.org) contains a variety of databases and analysis for lipid researchers.
3 Procedure
3.1 Sample preparation: non-polar lipid extraction
Perform extraction from biological tissues, biological fluids or cultured cells by following the steps in Options A, B or C respectively.
3.1.1 Option A: extraction from biological tissues (tumors/liver/brain/fat)
-
I.
Snap freeze tissue and smash/grind it for 1–2 min in tube with small pestle/tissue grinder on dry ice
-
II.
Note: Although crushing or grinding is sufficient in most cases, a bead beater or tissue lyser can be used to lyse the tissue. Add 200 µL of 1X PBS (RT) to fresh pulverized or ground frozen tissue piece(s) in a 12 mL glass vial
-
III.
Add 1.5 mL HPLC grade methanol and vortex for 1 min. Add 5 mL of MTBE and rock for 1 h at RT
-
IV.
Add 1.2 mL of water and vortex again for 1 min
-
V.
Spin for 10 min at 1,000 g (microcentrifuge, RT)
-
VI.
Collect the upper MTBE liquid phase containing the non-polar lipids
-
VII.
Re-extract the lower liquid phase with 2 volume parts of MTBE/methanol/water (10/3/2.5, v/v/v) by repeating steps v-vi
-
VIII.
Dry the combined MTBE phases in a SpeedVac or under a nitrogen stream
< PAUSE POINT > Dried lipid samples can be stored at −80 °C for several weeks.
3.1.2 Option B: extraction from biological fluids (serum/urine/cerebrospinal fluid):
-
i.
Centrifuge several hundred microliters at 14,000 g for 10 min at room temp to pellet solid particulates
-
ii.
Transfer 200 µL of the supernatant to 15 mL conical tube, if less fluid is available add 1X PBS to a total volume of 200 µL
-
iii.
Repeat steps iii–viii (Option A)
Note: If a higher volume of biological fluids is desired for extraction, adjust the volumes of all steps from the ii–viii (Option A) that the ratios stay the same (sample/methanol/MTBE/water: 1/7.5/25/6, v/v/v/v)
<PAUSE POINT > Dried lipid samples can be stored at −80 °C for several weeks.
3.1.3 Option C: extraction from adherent or suspension cell lines
-
i.
Change media of the 10 cm cell plate(s) 2 h prior to lipid extraction in order to be sure the cells are not lacking nutrients and in the same growth state at harvest
-
ii.
Harvest cells from plates at RT and transfer to 12 mL glass vial
Note: If necessary, trypsin or cell dissociation buffer may be used for adherent cells
-
iii.
Spin for 5 min at 1,000 g (microcentrifuge, RT) and remove supernatant
-
iv.
Add 200 µL of 1X PBS to vial
-
v.
Repeat steps iii – viii (Option A)
<PAUSE POINT > Dried lipid samples can be stored at -80 °C for several weeks.
Note Alternatively to MTBE, 2:1 chloroform:methanol can be used.
3.2 Chloroform:methanol lipid extraction procedure
-
I.
Collect tissue, cell pellet (human, yeast or bacteria) or biological fluid prepared according to the steps above
-
II.
Add with chloroform:methanol (2:1 ratio) to a final volume 20 times the volume of the biological sample (100 mg or 100 µL in 2 mL of solvent mixture) in a 12 mL glass vial
-
III.
Agitate the mixture for 30 min in an orbital shaker at RT
-
IV.
Add 0.2 volume parts of water (400 µL for 2 mL) and vortex for 1 min
-
V.
Let stand for 10 min and centrifuge at low speed (1000 g) to separate into three phases
-
VI.
Remove the upper aqueous phase and keep it (optional) to analyze small organic polar molecules. The middle layer contains protein, DNA and polar large molecules
-
VII.
Remove the lower phase containing non-polar lipids
-
VIII.
Evaporate under vacuum using a SpeedVac rotary evaporator or under a nitrogen stream
3.3 Preparing the instrument for acquisition
Create an instrument method in data dependent acquisition (DDA) mode according to manufacturer’s acquisition software. Enter the mass range for MS1 and MS2, acceptable charge states, resolution for both MS1 and MS2, number of MS2 scans per cycle and the appropriate collision energies for both positive ion mode and negative ion mode. It is necessary to utilize dynamic exclusion in order to maximize the number of M2 acquisitions on unique targets and reduce redundancy.
CRITICAL STEP: Be sure that the autosampler tray and lipid samples are at RT prior to and during analysis to maintain solubility and prevent lipid droplet formation
-
•
Check that the source conditions are suited to high flow rates from the HPLC. For the QExactive Plus, the source tuning parameters for both positive and negative ion mode:
Spray voltage (pos) | +4350 V |
Spray voltage (neg) | −4000 V |
Capillary temp. (pos or neg) | 330 V |
Sheath gas (pos or neg) | 35 |
Aux gas (pos or neg) | 12 |
Spare gas (pos or neg) | 3 |
Max spray current (pos or neg) | 100 |
Probe heater temp. (pos or neg) | 325 °C |
S-Lens RF level | 55 |
DDA acquisition parameters | |
MS1 (pos and neg) | 28 min. acquisition time |
1 mscan, AGC target value | 5e5 |
Max ion injection time | 65 msec |
Resolution | 70,000 |
Scan range (Profile) | m/z 200—m/z 1450 Da |
MS2 (pos and neg) | 28 min. acquisition time |
1 mscan, AGC target value | 1e6 |
Max ion injection time | 75 msec |
Resolution | 35,000 |
Scan range (Profile) | m/z 50 – m/z 2000 Da |
Isolation width | 1.7 Da |
Collision energy | 30 V |
DDA Loop count | Top 8 ions from MS1 scan |
DDA exclusion time | 60 s |
The above parameters represent a DDA experiment in both positive and negative ion mode whereby one MS1 scan in positive ion mode is followed by 8 MS2 scans of the highest intensity ions followed an identical negative ion mode cycle. The 16 ions targeted for MS2 (8 in pos mode and 8 in neg mode) are then put on an exclusion list for 60 s to allow for the MS2 targeting of less intense ions in the following cycles. In the cycle of pos to neg DDA, only one polarity switch occurs in the cycle. This is similar to DDA experiments commonly used in proteomics and allows for deep lipidome coverage (Tate et al. 2013; Breitkopf et al. 2015; Schwudke et al. 2011; Min et al. 2011; Houjou et al. 2005)
The Agilent 1100 HPLC buffer and gradient parameters used for each LC-MS/MS run are listed below. The reversed phase column is a Cadenza 150 mm x 2 mm C18 with 3 µm particle size (Imtakt USA). Set up the generic HPLC gradient as follows:
Flow-rate | ~200–270 µL/min (back pressure should not exceed ~3,000 psi at 32% (v/v) B) |
|---|---|
32% (v/v) B | 0.0 min |
32% B | 1.5 min |
45% B | 4.0 min |
52% B | 5.0 min |
58% B | 8.0 min |
66% B | 11.0 min |
70% B | 14.0 min |
75% B | 18.0 min |
97% B | 21.0 min |
97% B | 25.0 min |
32% B | 26.0 min |
32% B | 32.0 min |
Each buffer composition is as follows:
Buffer A: 60/40 acetonitrile (ACN)/water (H2O), 10mM ammonium formate, 0.1% formic acid
Buffer B: 90/10 isopropanol (IPA)/ACN, 10mM ammonium formate, 0.1% formic acid
Lipid sample preparation
For each lipid of interest, prepare and run a standard compound to be sure of the proper chromatographic elution time on your particular system. It should be prepared in 50% IPA/50% MeOH at a concentration of ~1 µM or less. Before running a real biological sample, one should inject 2–3 methanol blanks to condition the column followed by a freshly prepared “standard” sample to be run as the first sample preceding a biological sample set to be sure that the LC-MS system is operating efficiently (sufficient lipid separation on the HPLC column, well-calibrated MS with proper sensitivity). We use a mixture of 5 different lipids or lipid class mixtures that elute at various times across the chromatogram. The standard lipids are as follows:
-
L-a-phosphatidylinositol (PI),1 × 25 mg (Avanti Polar Lipids, 840042C)
-
Cholesterol, 1 g (Avanti Polar Lipids, 700000P)
-
Sphingomyelin, 2 × 100 mg (Avanti Polar Lipids, 860063)
-
L-a-lysophosphatidylcholine (LPC), 200 mg (Avanti Polar Lipids, 830071P)
-
Stearic Acid (C18:0), 1 g (Matreya, 1020)
-
Triglyceride Mixture (C2-C10), 100 mg (Sigma–Aldrich, 17810-1amp-s).
Make up the following stock solutions from the vendor bottles. TG mix (25 mg/mL), SM (25 mg/mL), LPC (1 mg/mL), SA (10 mg/mL), PI (10 mg/mL), Cholesterol (10 mg/mL)
Prepare the final lipid standard by mixing 1 µL of each stock and diluting to 200 µL in 50% IPA/50% MeOH and store in a glass 12 × 32 mm vial. Inject 5 µL of the standard mix onto LC-MS/MS. The final concentration of each standard is 125 µg/mL (125 ng/µL) for TG mix and SM, 5 µg/mL for LPC and 50 µg/mL for PI and cholesterol
The standard sample should produce a set of distinct peaks in the MS1 spectra from ~2 min to 28 min. Figures 1, 2 and 3 show examples of the peaks in the MS1 and MS2 ion chromatograms from both positive and negative ion modesCholesterol, 1 g (Avanti Polar Lipids, 700000P)
For lipid samples after extraction with MTBE or chloroform:methanol(2:1), add ~30 μL of 1:1 LC/MS grade isopropanol:acetonitrile methanol to resuspend each biological sample just prior to LC-MS/MS analysis
Inject ~5-8 μL of sample onto the LC-MS/MS system using an autosampler. The aim is to load a sample amount just below the column and mass spectrometer saturation point in order to avoid carryover but sufficient to obtain the highest possible number of lipid ions. (Do not inject more than half of the sample in case of a system failure and inject no more than ~5 μL if technical triplicate runs are desired).
In order to prevent carryover due to column saturation one can add a methanol blank LC-MS/MS run between each sample. In addition, one can run the methanol blank through the lipid identification software (below) to assess the level of column carryover. For sample containing very high lipid levels, multiple blanks should be run between samples.
At the end of each sample set, add 2-3 methanol blanks to prevent carry over followed by another 1-2 “standard” samples to assure the platform is still performing efficiently.

Lipidomics platform schematic. Overview of the untargeted LC-MS/MS experiment for lipid metabolite profiling via data dependent acquisition (DDA) using positive/negative switching from a single 28 min reversed phase column run while routinely identifying and quantifying more than 1000 compounds

LipidSearch software workflow. a RAW data files from the high resolution QExactive Plus are loaded into the LipidSearch software which then integrates the MS1 peak areas, and then proceeds to interpret the MS2 fragment ions for lipid ion identification based on an internal database query. The lipid molecules are then scored to accept the top scoring lipid and rated using a quality assessment of A-D. During the peak alignment step, the lipid ion peak areas are compared across the sample set and relative quantification is performed. b Lipids are identified based on the major structural units of lipid molecules including the lipid backbone, head group, fatty acid chain composition and adduct ions within each polarity mode. For this to be successful, high mass accuracy is necessary

Lipid standards and quantitative validation. a The bar plot with error bars from three replicate injections for the MS1 peak areasshow the quantitative accuracy of the lipid standards LPC, SM and TG mix from 4 different concentration dilutions (1:1, 1:2, 1:4 and 1:8); the 1:1 conc. = 125 µg/mL. b The SM lipid standard fatty acid profile with relative peak areas and error bars. c The relative peak areas correlate with the injected sample concentration on the level of single lipid ions as seen with the sphingomyelin lipid SM(16:0/24:1) which include CV values. d The same single ion quantification of the phosphatidylinositol PI(18:0/20:4)
3.4 Data analysis
Once the DDA data is acquired, MS1 peaks and MS2 fragment ion peaks must be integrated in order to generate both chromatographic peak areas used for alignment across the sample set and for identification of lipid molecules. Use a suitable software platform for identification and quantification such as LipidSearch version 4.1 or later from Thermo Fisher Scientific(Breitkopf et al. 2015; Peake et al. 2013). LipidSearch software first identifies lipid based on an internal library of masses and fragment ions from fatty acid chains and head groups from more than 18 main lipid classes and 66 sub classes. Lastly, the software can perform alignment of samples from patient or sample cohorts in order to accurately quantify the MS1 peak profiles
Note: Ideally, standard compounds from lipids of interest should be tested on the platform to assure proper chromatographic elution times and fragmentation in the optimized polarity mode.
-
•
For LipidSearch lipid identification and quantitation parameters, use the following:
Database | General and Q exactive |
|---|---|
Search type | Product |
Exp type | LC-MS |
Precursor tolerance | ~5–7 ppm |
Product tolerance | 7–10 ppm |
Intensity threshold | 1.0% |
height-Score threshold | 8.0 |
Execute Quantitation | On |
M/z tolerance (quantitation) | −/+ 5.0 ppm |
RT range (min.) (quantitation) | −/+ 0.5 |
Top rank filter | On |
Main node filter | Main isomer peak |
m-Score threshold | 5.0 |
c-Score threshold | 2.0 |
FA priority | On |
ID Quality Filter | Check A, B, and C or A-D for lower quality IDs |
Target class | Select ALL lipid classes |
Ion adducts (pos) | +H, +NH4 and +2 H |
Ion adducts (neg) | −H, +HCOO and −2H |
Note: The m-Score is calculated based on the number of matches with product ion peaks in the spectrum. A higher m-Score means higher identification reliability compared to the c-Score which is calculated from the total of the degrees of coincidence between each neutral loss and precursor scan peak for peaks of a given lipid. The Multi-Search allows the identification of peaks obtained from MS, product ion scanning or neutral loss scanning of class- or fatty acid-specific fragments on triple quadrupole instruments. The ID quality filter A-B type identifies all features including head group, glycerol backbone and both fatty acid chains whereby C-D type identifies the lipid class based on head group and backbone but may have missing information in the MS2 spectra for one of the fatty acid chains or have merged fatty acid chain information. D type identifications base Lipid IDs simply by mass or features such as fragment ions (water losses, etc.)
Select a representative control sample and manually scroll through each lipid peak to be sure that the expected retention time, peak width and masses are accurate. If there is an error with peak selection, one can investigate whether the problem lies within the chromatography or the mass spectrometry component
<CRITICAL STEP> It is important to run a standard compound to be sure of the proper chromatographic elution time on your particular system. This is essential when incorporating new standard compounds into your workflow. Previous knowledge of the compound of interest and the chromatography can also be used to identify the correct peak for identification and relative quantitation. Lipid spectra and information can be obtained from public mass spectral databases such as LipidMaps (http://www.lipidmaps.org/) or the Human Metabolome Database (HMDB) (http://www.hmdb.ca/)
After completing the lipid identification step, peak alignment should be performed in order to group each sample cohort and to quantify individual lipids across your dataset. The parameters used for Alignment in LipidSearch software are the following:
Search type | Product |
|---|---|
ExpType | LC-MS |
Alignment Method | Max (or Mean) |
R.T. Tolerance | 0.50 min (depends upon chromatographic reproducibility, typically ~0.25 min to 0.8 min) |
Calculate unassigned peak area | On |
Filter type | New filter |
Toprank filter | On |
Main node filter | Main isomer peak |
m-Score threshold | 5.0 |
ID quality filter | A, B and C (uncheck D) |
After alignment, the entire alignment file including individual lipid ion measurements for each of the samples can be exported into a .txt file and then opened by Excel. Charts of both lipid classes and fatty acid chains per lipid class can be viewed in the alignment profile of LipidSearch software. In some cases, a lipid ion will be identified in both positive and negative ion modes due to a different adduct for ion formation. When analyzing such data, be sure to quantify the lipid ion across samples using the same polarity mode as different polarity modes will yield different peak areas due to different ionization efficiencies
3.4.1 <Alternative Software> Identification and relative quantification using Elements
Elements (Proteome Software, Inc.) performs metabolite identification through MS and MS2 searches against multiple metabolite/lipid databases that include spectra libraries, such as the NIST, HMDB and LipidMaps. Note that only the NIST database contains sufficient MS2 spectral data (version 2014 contains 234,284 MS/MS spectra). Once imported, the MS data is analyzed for feature (peak) detection, searched for identifications by MS1 mass accuracy, MS2 data is then searched for fragmentation peak matches vs. existing spectral libraries and, if multiple samples are loaded, aligned according to retention time and mass(Figure S3B). Be aware that the Elements results are largely limited by the quality and size of the MS2 spectral libraries available to Elements. This can result in a significant difference in the output between LipidSearch and Elements.
The parameters used for Elements software are the following:
Parent mass tolerance | 7–10 ppm |
|---|---|
Fragment mass tolerance | 0.50 Da (MS2 library contains mostly low mass accuracy data) |
Mass range (M/z) | 50–1000 |
Max charge | 2 |
Ion mode | Mixed (Pos. and Neg.) |
Noise threshold | 0.1% of max signal |
Adducts | [M + NH4]+; [M + H]+; [M-H]- |
Library 1 | hmdb_library_elements.libdb |
Library 2 | nist.libdb (other libraries can be used such as LipidMaps) |
ID Score threshold | 0.6 |
The ID score is the backbone scoring system within Elements and consists of a weighted average of the highest value for each of three match scores, calculated over the set of features that are associated with the given molecule. These match scores include a mass accuracy score, an MS2 fragmentation score and an isotopic distribution score. The ID Score is then adjusted according to the number of ion forms matched.
3.4.2 Troubleshooting
-
•
The dried lipid pellet appears to be very large and fluffy. This may indicate contamination with large molecules (protein, DNA, etc.). Be sure that only the upper non-polar lipid phase was extracted after sufficient MTBE phase separation (or the lower phase if using a chloroform:methanol extraction). Allow sufficient incubation time at RT with MTBE and be sure to spin the entire liquid–liquid extraction solution containing methanol and water at 1,000 g for a minimum of 10 min. One can spin for a longer time if the phases do not clearly separate after 10 min. The final pellet should be less than a 20 µL liquid equivalent in volume according to a 1.5 mL microcentrifuge tube
-
•
The mass spectrometer is not showing any peaks above baseline. This would indicate that not enough lipid content is on the HPLC column during the acquisition. Be sure that HPLC buffers are filled, that the nitrogen gas flow rate for collision and source is sufficient, and that the column or tubing is not leaking. How many cells or how much tissue was used? It is possible that more sample quantity is needed (equivalent of >1–2 million cells or >5–10 mg of tissue). In addition, the ion source area has a tendency to get dirty so routine source cleaning is necessary
-
•
The lipid peaks are either not being retained or eluting very late on the HPLC column. Are your HPLC buffer concentrations and pH correct? Are the HPLC A and B pumps properly purged to remove air in pumps? If so, this may indicate that either your pre-column filter is clogged or that you need to change or clean your HPLC column due to contamination. First, try changing the pre-column filter followed by an analytical column change
-
•
Some lipids are never detected no matter what I change or what concentrations I inject. It is possible that the lipid conditions are not optimal or that the collision energy selected is not ideal for the specific lipid compound of interest. Also, the particular lipid of interest may exist in very low concentrations in your biological matrix or degrades quickly. For example, PIP2 and PIP3 lipids involved in kinase signaling are present in very low levels and are sticky so they are more difficult to detect than other lipids. They may require direct infusion rather than column separation. Other possibilities are that MTBE or chloroform/methanol liquids are not ideal for the lipid extraction or the chromatography used is not suitable for the compound of interest. If you are using a database search method via Elements, it is possible that your lipid of interest is not present in the spectral library
-
•
Multiple instances of unique lipid species are showing up in the LipidSearch analysis In some cases, if the retention time window during the alignment step is not set wide enough to encompass the lipid ion’s elution profile or if some lipid peaks are very wide, contain shoulders, tails, etc., the LipidSearch software may interpret identical lipid ions as multiple peaks and list them multiple times. In these cases, one can either eliminate the duplicate results showing lower peak area values since the main peak component shows a much higher peak area, or one can sum all instances of the same lipid ion. One can also attempt to re-align the dataset using a wider retention time window. Before attempting this, be sure that the ions are truly identical since they may be due to different charge states, polarity modes or different degrees of fatty acid chain saturation
-
•
The spray needle is prone to clog periodically. Lipid molecules can be sticky and the buffers contain some salt so if the spray needle is not flushed with clean solvent, it can clog and require replacement. To help avoid this, run a low flow (0.002 mL/min) of 1:1 methanol/water when the instrument is not in use or between large datasets
3.4.3 Timing
-
From tissue/cells/fluid harvest to sample injection metabolite extract takes ~4–6 h mainly depending upon the drying speed of the SpeedVac/lyophilizer
-
The total LC-MS/MS run time is 33 min (28 min MS acquisition) from inject to inject
-
Peak identification, alignment, quantification and verification of raw data takes ~1–3 h per dataset depending upon the number of samples
Data analysis using various bioinformatics software tools can vary depending upon the degree of interpretation.
3.4.4 Anticipated results
The above parameters on most MTBE or CHCl3:MeOH lipid extractions from our 30 min LC-MS/MS QExactive Plus platform can produce a range of ~750–1750 unbiased lipid ion identifications per run, depending upon sample complexity and sample abundance. These identifications are split between both positive and negative modes, depending upon the lipid type. LipidSearch software can also align datasets and group different cohorts for relative quantification of lipids based on the user’s customized needs for data analysis. Figure 1 shows the workflow for the lipidomics platform from lipids extracted from 2 to 3 million TSC2-/- MEF cells, 30 mg of mouse lung tissue or 150 µL of human blood plasma from a MTBE or chloroform:methanol extraction that was injected onto our Agilent 1100-Thermo QExactive Plus-Thermo LipidSearch lipidomics platform. Approximately 1/6 of the total lipid extraction was injected onto the LC-MS/MS system (5 of 30 µL) after a dry-down of the extraction and solubilization in 50% IPA/50% MeOH. The platform is capable of successfully profiling far lower sample amounts at the expense of less lipid identifications. For example, a 10 mg extraction of mouse breast tumor with a 1/6 injection volume yields approximately 750 lipid identifications and a 25 µL serum extraction at 1/6 injected amount yields approximately 400 lipid identifications. The MS1 ion chromatogram from both the positive and negative ion modes are shown in addition to a scatterplot (Log2 Ratio vs. Log2 Intensity) and heat map of the quantitative results. The spectrum is also annotated to show the regions that are typically enriched for certain lipid classes in the C18 chromatogram. This workflow represents a typical untargeted high resolution LC-MS/MS experiment that is subjected to LipidSearch software and downstream data analysis. To control for proper identification and relative quantification, a set of known lipid standards at different concentrations can be injected on the platform. Figure 2a shows the LC-MS/MS ion chromatogram, MS/MS spectrum and scoring charts for the phosphatidylinositol lipid PI (17:0/18:2) in negative ion mode. The figure also shows a list of top scoring lipid ions identified from the precursor mass and fragment ions using LipidSearch software as well as the fragment ion match details for MS2 ions in the HCD spectrum. One can also observe that the PI lipid is present in different quantitative levels across four different sample conditions and elutes in a similar chromatographic region of the LC-MS chromatogram, as expected. Figure 2b shows how a lipid molecule can be assembled with the different fragments that LipidSearch uses in its library for identification. In our buffer/column system, the most common ion adducts for positive mode are +H+, +HCOO+ and +NH4 + and for negative mode it is deprotonation, -H−. Figure S1A shows the FT-MS spectra and FT-HCD fragmentation spectra from 1552 unique lipid ions extracted from human blood plasma (Supporting Dataset 5). The data shows that acquiring lipid data in both positive ion and negative ions modes is necessary since different lipids are prone to produce ions in either mode. The data in Figure S2B shows the MS1 base peak trace from a mixture of six lipid standards that included a triglyceride mix (C2-C10) (TG), sphingomyelin (SM), lysophosphatidylcholine (LPC), stearate (C18:0), L-a-phosphatidylinositol (PI) and cholesterol in both positive and negative ion mode with each peak or set of peaks labeled. Notice that the chromatographic elution times for some different lipid compounds vary within the same lipid class. This data shows that the fatty acid composition in addition to the head group is an important factor in lipid chromatography. Figure 3 shows the reproducibility of our platform from three lipids (LPC, SM and one TG) within our standard sample mixture. The lipid standards were injected in technical triplicate at four different concentrations (1:1 (125 µg/mL), 1:2 (62.5 µg/mL), 1:4 (31.25 µg/mL), 1:8 (15.63 µg/mL)) and the average peak areas of the three lipid classes LPC, SM and TG were plotted (Fig. 3a). The data shows the accurate correlation between the measured intensity of the lipid class according to the injected concentration. Figure 3b provides the SM lipid fatty acid profile and clearly shows that the relative peak area correlates with the injected sample concentration. This is true for the individual lipid ions such as SM (16:0/24:1) (Fig. 3c) and PI (18:0/20:4) (Fig. 3d). CV values were also calculated for each concentration of the SM and PI lipids and combined with the error bars, show the relative quantitative accuracy and reproducibility of the platform (Fig. 3c, d). The LipidSearch output for the standard mix dataset can be found in Supporting Dataset 1 and includes all identified lipid ions as well as their composition, mass, retention time and peak area, etc.
To test the platform from cells with a genetic background known to influence lipid metabolism, we performed an experiment to study the lipid profiles of paired mouse embryonic fibroblasts (MEF) with wild type or knockout of the tumor suppressor tuberous sclerosis complex 2 (Tsc2) using three biological replicates per sample condition. TSC2 is a component of the TSC complex, which is a negative regulator of mTORC1(Kwiatkowski and Manning, 2014). As a major regulator of cell growth and proliferation, mTORC1 has been found to alter lipid metabolism in many settings (Ricoult and Manning 2013). Members of the sterol regulatory element binding protein (SREBP) family of transcription factors, which activate the transcription of many genes required for de novo fatty acid and sterol synthesis (Horton et al. 2002), are downstream transcriptional effectors of mTORC1 under both physiological and pathological setting (Duvel et al. 2010; Porstmann et al. 2008; Yecies et al. 2011; Ricoult et al. 2015). Wild-type MEFs were compared to littermate-derived Tsc2-null MEFs, which have growth factor-independent activation of mTORC1 signaling, and the role of mTORC1 and SREBP were assessed through use of the mTORC1 inhibitor rapamycin (20nM, 16 h) and siRNA-mediated knockdown SREBP (72 h), respectively. Lipids from cells grown in the absence of exogenous growth factors were analyzed from triplicate extractions from four different biological conditions (12 total samples). Each sample yielded between 1150 and 1312 lipid ion identifications and, after alignment, a total of 2,493 unique lipid ion identifications across 12 total samples. 1,756 were identified in positive ion mode and 737 from negative ion mode. “D” quality identifications were omitted from the LipidSearch alignment with A, B and C graded lipid identifications accepted for further analyses.
Analysis of the data showed that the most abundant lipid classes identified from MEFs were phospholipids. Phospholipids are a major component of membranes, which explains their abundance. The lipid class profile generated from LipidSearch software (Fig. 4a) shows that phosphatidylcholine (PC) is four times more abundant than the next lipid class for all conditions tested. Abundant lipid classes from MEFs also include phosphatidylethanolamine (PE), sphingomyelin (SM), and triglyceride (TG). The comparative results show that the TG class displays significant up-regulation in Tsc2 −/− cells treated with rapamycin for 16 h. While this lipidomics platform can detect differences among lipid classes across different biological conditions, the real power in using an unbiased approach with LipidSearch software is the ability to detect individual intact lipid molecules that are regulated. In order to further investigate the TG lipids, each lipid species within this class was plotted. Figure 4b shows the fatty acid profile that was detected for the triglyceride lipid class from Tsc2 −/− MEFs. Of the more than 200 TGs, many contain the basic fatty acid building blocks of palmitate (C16:0), palmitoleate (C16:1), stearate (C18:0) and oleate (C18:1). The fatty acid profile for the most abundant diacylglycerides (DG) also shows the basic building blocks (C16:0, C18:1, C18:0, C16:1), although they also show longer fatty acid chain lengths. Interestingly, rapamycin-treatment increased nearly all species of TGs, relative to vehicle-treated Tsc2 −/−cells. Unlike TGs, other lipid classes show little class-wide regulation between vehicle and rapamycin-treated Tsc2 −/− cells (Fig. 4a). The LipidSearch output for the Tsc2 MEFs dataset can be found in Supporting Dataset 2 and includes all identified lipid ions as well as their composition, mass, retention time and peak area, etc.

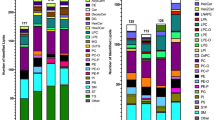
Lipid class and fatty acid profiles. a The overall lipid class profile across all identified lipids from extracts (~5 × 105 cells injected) of TSC2-/- MEF cells from three biological replicates per condition. PC lipids are the most abundant in MEFs followed by PE, SM, TG, DG, PI and PS. Across the four cellular conditions, TG was the most responsive by increasing upon stimulation with 20nM of the TOR inhibitor Rapamycin (condition S3) as compared to untreated TSC2-/- cells (condition S1). b The fatty acid profile for the TG lipid class showing that the majority of the fatty acid chains in triglycerides in MEFs contain the basic lipid building blocks of palmitate, palmitoleate, oleate and stearate. The data also shows that the majority of the 249 TG lipid ions are elevated when TSC2-/- cells are treated with Rapamycin
In addition to LipidSearch software, free online MetaboAnalyst 3.0 software was used for quality control of the lipidomics data, to generate heat maps, PCA clustergrams, K-means clustering, and to normalize the data. The LipidSearch alignment data was exported to .xls format and the peak area data were used to create the .csv input for MetaboAnalyst. Figure 5a shows the PCA clustering plot (PC1 vs. PC2) for the four MEF conditions described in Fig. 4. The biological triplicates for each cellular condition cluster separately, although the SREBP-knockdown cells show the most variance, perhaps due to varying efficiency of knockdown or cellular stress caused by loss of SREBP. Figure 5b shows a Ward clustergram, and consistent with reproducibility within each condition, the biological triplicates clusters separately. Compared to the vehicle-treated Tsc2-null MEFs with constitutive mTORC1 activation, the rapamycin-treated Tsc2-null cells cluster more closely with the Tsc2-WT MEFs, with inactive mTORC1. Figure 5c shows the basic biological pathway from mTOR signaling to lipid biosynthesis via SREBP, ACC, FAS, etc. A heat map of the most regulated 150 lipid species from this analysis shows those lipids most significantly increased or decreased between conditions (Fig. 5d). One can observe that most triglyceride (TG) lipid ions are significantly up-regulated in rapamycin- treated Tsc2 −/− cells. Interestingly, these results mirror, at the cellular level, the observation that human patients on rapamycin frequently develop hypertriglyceridemia (Morrisett et al. 2002; Kasiske et al. 2008). Consistent with the dependence of Tsc2 −/− cells on SREBP for lipid synthesis (Duvel et al. 2010), most lipid species are reduced in these cells with siRNA-mediated knockdown of SREBP.

Biostatistical analysis of TSC2 cell lipidomics data. a PC1 vs. PC2 PCA clustering analysis of TSC2+/+ mouse embryonic fibroblast cells, TSC2-/- cells, TSC2-/- with siSREBP cells and TSC2-/- with Rapamycin treated cells. The different sample types cluster distinctly and the biological replicates cluster tightly. b Sample clustering using the WARD method showing that each TSC2 sample type and replicates cluster as expected. c The basic lipid biosynthesis showing the major enzymes and pathways including the metabolic pathways and signaling pathways via mTOR leading to fatty acid synthesis via ACC, FAS and SREBP regulation. d A heat map of the same biological sample conditions as in A and B showing specific groups of lipid classes and their regulation
In addition to cultured cells, we also extracted lipids from solid tissue of normal mouse lung and mouse lung tumor. The mice were c57 black mice (Jackson Laboratory) and the tumors contained the T790M epidermal growth factor receptor (EGFR) mutation. Figure 6 shows lipidomics profiling data from 30 mg of lung tumor vs. normal lung tissue. A scatterplot of Log2 ratio (Tumor/Normal) vs. Log10 (Peak Area intensity) was generated (Fig. 6a) as well as a bar plot showing the most highly regulated lipid ions in the lung tumor tissue vs. normal lung (Fig. 6b). More than 2000 total unique lipid ions were identified in the analysis of both the tumor and normal tissue. Interestingly, up-regulated lipids in the EGFR lung tumor harbor TGs and phosphatidylmethanol (PMe) lipid ions while the down-regulated lipids contain a variety of phospholipids such as PS, PE, PI, and PG as well as some cardiolipins and cholesterol esters. The LipidSearch output for the lung tumor dataset can be found in Supporting Dataset 3.

Global lipid regulation in lung tumor tissue. a A scatterplot (Log10 Intensity vs. Log2 Ratio) showing the distribution of 2,062 identified lipid ions via LipidSearch from lipids extracted from three biological replicates of both T790M EGFR driven mouse lung tumor and normal mouse lung tissue extracts (~5 mg injected). b A bar plot of the most regulated lipid ions from the lung tumor and normal lung tissue experiment. The most regulated lipids suggest that a higher concentration of TG lipids is up-regulated while more phospholipids are down-regulated
In addition to LipidSearch, Elements software from ProteomeSoftware, originally developed for metabolomics can be used to identify lipid ions provided that lipids exist in spectral databases from NIST, HMDB, LipidMaps, etc. Figure S2A shows an example of the Elements output for a profiling experiment of biological triplicates of mouse lung tumor vs. normal lung tissue. Several scores are shown including an identification score, mass accuracy score, isotopic distribution score and a MS2 score. The quantitative values are shown as Log2 Intensity and one can choose whether normalization is utilized before exporting the data in Excel format as an array of lipid ions, scores and MS1 intensity values. Figure S2B shows an example of the identified lipid molecule LysoPE(16:0) as the MS/MS spectrum and molecular structure from the NIST database. Figure S3 shows lipidomics profiling data from 30 mg of a T790M lung tumor vs. normal lung tissue generated with Elements. Figure S3A displays a bar plot showing the most highly regulated lipid ions identified from a lung tumor tissue and normal lung lipidomics experiment using Elements software as well as a Log2 ratio (Tumor/Normal) vs. Log10 (Peak Area intensity) scatterplot from 762 identified unique lipid ions. Data were derived from Supporting Dataset 4. Figure S3B shows the workflow of the Elements software from the point of .RAW data collection to the final results file output as displayed in Figure S2A. It is important to note that Elements software is completely dependent upon the population of the small molecule fragmentation spectral libraries which are generally underpopulated to date so the results do not overlap to a great extent with LipidSearch, which is a database assisted de novo identification strategy.
4 Discussion
The LC-MS/MS based lipidomics platform based on QExactive Orbitrap technology and LipidSearch software can be used to identify and quantify lipid molecules in a non-targeted manner with high resolution mass spectrometry. The platform requires no chemical modifications of the lipid molecules and can be extracted from any biological source. The platform does not rely on an existing spectral database for lipid ion identification but an internal database of known spectral patterns for each lipid class. Keep in mind that structural isomers of identical mass with similar fragmentation patterns may not be discerned using this approach unless they can be chromatographically separated. We outline the parameters for sample preparation, instrumental analysis and data analysis. The combination of high resolution MS and LipidSearch software provides a powerful platform for high-throughput lipidomics profiling for biomarker discovery, relative quantification of lipid class, fatty acids, etc. In addition, the use of Elements software based on a fragmentation spectral library search can be used to identify lipids that are not covered by the main LipidSearch classes but Elements can also be limited by both the number of library entries and the quality of the fragmentation spectra. While LipidSearch can currently only be used with .RAW files from Thermo style high resolution mass spectrometers, Elements can be used by a wide variety of vendors since it uses freeware ProteoWizard’s MSConvert tool to generate spectral peak lists from the raw datafiles. The techniques presented require minimal sample handling and the instrument and software is automated after parameter setup. Both LipidSearch and Elements can be used in a complimaentarty fashion to both identify novel lipid ions and to match known lipid ions from spectral databases. We showed that biological differences between various different cell types and genetic background can be profiled and quantified using the untargeted LC-MS/MS platform.
References
AHMED, Z., MAYR, M., ZEESHAN, S., DANDEKAR, T., MUELLER, M. J., & FEKETE, A. (2015). Lipid-Pro: A computational lipid identification solution for untargeted lipidomics on data-independent acquisition tandem mass spectrometry platforms. Bioinformatics (Oxford, England), 31, 1150–1153.
Asara, J. M., Xu, Y., Breitkopf, S. B., Yuan, M., Ricoult, S. J. & Manning, B. D. (2016). Preparing Biological Samples for Metabolomics and Lipidomics, Can We Start with Just One Sample? Association of Biomolecular Resource Facilities. Ft. Lauderdale.
Ben-Sahra, I., Howell, J. J., Asara, J. M., & Manning, B. D. (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science, 339, 1323–1328.
Bilgin, M., Born, P., Fezza, F., Heimes, M., Mastrangelo, N., Wagner, N., Schultz, C., Maccarrone, M., Eaton, S., Nadler, A., Wilm, M., & Shevchenko, A. (2016). Lipid Discovery by Combinatorial Screening and Untargeted LC-MS/MS. Scientific Reports, 6, 27920.
Bird, S. S., Marur, V. R., Sniatynski, M. J., Greenberg, H. K., & Kristal, B. S. (2011). Serum lipidomics profiling using LC-MS and high-energy collisional dissociation fragmentation: focus on triglyceride detection and characterization. Analytical Chemistry, 83, 6648–6657.
Breitkopf, S. B., Yuan, M., Helenius, K. P., Lyssiotis, C. A., & Asara, J. M. (2015). Triomics analysis of imatinib-treated myeloma cells connects kinase inhibition to RNA processing and decreased lipid biosynthesis. Analytical Chemistry, 87(21), 10995–11006
Breitkopf, S. B., Yuan, M., XU, Y., & Asara, J. M. 2016. A quantitative positive/negative ion switching method for shotgun lipidomics via high resolution LC-MS/MS from any biological source. San Antonio: American Society for Mass Spectrometry
Cajka, T., & Fiehn, O. (2014). Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends In Analytical Chemistry, 61, 192–206.
Chou, C. H., Chang, W. C., Chiu, C. M., Huang, C. C., & Huang, H. D. (2009). FMM: A web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Research, 37, W129–W134.
Collins, J. R., Edwards, B. R., Fredricks, H. F., & van MOOY, B. A. (2016). LOBSTAHS: An adduct-based lipidomics strategy for discovery and identification of oxidative stress biomarkers. Analytical Chemistry, 88, 7154–7162.
COOMBS, K. M. (2011). Quantitative proteomics of complex mixtures. Expert Review of Proteomics, 8, 659–677.
COX, J., & MANN, M (2011). Quantitative, high-resolution proteomics for data-driven systems biology. Annual Review of Biochemistry, 80, 273–299.
Duvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., Triantafellow, E., Ma, Q., Gorski, R., Cleaver, S., Vander Heiden, M. G., Mackeigan, J. P., Finan, P. M., Clish, C. B., Murphy, L. O., & Manning, B. D. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell, 39, 171–183.
Fahy, E., Sud, M., Cotter, D., & Subramaniam, S. (2007). LIPID MAPS online tools for lipid research. Nucleic Acids Research, 35, W606–W612.
Fauland, A., Kofeler, H., Trotzmuller, M., Knopf, A., Hartler, J., Eberl, A., Chitraju, C., Lankmayr, E., & Spener, F. (2011). A comprehensive method for lipid profiling by liquid chromatography-ion cyclotron resonance mass spectrometry. Journal of Lipid Research, 52, 2314–2322.
Folch, J., Lees, M., & Sloane Stanley, G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. The Journal of Biological Chemistry, 226, 497–509.
Godzien, J., Ciborowski, M., Martinez-Alcazar, M. P., Samczuk, P., Kretowski, A., & Barbas, C. (2015). Rapid and reliable identification of phospholipids for untargeted metabolomics with LC-ESI-QTOF-MS/MS. Journal of Proteome Research, 14, 3204–3216.
Han, X., Yang, K., & Gross, R. W. (2012). Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrometry Reviews, 31, 134–178.
Hara, H., Uchimura, T., Akashi, N., Naganuma, T., Aizawa, T., Nagae, Y., & Masuda, N. (2004). Simultaneous analytical method for the determination of TCH346 and its four metabolites in human plasma by liquid chromatography/tandem mass spectrometry. Rapid Communications in Mass Spectrometry, 18, 377–384.
Hartler, J., Trotzmuller, M., Chitraju, C., Spener, F., Kofeler, H. C., & Thallinger, G. G. (2011). Lipid data analyzer: Unattended identification and quantitation of lipids in LC-MS data. Bioinformatics (Oxford, England), 27, 572–577.
Hein, E. M., Bodeker, B., Nolte, J., & Hayen, H. (2010). Software tool for mining liquid chromatography/multi-stage mass spectrometry data for comprehensive glycerophospholipid profiling. Rapid Communications in Mass Spectrometry, 24, 2083–2092.
Hopperton, K. E., Duncan, R. E., Bazinet, R. P., & Archer, M. C. (2014). Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Experimental Cell Research, 320, 302–310.
Horton, J. D., Goldstein, J. L., & Brown, M. S. (2002). SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of Clinical Investigation, 109, 1125–1131.
Hou, W., Zhou, H., Bou Khalil, M., Seebun, D., Bennett, S. A., & Figeys, D. (2011). Lyso-form fragment ions facilitate the determination of stereospecificity of diacyl glycerophospholipids. Rapid Communications in Mass Spectrometry, 25, 205–217.
Houjou, T., Yamatani, K., Imagawa, M., Shimizu, T., & Taguchi, R. (2005). A shotgun tandem mass spectrometric analysis of phospholipids with normal-phase and/or reverse-phase liquid chromatography/electrospray ionization mass spectrometry. Rapid Communications in Mass Spectrometry, 19, 654–666.
Junot, C., Fenaille, F., Colsch, B., & Becher, F. (2014). High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrometry Reviews, 33, 471–500.
Kasiske, B. L., De Mattos, A., Flechner, S. M., Gallon, L., Meier-Kriesche, H. U., Weir, M. R., & Wilkinson, A. (2008). Mammalian target of rapamycin inhibitor dyslipidemia in kidney transplant recipients. American Journal of Transplantation, 8, 1384–1392.
Kluger, B., Bueschl, C., Neumann, N., Stuckler, R., Doppler, M., Chassy, A. W., Waterhouse, A. L., Rechthaler, J., Kampleitner, N., Thallinger, G. G., Adam, G., Krska, R., & Schuhmacher, R. (2014). Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching LC-ESI-HRMS. Analytical Chemistry, 86, 11533–11537.
Kofeler, H. C., FAULAND, A., RECHBERGER, G. N., & TROTZMULLER, M. (2012). Mass spectrometry based lipidomics: an overview of technological platforms. Metabolites, 2, 19–38.
Koyama, J., Taga, S., Shimizu, K., Shimizu, M., Morita, I., & Takeuchi, A. (2011). Simultaneous determination of histamine and prostaglandin D2 using an LC–ESI–MS/MS method with positive/negative ion-switching ionization modes: Application to the study of anti-allergic flavonoids on the degranulation of KU812 cells. Analytical and Bioanalytical Chemistry, 401, 1385–1392.
Kwiatkowski, D. J., & Manning, B. D. (2014). Molecular basis of giant cells in tuberous sclerosis complex. The New England Journal of Medicine, 371, 778–780.
Lam, S. M., & Shui, G (2013). Lipidomics as a principal tool for advancing biomedical research. Journal of Genetics and Genomics, 40, 375–390.
Li, M., Yang, L., Bai, Y., & Liu, H. (2014). Analytical methods in lipidomics and their applications. Analytical Chemistry, 86, 161–175.
Linden, M. A., Lopez, K. T., Fletcher, J. A., Morris, E. M., Meers, G. M., Siddique, S., Laughlin, M. H., Sowers, J. R., Thyfault, J. P., Ibdah, J. A., & Rector, R. S. (2015). Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Applied Physiology, Nutrition, and Metabolism, 40, 1038–1047.
Ma, S., & Chowdhury, S. K. (2013). Data acquisition and data mining techniques for metabolite identification using LC coupled to high-resolution MS. Bioanalysis, 5, 1285–1297.
Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A., & Schwudke, D. (2008). Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. Journal of Lipid Research, 49, 1137–1146.
Menendez, J. A., & Lupu, R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature Reviews, 7, 763–777.
Min, H. K., Lim, S., Chung, B. C., & Moon, M. H. (2011). Shotgun lipidomics for candidate biomarkers of urinary phospholipids in prostate cancer. Analytical and Bioanalytical Chemistry, 399, 823–830.
Morrisett, J. D., Abdel-Fattah, G., Hoogeveen, R., Mitchell, E., Ballantyne, C. M., Pownall, H. J., Opekun, A. R., Jaffe, J. S., Oppermann, S., & Kahan, B. D. (2002). Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. Journal of Lipid Research, 43, 1170–1180.
Narvaez-Rivas, M., & Zhang, Q (2016). Comprehensive untargeted lipidomic analysis using core–shell C30 particle column and high field orbitrap mass spectrometer. Journal of Chromatography A, 1440, 123–134.
Nicolay, B. N., Gameiro, P. A., Tschop, K., Korenjak, M., Heilmann, A. M., Asara, J. M., Stephanopoulos, G., Iliopoulos, O., & Dyson, N. J. (2013). Loss of RBF1 changes glutamine catabolism. Genes and Development, 27, 182–196.
Peake, D. A., Yokoi, Y., Wang, J. & Huang, Y. (2013). A New Lipid Software Workflow for Processing Orbitrap-based Global Lipidomics Data in Translational and Systems Biology Research. http://www.thermoscientific.com.
Porstmann, T., Santos, C. R., Griffiths, B., Cully, M., Wu, M., Leevers, S., Griffiths, J. R., Chung, Y. L., & Schulze, A. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metabolism, 8, 224–236.
Ricoult, S. J., & Manning, B. D. (2013). The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Reports, 14, 242–251.
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I., & Manning, B. D. (2015). Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene, 35(10), 1250–1260
Rodamer, M., Elsinghorst, P. W., Kinzig, M., Gutschow, M., & Sorgel, F. (2011). Development and validation of a liquid chromatography/tandem mass spectrometry procedure for the quantification of sunitinib (SU11248) and its active metabolite, N-desethyl sunitinib (SU12662), in human plasma: Application to an explorative study. Journal of Chromatography B, 879, 695–706.
Rolim, A. E., Henrique-Araujo, R., Ferraz, E. G., Dultra, F. K. D. A. A., & Fernandez, L. G. (2015). Lipidomics in the study of lipid metabolism: Current perspectives in the omic sciences. Gene, 554, 131–139.
Sajic, T., Liu, Y., & Aebersold, R. (2015). Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clinical Applications, 9, 307–321.
Schwudke, D., Schuhmann, K., Herzog, R., Bornstein, S. R., & Shevchenko, A. (2011). Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harbor Perspectives in Biology, 3, a004614.
Tate, S., Larsen, B., Bonner, R., & Gingras, A. C. (2013). Label-free quantitative proteomics trends for protein–protein interactions. Journal of Proteomics, 81, 91–101.
Theodoridis, G. A., Gika, H. G., Want, E. J., & Wilson, I. D. (2012). Liquid chromatography-mass spectrometry based global metabolite profiling: A review. Analytica Chimica Acta, 711, 7–16.
Tyurina, Y. Y., Domingues, R. M., Tyurin, V. A., Maciel, E., Domingues, P., Amoscato, A. A., Bayir, H., & Kagan, V. E. (2014). Characterization of cardiolipins and their oxidation products by LC-MS analysis. Chemistry and Physics of Lipids, 179, 3–10.
van der Kloet, F. M., Hendriks, M., Hankemeier, T., & Reijmers, T. (2013). A new approach to untargeted integration of high resolution liquid chromatography-mass spectrometry data. Analytica Chimica Acta, 801, 34–42.
Vaz, F. M., Pras-Raves, M., Bootsma, A. H., & van Kampen, A. H. (2015). Principles and practice of lipidomics. Journal of Inherited Metabolic Disease, 38, 41–52.
Wang, C., Wang, M., & HAN, X. (2015). Applications of mass spectrometry for cellular lipid analysis. Molecular Biosystems, 11, 698–713.
Wishart, D. S., Knox, C., Guo, A. C., Eisner, R., Young, N., Gautam, B., Hau, D. D., Psychogios, N., Dong, E., Bouatra, S., Mandal, R., Sinelnikov, I., XIA, J., Jia, L., Cruz, J. A., Lim, E., Sobsey, C. A., Shrivastava, S., Huang, P., Liu, P., Fang, L., Peng, J., Fradette, R., Cheng, D., Tzur, D., Clements, M., Lewis, A., de Souza, A., Zuniga, A., Dawe, M., Xiong, Y., Clive, D., Greiner, R., Nazyrova, A., Shaykhutdinov, R., LI, L., VOGEL, H. J., & FORSYTHE, I. (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Research, 37, D603–D610.
Xia, J., Psychogios, N., Young, N., & Wishart, D. S. (2009). MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Research, 37, W652–W660.
Xia, J., & Wishart, D. S. (2011). Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nature Protocols, 6, 743–760.
Xiao, J. F., Zhou, B., & Ressom, H. W. (2012). Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trends in Analytical Chemistry, 32, 1–14.
Yamada, T., Uchikata, T., Sakamoto, S., Yokoi, Y., Fukusaki, E., & Bamba, T. (2013a). Development of a lipid profiling system using reverse-phase liquid chromatography coupled to high-resolution mass spectrometry with rapid polarity switching and an automated lipid identification software. Journal of Chromatography A, 1292, 211–218.
Yamada, T., Uchikata, T., Sakamoto, S., Yokoi, Y., Nishiumi, S., Yoshida, M., Fukusaki, E., & Bamba, T. (2013b). Supercritical fluid chromatography/Orbitrap mass spectrometry based lipidomics platform coupled with automated lipid identification software for accurate lipid profiling. Journal of Chromatography A, 1301, 237–242.
Yang, K., Jenkins, C. M., Dilthey, B., & Gross, R. W. (2015). Multidimensional mass spectrometry-based shotgun lipidomics analysis of vinyl ether diglycerides. Analytical and Bioanalytical Chemistry, 407, 5199–5210.
Yecies, J. L., Zhang, H. H., Menon, S., Liu, S., Yecies, D., Lipovsky, A. I., Gorgun, C., Kwiatkowski, D. J., Hotamisligil, G. S., Lee, C. H., & Manning, B. D. (2011). Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metabolism, 14, 21–32.
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., Locasale, J. W., Son, J., Zhang, H., Coloff, J. L., Yan, H., Wang, W., Chen, S., Viale, A., Zheng, H., PAIK, J. H., LIM, C., Guimaraes, A. R., Martin, E. S., Chang, J., Hezel, A. F., Perry, S. R., Hu, J., Gan, B., Xiao, Y., Asara, J. M., Weissleder, R., Wang, Y. A., Chin, L., Cantley, L. C., & Depinho, R. A. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell, 149, 656–670.
Yuan, M., Breitkopf, S. B., Yang, X., & Asara, J. M. (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nature Protocols, 7, 872–881.
Zhang, Y., Ren, Y., Jiao, J., & Li, D. (2011). Ultra high-performance liquid chromatography-tandem mass spectrometry for the simultaneous analysis of asparagine, sugars, and acrylamide in Maillard reactions. Anal Chem, 83, 3297–3304.
Acknowledgements
We thank the Daniel Tenen lab at BIDMC for providing frozen lung tissue. We also thank Simon Dillon and Towia Libermann at BIDMC for providing human plasma samples. This study was funded by grants from the National Institutes of Health 5P01CA120964 (B.D.M. and J.M.A.), 5P30CA006516 (J.M.A.), and R35CA197459 (B.D.M.), from the National Science Foundation DGE-1144152 (S.R.), and the BIDMC Research Capital Fund for funding the mass spectrometry instrumentation (J.M.A.).
Author Contributions
J.M.A., Y.X., S.B.B., S.R. and D.P. developed the platform. J.M.A., MY., S.R. and S.B.B. wrote the protocol. D.P. and B.M. edited the protocol and provided insight. Y.X, S.R., S.B.B. and M.Y. prepared biological samples for testing the protocol. J.M.A., M.Y., S.B. and S.R. analyzed data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing financial interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. Mouse tissue and human plasma samples were previously acquired and stored frozen by other laboratories.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Breitkopf, S.B., Ricoult, S.J.H., Yuan, M. et al. A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 13, 30 (2017). https://doi.org/10.1007/s11306-016-1157-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-1157-8