
Abstract
Volatile compounds are together with sugars and organic acids the main determinants of tomato fruit flavour and are therefore important for consumer acceptance. Consequently, in the last years many studies have been performed using different volatile analytical techniques on a large diversity of tomato fruits, aimed mainly at detecting the compounds affecting flavour or at the identification of QTLs and key genes involved in fruit volatile contents. The comparison of three of the analytical methods most commonly applied (headspace, solid phase microextraction, adsorption on Tenax followed by thermal desorption) revealed not only differences in sensitivity, but also dramatic variations in the volatile profile obtained by each of these techniques. The volatile profile was also largely influenced by the way samples were processed before analysis. Four widely used sample processing methods were compared (whole tomato, sliced fruit and two different types of fruit paste), each one producing a characteristic volatile pattern. Therefore, great care should be taken when comparing results available from the literature obtained by means of different methods, or when using the volatile levels obtained in an experiment to predict their influence on tomato flavor or consumer preference, or to assess the success of breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Tomato flavour is mainly the result of the interaction of aroma and taste, together with other sensory inputs such as texture, temperature or mouth feel (Goff and Klee 2006), and it has been the object of many studies in the last two decades. Volatile organic compounds (VOCs) play an important role in the flavour, as they not only define the aroma but also modify other perceptions related to taste, like sweetness or sourness (Baldwin et al. 1998, 2004; Vogel et al. 2010; Tieman et al. 2012). A large number of volatile compounds have been described in fresh tomato or tomato products (Petró-Turza 1987), but most of them seem to have no effect on our perception of tomato flavour. Thus, based on quantitative results of volatile composition of ripe tomato fruits, extracted from a fresh tomato paste, a list of 16 compounds was proposed (Buttery 1993) to presumably affect fresh tomato flavour and aroma including how relevant each of them should be for flavour perception. The contribution of each individual volatile compound was estimated by determining the odour threshold values and checking whether or not the detected levels in red fruits were above or below these thresholds (Buttery 1993). However, more recent research revealed that some compounds not present in that list were also important for flavour perception at least in some varieties (Causse et al. 2002; Zanor et al. 2009; Tikunov et al. 2010, 2013; Selli et al. 2014), and the relevance of some compounds that had been considered very important for tomato flavour has been questioned (Tieman et al. 2012). The difficulty in defining the volatiles most relevant for flavour is aggravated by the five orders of magnitude variation in the sensitivity to the same compound between individuals due to genetical variation for olfactory receptor genes (Jaeger et al. 2013; McRae et al. 2013). These issues are still under debate and have been discussed in detail in a recent review (Rambla et al. 2014).
Considering its importance on consumer preference, the identification of different factors affecting volatile profiles has been object of many studies. Thus, different studies have evaluated the changes in volatile profiles associated to climatic and cultural conditions (Cebolla-Cornejo et al. 2011), physiological maturity at harvest (Baldwin et al. 1991; Maul et al. 1998) or postharvest handling (McDonald et al. 1996; Maul et al. 2000; Renard et al. 2013). Considerable efforts have also been carried out to map the genetic regions controlling volatile emission (Causse et al. 2002; Tadmor et al. 2002; Tieman et al. 2006a; Mathieu et al. 2009; Zanor et al. 2009) or to identify the genes involved in the biosynthetic pathways of key volatiles (Chen et al. 2004; Simkin et al. 2004; Tieman et al. 2006b, 2007, Tieman et al. 2010; Goulet et al. 2012; Mageroy et al. 2012; Tikunov et al. 2013).
Different methods for the sampling of tomato volatiles have been used in the literature, such as headspace (Baldwin et al. 1991, 1998, 2004; Maul et al. 2000; Tandon et al. 2003), headspace solid phase microextraction (Tikunov et al. 2005, 2013; Zanor et al. 2009; Ortiz-Serrano and Gil 2010), adsorption in a Super Q resin followed by elution in an organic solvent (Tieman et al. 2006a; Mathieu et al. 2009; Goulet et al. 2012; Mageroy et al. 2012), liquid–liquid extraction with an organic solvent (Aubert et al. 2005; Selli et al. 2014), purge-and-trap in a Tenax sorbent followed by thermal desorption (Ruiz et al. 2005), or purified air or dry nitrogen passed over the sample so that the extracted volatiles are retained in a Tenax trap and then extracted with an organic solvent (Buttery et al. 1987, 1988; Buttery 1993; Beltran et al. 2006), just to cite some of them. All these methods are capable of collecting a subset of volatiles from the fruit samples (some of them already present in the whole intact fruit, others produced during the experimental procedure), but no systematic comparative study had been performed to compare them.
Additionally, different alternatives can be found in the literature regarding the way tomato samples are processed before analysis. The method most frequently used is to process the fruit to form a paste, with some variations in the preparation between research groups (Buttery et al. 1987; Baldwin et al. 1991; Ruiz et al. 2005; Tikunov et al. 2005; Ortiz-Serrano and Gil 2010), while some researchers perform the analysis on sliced/chopped fruit (Tieman et al. 2006a; Mathieu et al. 2009; Vogel et al. 2010) or even the whole intact fruit (Carbonell-Barrachina et al. 2006). The results obtained show a wide range of different detected compounds, and a large sample to sample variability for a given compound (Carbonell-Barrachina et al. 2006). This variation is in part, but not completely, due to the high degree of biological variability existing in the levels of volatile compounds between different tomato cultivars (Tieman et al. 2012; Rambla et al. 2014).
The present work unravels the variability in the tomato fruit volatile profile obtained by comparing a number of volatile capturing and sample processing protocols that were chosen among those more frequently reported in the literature. The aim of the present work is to find out to what extent the different methodologies employed affect the volatile profile obtained and also to shed some light on which method is preferable depending on the objective of the study.
2 Materials and methods
2.1 Plant material
Tomato fruits (Solanum lycopersicum L. cv. Moneymaker) were grown under standard cultural practices in a greenhouse at Experimental Station La Mayora in Málaga (Southern Spain) during the spring season. Red ripe fruits were collected in early June.
2.2 Chemicals and compound identification
Compounds in the chromatograms were identified based on the coincidence of both their mass spectra and retention times with those of authentic standards injected under the same analytical conditions. All the standards were injected with each of the techniques used (thermal desorption, headspace and solid phase microextraction) due to small shifts in retention times depending on the technique used. The retention times indicated in Tables 1 and S1 correspond to those obtained from thermal desorption. All the compounds used as standards were purchased from Sigma-Aldrich (Madrid, Spain) except for 1-nitro-2-phenylethane that was acquired from Apin Chemicals Ltd. (Abingdon, UK). Dihydrate calcium chloride was purchased from Merck (Darmstadt, Germany). EDTA was obtained from Panreac Química (Barcelona, Spain). Milli-Q water was used throughout the study.
2.3 Fruit samples
Samples were processed following four different methods. In each method four biological replicates were analyzed. The four sample processing methods used were: whole tomato fruit, sliced fruit and two types of tomato paste (A and B). For whole fruit analysis, each replicate consisted of 4 tomato fruits of about 50 g each (total weight about 200 g). For sliced fruit, tomatoes were cut in 16 half-wedges (1 transversal cut and 4 longitudinal cuts through the middle of the fruit). Each piece weighed about 3 g (total weight about 50 g). Tomato paste A was prepared following essentially the procedure of Buttery et al. (1987). Roughly, fresh tomatoes were homogenized at room temperature (25 ± 1 °C) for 30 s in a Waring blender. The purée obtained was allowed to stand for 3 min, and then the same volume of a saturated CaCl2 solution was added, and the sample was thoroughly mixed, stored in 50 mL aliquots and frozen at −20 °C until analysis. Immediately before analysis, the sample was thawed at room temperature (25 ± 1 °C) and transferred to the corresponding vial. Tomato paste B preparation followed essentially the procedure of Tikunov et al. (2005) with minor modifications. Roughly, fresh tomatoes (two in each replicate) were cut into pieces and flash frozen in liquid nitrogen. The frozen material was homogenized in a cryogenic mill, and stored at −80 °C until analysis. Immediately before analysis, 1 g of the resulting powder was weighed in a 7 mL vial, incubated for 10 min at 37 °C, and then 2.2 g of CaCl2·2H2O and 1 mL of a 100 mM EDTA-NaOH pH 7.5 solution were added, gently mixed and sonicated for 5 min. Finally, 2 mL of the resulting paste were transferred to a 22 mL crimp cap vial for analysis.
2.4 Tenax adsorption followed by thermal desorption (TD) procedure
Fifty grams of sample, either fresh fruit or fruit paste (except for the whole fruit, where about 200 g were used), were placed in a glass tray inside a 1.3 L glass cylinder (45 cm length, 6 cm diameter). A zero air flow of 100 mL/min passed through the cylinder with the tray during 30 min at room temperature (25 ± 1 °C) and the volatiles emitted by the sample were adsorbed in a Tenax TA stainless steel TD tube (OD × L. 1/4 in. × 3 ½ in.) (Supelco, Pennsylvania, USA) located at the end of the glass cylinder. This acquisition procedure is similar, although not identical, to that described in Tieman et al. (2006a). Desorption was performed thermally in a TurboMatrix TD Thermal Desorber (Perkin Elmer, Wellesley, USA) with the following parameters: primary desorption temperature 300 °C, desorb flow 100 mL/min, desorption time 10 min, inlet split 3 mL/min, outlet split 20 mL/min, Tenax trap temperature −30/300 °C.
2.5 Static headspace trap (HS) procedure
Twenty-two millilitre crimp cap vials (Perkin Elmer) were used, each vial containing 4 mL of tomato paste A. Analyses were performed in a TurboMatrix 40 Trap Headspace Sampler (Perkin Elmer). The analysis conditions were: equilibration time, 80 min; split 1/13; equilibration temperature, 37, 50 or 80 °C, as specified in each case.
2.6 Headspace solid phase microextraction (SPME) procedure
For SPME analysis, 2 mL of tomato paste A were introduced in a 22 mL crimp cap vial, and pre-incubated at either 37, 50 or 80 °C for 10 min. Then a 65 µm PDMS/DVB fiber (Supelco) was exposed to the headspace for another 10 min at 37, 50 or 80 °C respectively. During both pre-incubation and extraction, samples were agitated in a ThermoMixer Comfort agitator (Eppendorf) at 300 rpm. The volatiles trapped on the fiber were desorbed for 1 min at 250 °C in the injection port of the GC/MS and then the fiber was cleaned by exposing it for 5 min at 250 °C in another injection port to prevent cross-contamination. The mode of injection was splitless. Sampling was performed manually.
2.7 Gas chromatography/mass spectrometry conditions
In all cases, volatile organic compounds were analyzed by GC/MS using a Clarus®500 GC/MS from PerkinElmer, equipped with ZB-5 capillary column (30 m, 0.25 mm, 0.25 µm) (Phenomenex, Torrance, CA, USA). Oven programming conditions were 40 °C for 3 min, 3 °C/min ramp until 60 °C, 6 °C/min ramp until 160 °C, 12 °C/min ramp until 260 °C, then 5 min at 260 °C. Helium was used as the carrier gas: for SPME analysis, with a 1.2 mL/min constant flow; for TD ad HS analysis, at a constant pressure of 19.5 psi. Mass spectra in the electron impact (EI) mode were generated at 70 eV. Chromatograms were recorded in scan mode in the m/z range 35–300. Chromatograms and spectra were recorded with GC/MS TurboMass software version 5.0 (Perkin Elmer).
To calculate compound abundance, firstly a specific ion was selected for each compound and the resulting peak areas were integrated. Next, the corresponding total ion count (TIC) area of each particular peak was calculated considering the relative abundance of the selected ion in relation to the mass spectrum of that compound. Finally, the Total Ion Count area of all peaks in the chromatogram was integrated. The results are expressed as area of each peak relative to the total area of all compounds in the chromatogram.
2.8 Statistical analysis
In order to test the effect of the capturing method, data were arranged in a matrix with 26 rows (4 replicates × 7 experimental conditions: TD, HS37, HS50, HS80, SPME37, SPME50 and SPME80) by 49 compounds in columns. For SPME at 50 and 80 °C only 3 replicates were analyzed. In order to study the effect of sample processing experiment, another matrix was arranged containing 16 rows (4 types of sample processing x 4 replicates) by 26 columns (volatile compounds). Each row can be regarded as the volatile profile of the experimental trial. In both cases, data in columns were mean-centered and scaled to unit variance and, next, Principal Component Analysis (PCA) was applied using the software SIMCA-P 10.0 (Umetrics, Sweden) in order to highlight the differences among experimental conditions. Principal components are directions of maximum data variance obtained as linear combinations of the original variables. The contributions of variables (compounds) in the formation of a given component are called loadings, p[1] being the loadings in the formation of the first principal component (PC1); p[2], the loadings of PC2; and so on. The projections of observations (experimental trials in this case) over the directions determined by PC1 and PC2 are called t[1] scores and t[2] scores, respectively. A scatter plot of loadings or scores corresponding to two different components is referred to as loading plot or score plot, respectively.
For hierarchical cluster analysis (HCA), the mean of the data for each capturing method (TD, HS37, HS50, HS80, SPME37, SPME50 and SPME80) was used after square root normalisation. HCA was performed by means of Acuity 4.0 software (Axon Instruments; Union City, CA, USA) with the distance metrics based on the Pearson correlation. The normalised data was represented as a heatmap by means of the same software. For both PCA and HCA, the compounds not detected (either below the detection threshold or not present) were assigned a value of 0.
3 Results and discussion
3.1 Effect of the capturing method on the volatile profile
A large sample of tomato fruit was processed as described for tomato paste A, and different aliquots of it were subjected to volatile capture/release by means of three different techniques. The first was a Tenax adsorption-thermal desorption (TD) method. Volatiles emitted at room temperature (25 °C) from 50 mL of tomato paste located on a glass tray inside of a glass cylinder were adsorbed on a Tenax tube by flushing purified air and the trapped volatiles were desorbed thermally in line with the GC/MS. The second was a headspace (HS) method. Volatiles were partitioned between the matrix and the headspace at three different temperatures: 37, 50 and 80 °C. According to this method, 4 mL of sample in a 22 mL vial emitted volatile compounds until equilibrium, and all the volatiles in the headspace were then concentrated in a cold trap and thermally desorbed for analysis. The third method was based on headspace solid phase microextraction (SPME). Analyses were also performed at three different temperatures: 37, 50 and 80 °C. In this method, the volatiles emitted by 2 mL of sample to the headspace of a 22 mL vial were captured by a PDMS/DVB coated fiber, and the retained volatiles were thermally desorbed in the injection port of the gas chromatograph for analysis.
3.1.1 Capturing by SPME allows the detection of more complex volatile profiles
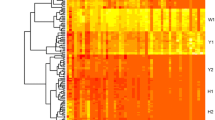
A few hundred compounds were detected in the samples analysed, many of which were present at low levels. A total number of 49 volatile compounds were unequivocally identified. Only this set of compounds was used in the comparative study, although not all were detectable by all trapping techniques. Headspace solid phase microextraction (SPME) seemed to produce the richest profile of all the methods tested. SPME allowed the detection of about 10–15 compounds more than headspace-trap (HS) or Tenax adsorption-thermal desorption (TD) methods. In total, 40–41 compounds were unequivocally identified after SPME, depending on the temperature of volatile acquisition, whilst only 26–31 were detected after HS, again depending on temperature, and only 25 compounds were detected following TD (Fig. 1, Table S1). Compounds that were only tentatively identified were not considered in our study (data not shown). Figure S1 shows a representative chromatogram obtained by each of the capturing methods.

Hierarchical Cluster Analysis from the average data of both analytical methods and volatile compounds. Values are represented as a heatmap according to the scale below. Black colour corresponds to compounds not detected; blue colour corresponds to compounds with very low abundance; pink corresponds to the maximum relative abundance values. Data were square root normalized (Color figure online)
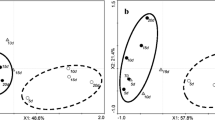
In order to highlight the differences among the resulting volatile compound profiles associated to each capturing method, Principal Component Analysis (PCA) was applied to the dataset matrix. PC1 explained 47.3 % of the total data variability, and the score plot (Fig. 2a) revealed that SPME produced the most distinctive volatile profiles. In order to better characterize the differences between TD and HS, the PCA was repeated after removing SPME values from the dataset (Fig. 3a). From both Figs. 2a and 3a it is evident that each of the capturing methods resulted in a differentiated volatile profile.

Principal component analysis score plot (a) and loading plot (b) of the volatile profiles obtained by different analytical methods. TD thermal desorption, HS headspace at 37, 50 or 80 °C, SPME headspace solid phase microextraction at 37, 50 or 80 °C. Observations corresponding to different replicates are joined with solid lines. Compound codes in (b) are as in Fig. 1 and Table S1. Triangles are coloured according to the molecular weight (MW) of each compound: white MW ≤ 100 Da, gray 130 Da ≥ MW > 100 Da, black MW > 130 Da

Principal component analysis score plot (a) and loading plot (b) of the volatile profiles obtained by thermal desorption (TD) or headspace (HS) at 37, 50 or 80 °C. Observations corresponding to different replicates are joined with solid lines. Compound codes in (b) are as in Fig. 1 and Table S1
The specific compounds that allowed us to discriminate among the different volatile profiles according to the VOC capturing method can be deduced from the PCA loading plots (Figs. 2b and 3b). It turned out that the profile obtained by SPME analysis was markedly enriched in higher molecular weight compounds with lower volatility, and particularly in compounds from C8 to C13. Thirteen compounds were exclusively detected by SPME: relatively long-chain compounds such as decanal, (E,E)-2,4-decadienal, geranial, 2-pentylfuran, acetophenone, benzophenone, benzylnitrile, 1-nitro-2-phenylethane, methyl salicylate and eugenol, and also high polarity compounds such as 3-methylbutanoic, pentanoic and octanoic acids. All these compounds apparently fell below the detection limit of the other techniques used. This is consistent with the higher sensitivity of SPME as compared to HS for the analysis of volatile compounds in food products previously reported (Gamero et al. 2013). Additionally, the volatile profiles obtained after SPME were characterized by higher levels of other compounds with low or relatively low volatility such as β-ionone, β-damascenone, geranylacetone, linalool, α-terpineol, benzaldehyde, phenylacetaldehyde, 2-phenylethanol, 2-isobutythiazole, hexanoic acid, 2-ethylhexanoic acid, nonanal, octanal, (E)-2-octenal, (E)-2-heptenal, (Z)-3-hexenol or 6-methyl-5-hepten-2-one when compared to those profiles obtained by either TD or HS.
3.1.2 Capturing by TD or HS provides better sensitivity for highly volatile molecules
On the other hand, both TD and HS revealed to be more sensitive for the detection of highly volatile compounds. These techniques allowed the determination of four short-chain compounds that were not detected by SPME: 2-methylpropanal, 2-methylpropanol, 3-methylbutanal and 2-methylbutanal. Additionally, the profiles of both TD and HS were enriched in C4 and C5 compounds, and also a few C6 volatiles, including butanol, 2-methylbutanol, (E)-2-methyl-2-butenal, 3-methylbutanenitrile, pentanal, (E)-2-pentenal, 1-penten-3-ol, 1-penten-3-one, hexanal, (Z)-3-hexenal and 2-ethylfuran. All these compounds show low affinity for the PDMS/DVB fiber coating used, and consequently they were poorly retained.
When the results obtained by HS with different temperatures of incubation and TD were compared, the PCA score plot (Fig. 3a) revealed that each of them also produced a characteristic profile. PC1, accounting for 39.5 % of the total variability, separated the different variations of HS technique according to the temperature used for the collection of volatiles. PC2 accounted for 25.6 % of the variability, and separated the samples acquired by means of TD from those acquired through any variation of HS. The loading plot revealed that acquisition by means of TD produced profiles enriched in (Z)-3-hexenal, (E)-2-heptenal, benzaldehyde, 1-penten-3-one, 3-methylbutanol, 2-methylbutanal, 2-methylpropanol and 2-isobutylthiazole. On the contrary, when the samples were acquired by means of HS, the profile obtained had lower relative levels of these compounds and higher levels of hexanal, (E)-2-hexenal, pentanal, 2-ethylfuran and 6-methyl-5-hepten-2-one (Fig. 3b).
3.1.3 Effect of the temperature
Three different temperatures were used for capturing volatiles in both HS and SPME: 37, 50 and 80 °C. It was decided to use 37 °C because this is the temperature at which volatiles are expected to be released in people’s mouth when eating tomato. Similarly low temperatures have also been used in headspace analysis of volatiles in other fruit species (Allwood et al. 2014). The temperatures of 50 and 80 °C have been widely used in the literature for volatile analysis of tomato (Maul et al. 2000; Tandon et al. 2003; Tikunov et al. 2005; Baldwin et al. 2008; Zanor et al. 2009). Relatively high temperatures are often used for the analysis of volatile compounds in methods based on compound volatilization in the headspace in order to increase analytical sensitivity (Nongonierma et al. 2006).
Figure 2a reveals that temperature has a major effect on the volatile profile obtained, most notably in SPME, where the volatile profiles at the highest temperature evaluated, 80 °C, were very different to those obtained by any of the other analytical conditions evaluated. Results obtained for SPME at 50 °C were intermediate to those at 37 and 80 °C, although much more similar to the former. The increase of the sample incubation temperature affected the volatile profile after SPME capture by increasing the relative levels of a number of relatively long-chain semi-volatile compounds, and most remarkably β-ionone, β-damascenone, decanal, 2-ethylhexanoic, hexanoic and octanoic acids, 2-pentylfuran, (E,E)-2,4-decadienal, geranylacetone, 2-isobutylthiazole, 2-phenylethanol, phenylacetaldehyde, 1-nitro-2-phenylethane, benzylnitrile, linalool, α-terpineol, geranial and benzophenone (Fig. 2b). The latter compound was only detected with SPME at 80 °C.
The temperature of incubation barely affected the number of compounds detected after SPME capture, which yielded 41, 40 and 41 compounds at 37, 50 and 80 °C respectively. The main effect was found on the relative abundance of low volatility compounds, all of which increased their levels with the temperature. As a consequence, the maximum levels of organic acids and long chain compounds (C10 and over) were observed at 80 °C.
The effect of temperature on HS analysis can be clearly observed in Fig. 3a. PC1 (39.5 % of total variability) shows the effect of the temperature on the profiles obtained. As we have previously described for SPME, incubation at 37 and 80 °C produced clearly distinct profiles, whilst incubation at 50 °C produced intermediate results closer to those obtained at 37 °C. Incubation at high temperatures increased the levels of many semi-volatile compounds we have previously described for SPME such as phenylacetaldehyde, 2-phenylethanol, geranylacetone, hexanoic and 2-ethylhexanoic acids and β-damascenone, but also increased the levels of a number of short-chain compounds including 1-pentanol, (E)-2-pentenal, (E)-2-methyl-2-butenal, 2-methylpropanol, 1-penten-3-ol and 2-ethylfuran (Fig. 3b). Among these compounds, 2-ethylhexanoic acid and geranylacetone were only detected after HS at the highest temperature. On the other hand, several compounds detected by HS after incubation at 37 and 50 °C failed to be detected at 80 °C. This is the case of a set of branched-chain small molecular weight compounds such as 3-methylbutanenitrile, 3-methylbutanal, 3-methylbutanol, or 2-methylpropanol, the linear molecules pentanal and (E)-2-octenal, and also 6-methyl-5-hepten-2-one. This could be due to degradation of these compounds during the extended time required for equilibrium in the gas phase -80 min- at this high temperature.
The total number of compounds detected by HS at 37, 50 and 80 °C were 27, 31 and 26 respectively (Fig. 1, Table S1). Therefore, for headspace analysis, an intermediate temperature such as 50 °C is apparently the most appropriate for the detection of a higher number of volatile compounds, as an increase in the incubation temperature favours the volatility of the compounds, but too high temperatures would accelerate the processes of degradation of many volatiles.
In conclusion, SPME was revealed to be by far the most sensitive of the three trapping techniques evaluated, yielding the most complex volatile profiles in our tomato samples. Therefore, it seems to be the best acquisition technique for approaches where a high-throughput volatile metabolomics analysis is necessary. Additionally, SPME should be the capturing method of choice when there is an interest on semi-volatile compounds, as in our hands it was the only technique which allowed the detection of most of them. An additional advantage of this technique is the low amount of biological material required (only 1 g of tomato fruit) to obtain a good sensitivity. The main limitation of SPME as performed in our experiments was the low sensitivity for highly volatile compounds, which were not detectable in our assays. This limitation could possibly be overridden by the use of a fiber coating with higher affinity for those compounds, such as divinylbenzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS). TD and HS, although less sensitive for a wide range of compounds, were very efficient for the detection of short-chain highly volatile compounds and would probably be useful techniques in those cases where this type of volatiles are of particular interest.
Regarding temperature of analysis, a moderately elevated temperature such as 50 °C seems to be the most adequate, as it favours the emission and therefore the detection of semi-volatile compounds while minimizing the degradation of the sample.
3.2 Effect of sample processing on the volatile profile
In order to assess the effect of the sample processing method on the volatile profile, we compared the results obtained after processing the same biological sample (a pool of red ripe fruits) following four different methods. These were selected among those most commonly reported in the literature: tomato paste A (first processed, then frozen), tomato paste B (first frozen, then processed and pH adjusted), sliced fresh tomato fruit, and the whole intact unprocessed fresh fruit. After having processed the samples according to each method, the emitted volatiles were captured with a Tenax trap system and analyzed by thermal desorption coupled to gas chromatography and mass spectrometry, as described in the corresponding Sect. 2. This capturing method was used because it allowed the handling of the different types of processed samples analyzed, as the size of sliced and whole fruits did not allow these samples to be introduced inside the vials used for HS or SPME. Representative chromatograms obtained by each of the sample processing methods are shown in Figure S2.
Out of the 49 volatile compounds previously described in this paper (Fig. 1, Table S1), only 26 were detected by Thermal Desorption. The effect of sample processing on these compounds was so dramatic that only 3 of the 26 identified compounds (2-methylbutanol, 3-methylbutanenitrile and hexanal) were detected in all four types of samples. Table 1 shows the relative abundance of compounds expressed as the percentage area of each particular compound relative to the area of all peaks in the chromatogram. It turned out that only 6 of these compounds were detected in the whole fruit, whilst 25 were detected in paste A. Eighteen volatiles were present in both paste B and the sliced fruit, although they are not all the same compounds.
PCA was applied to the data and PC1 and PC2 explained 45.9 and 30.9 % of the total variability respectively. The score plot (Fig. 4a) revealed the dramatic effect of sample processing on the pattern of volatile compound emission. Three completely separated groups could be easily identified that correspond to each type of processing method: (1) whole fruit, (2) sliced fruit, and (3) both tomato pastes. This result indicates how dependable the volatile compound profiles and composition are on the processing method used. The two methods applied to process the pastes (A and B) resulted in the most similar volatile patterns, which were discriminated by PC1 but not so clearly by PC2 (Fig. 4a). Nevertheless, paste A produced the richest volatile profile, emitting 7 detectable compounds more than paste B. The most remarkable case is (E)-2-heptenal, which is relatively abundant in the first method but not detected in the latter. In any case, results reported here highlight the remarkable effect variations in the protocol used to prepare the sample have on the profiles of volatile compounds obtained.

Principal component analysis of volatile profiles obtained with different methods of sample processing: score plot (a) and loading plot (b). Observations corresponding to different replicates are joined with solid lines. Compound codes in (b) are as in Table 1
The PCA loading plot (Fig. 4b) permits to identify a series of compounds that are more characteristic of each sample cluster observed in the corresponding score plot. It turned out that compounds also tended to group together, which implies that each sampling method tends to produce a distinct, non-overlapping set of volatiles. PC1 discriminates both pastes A and B with respect to the other methods. Both pastes produced considerably higher levels of a set of short-chain fatty acid-derived volatiles: C6 compounds (Z)-3-hexenal, hexanal and (E)-2-hexenal; C5 compounds 1-penten-3-one, 1-penten-3-ol, 1-pentanol, (E)-2-pentenal and pentanal; and also C4 compound butanol. Moreover, (E)-2-heptenal was detected exclusively in paste A, but not in paste B. The levels of most of these compounds, although markedly lower, were also induced in the sliced fruit samples when compared to the whole fruit. It has been reported that both biotic and abiotic stresses including physical damage induce the production of a variety of volatile compounds. In plant material containing intact living cells, such as the sliced fruit, the stress associated to cutting the fruit would activate gene responses and the biosynthesis of wound stress-related metabolites, including volatile compounds (Niinemets et al. 2013). Additionally, the intact enzymatic machinery in living cells could modify some of the compounds produced by neighbouring injured cells, as the conversion of wounded cell-produced (Z)-3-hexenal into the corresponding alcohol and acetyl ester by neighbouring intact cells (Matsui et al. 2012). In the fruit pastes such response would not take place, because this response requires maintaining homeostasis and this is not happening after homogenization. In the homogenized samples, gene expression is not operational and the only mechanisms altering the volatile composition would be either chemical or enzymatic involving preformed molecules. In fact, it has been described that the homogenization of the fruit would facilitate the contact between enzymes and substrates otherwise localized in different cellular compartments in the living cells, and this would be responsible for the burst of many volatiles, including many fatty acid derivatives (the so-called green leaf volatiles) and several phenylpropanoids (Chen et al. 2004; Granell and Rambla 2013; Shen et al. 2014). The more complete the homogenization (in the paste much more than in the sliced fruit), the higher the production of these compounds. Our results indicate that the latter process rather than the first has a major quantitative effect on the volatile profile obtained (Fig. 4; Table 1).
Although the volatile profiles produced by both tomato pastes produced the most similar volatile patterns (Fig. 4a), the particular way the tomato paste was produced also had a substantial effect on the volatile profile. Interestingly, paste A (first homogenized and incubated, then frozen) allowed the detection of more compounds than paste B (first frozen, then homogenized and finally thawed and incubated). Octanal, nonanal, (E)-2-heptenal, 2-methylpropanal, 6-methyl-5-hepten-2-one, phenylacetaldehyde and 2-isobutylthiazole were detected in tomato paste A but not in paste B. This probably indicates that most volatiles detected in tomato samples are mainly produced when precursors and biosynthetic enzymes of those volatile compounds meet each other after tissue disruption, as it has been documented for several biosynthetic pathways, either by de novo biosynthesis (Chen et al. 2004; Shen et al. 2014) or by the release of volatile aglycones accumulated as conjugates (Tikunov et al. 2013). After tissue disruption, volatile analysis protocols usually include some time of incubation so that detectable levels of the volatiles are produced. In the case of tomato paste, a high amount of calcium chloride is usually added after a few minutes of incubation in order to inhibit further reactions and stabilize the volatile profile during the time of analysis. In tomato paste A, incubation took place immediately after homogenization of the fresh tomato. In paste B, the sample was flash frozen with liquid nitrogen before incubation. This would probably produce a partial or total inactivation of some of the participating enzymes, as has been previously reported (Díaz de León-Sánchez et al. 2009), causing some volatile compounds to either fall below detection levels or even not be present at all.
In accordance to this, the whole unprocessed fruit should produce a very poor volatile profile, which is precisely the case. Under our analytical conditions, only six of the previously identified compounds were present at detectable levels, most of which appear grouped together at the bottom right of the loading plot with the only exception of hexanal (Fig. 4b). The whole fruit profile was basically composed of short branched-chain amino acid-related volatiles (2-methylpropanal, 2-methylbutanol, 3-methylbutanenitrile and 2-isobutylthiazole), one fatty acid derivative (hexanal), and also small amounts of α-pinene, a monoterpene which often accumulates in glandular trichomes, that in tomato are present in both leaves and fruit (Schilmiller et al. 2010). Interestingly, 2-isobutylthiazole is a highly potent odorant and it recalls the smell of tomato leaves. We also have to consider that, in the whole fruit, diffusion of volatiles through the cuticle is extremely slow even for compounds as short as ethane, whilst resistance to diffusion through the sepals/nectary abscission scar is about three orders of magnitude lower (Cameron and Yang 1982). It is therefore likely that a sound tomato fruit needs to drop off the plant in order to release volatiles through the abscission scar.
Finally, the sliced fruit, when compared to the fruit pastes, produced considerably lower levels of many fatty acid derivatives including (Z)-3-hexenal and undetectable levels of (E)-2-hexenal, (E)-2-pentenal, 1-penten-3-one or 1-pentanol, but similar levels of others such as hexanal, pentanal or (Z)-3-hexenol. The loading plot (Fig. 4b) indicates a group of 11 compounds that yielded the highest values with the sliced samples. Interestingly, higher levels of all branched-chain amino acid-related volatiles such as 3-methylbutanal, 3-methylbutanol, 2-methylbutanal, 2-methylbutanol, 3-methylbutanenitrile, (E)-2-methyl-2-butenal, 2-methylpropanal, 2-methylpropanol, and 2-isobutylthiazole were produced, compared with the pastes. This observation suggests that branched-chain amino acid related compounds are readily produced in the intact fruit, and unlike observed for most fatty acid derivatives, fruit homogenization does not contribute to enhance their levels; on the contrary, their levels seem to be reduced after homogenization.
3.3 Possible consequences and limitations for metabolomic studies derived from the variability in VOCs introduced by processing and capture methods
The comparison of these analytical methods revealed that both the sample processing and the technique used for the capture of volatiles have a dramatic effect on the compounds detected and their abundance in a particular sample. This fact, together with the wide range of biological variation observed among tomato cultivars (Tieman et al. 2012; Rambla et al. 2014), explains the high degree of variability in the abundance of volatile compounds in tomato fruit reported in the literature.
There is no single methodology that could be claimed as ‘the best’, but the comparative results here described can be used as a guide to select the most suitable approach for a particular experiment, depending on its objective or technical limitations. Regarding sampling processing, paste A yielded the highest number of detected compounds. Paste B produced a lower number of detectable volatiles, but it has the advantage that the same flash frozen material used for the volatile analysis can be stored at −80 °C until further use or shipped to other labs for determination of other metabolites, proteomic analysis or even gene expression analysis, and therefore it is compatible with a multi-omics approach of a given biological sample. Sliced and whole fruit procedures have the disadvantage that the analysis requires to be performed on the fresh sample, and a complex set up is needed when parallel simultaneous acquisition of volatiles from a high number of samples is required, such as when profiling breeding collections which are highly dependent on harvest time (Tieman et al. 2012). Nevertheless, sliced fruit samples produce a reasonably rich volatile profile, with particularly high levels of short branched-chain compounds, and therefore this sample processing method has been useful for a large number of studies (Tieman et al. 2006b, 2007, 2010, 2012; Mathieu et al. 2009; Vogel et al. 2010; Goulet et al. 2012; Mageroy et al. 2012; Shen et al. 2014). In fact, this sample processing technique is particularly useful when these volatiles are of particular interest, since it allows the detection of a higher number of such compounds. The whole fruit produced a very poor profile, but it could be useful in assays to determine how odour-attractive intact fruits are for seed dispersers. Regarding the technique used for volatile acquisition, each of the evaluated techniques has its own advantages, although SPME was the one that provided the largest number of compounds with a very low requirement of sample (only 1 g).
Considering that the high degree of variation in the volatile profiles depends largely on the method of analysis used, a concern arises about the limitations of each analytical study. The quantitative results obtained by different research groups using a range of methodological conditions cannot be directly compared. For example, the quantitative amount of volatiles of a particular variety analyzed, e.g. from a paste, cannot be directly compared to results obtained from another cultivar analyzed in a different laboratory from e.g. sliced fruit, because a very relevant part of the differences observed would not be due to the effect of the variety, but to that of the sampling procedure. Obviously, this does not mean that the analyses of volatile compounds in tomatoes are unreliable. Far from that, the comparison of different sets of samples using the same procedure has produced fruitful results, including the effect of different treatments to the volatile profile (Baldwin et al. 1991; McDonald et al. 1996; Maul et al. 1998, 2000; Cebolla-Cornejo et al. 2011; Renard et al. 2013), the identification of genomic regions responsible of particular traits (Causse et al. 2002; Tadmor et al. 2002; Tieman et al. 2006; Mathieu et al. 2009; Zanor et al. 2009) or the identification of genes implied in biosynthetic pathways (Chen et al. 2004; Simkin et al. 2004; Tieman et al. 2006b, 2007, 2010; Goulet et al. 2012; Mageroy et al. 2012; Tikunov et al. 2013). Thus, the identification of the QTL and gene underlying the accumulation of smoky flavour in tomatoes resulted in the cloning of NSGT1; this hallmark in the way volatiles are kept/released by higher level glycosilation could have been more difficult to be unveiled should the authors had used whole or sliced fruits, since incubation of the extracts and activity of the glycosidases to release the volatile was consubstantial with its discovery. Therefore, a sample treatment which involved homogenization of the tissue and allowed the glycosidases in the extract to act on the non-volatile glycoside form so as to liberate or not the volatile was necessary (Tikunov et al. 2010, 2013). In summary, the identification of gene/gene products depends on the volatile profile obtained during the screening methods and this, as we have demonstrated, is highly influenced by the capture and processing method. Therefore, the identification of genes involved in volatile biosynthesis (Chen et al. 2004; Simkin et al. 2004; Tieman et al. 2006b, 2007, 2010; Goulet et al. 2012; Mageroy et al. 2012; Tikunov et al. 2013) has been so far highly dependent on the conditions the volatiles were released and analyzed. Our results obtained here indicate that each methodological approach has its pros and cons, and great care should be taken when trying to compare different data from the literature, as they have been obtained under different methodological conditions.
The present study reveals that the ‘quantitative’ results obtained for the fruit of a given variety of tomato when analyzed by means of a particular technique may in some way be more representative of the sampling procedure utilized than on the variety itself, as a consequence of the strong effect of the method used on the volatile profile obtained. This issue has important implications when trying to translate the quantitative results (profile of volatile compounds) obtained for each volatile compound in a cultivar in terms of flavour and aroma. The most widely accepted approach to characterize tomato aroma is based on odour units, although it is a simplistic approach with rather limitations, as discussed recently (Tieman et al. 2012; Rambla et al. 2014). Basically, a threshold of human perception is determined for each volatile compound dissolved in water, and then compared to the quantitative results obtained from a tomato sample. In theory, only those compounds produced at levels above the threshold would participate in our perception of flavour and aroma. The odour units currently reported for tomato are based on red ripe tomato fruit samples processed exactly as paste A. Buttery (1993) quantified 16 compounds over the respective threshold levels, and they were considered thereafter as responsible of tomato flavour and aroma. Our results suggest that a different list of compounds would be obtained if a different sampling procedure (such as paste B or the sliced fruit) was used. Therefore, depending on the precise way how the tomato fruit is manipulated, different amounts of each volatile compound will be released, either above or below our detection threshold. An important difficulty to relate the volatile content with consumer liking is that none of the sampling procedures used up to date resembles much the ‘procedure’ that takes place in the consumer´s mouth (chewing for a short time, insalivating, heating at mouth temperature…). In this sense, new sampling procedures have been recently developed (Farneti et al. 2013) with the objective to obtain less artifactual analytical results, which would be more easily translatable to our perception of tomato flavour.
4 Concluding remarks
Our results clearly show that both the process of sample preparation and the technique used for capturing the volatiles have a dramatic effect on the volatile profiles obtained, and they contribute to the wide range of variability in volatile profiles reported in the literature for tomato fruit. Different protocols provide different views of the volatile content which are not readily comparable, therefore suggesting that although each method can be suitable for a specific purpose, great care should be taken when comparing results between experiments using different volatile technologies, or when using the resulting volatile levels to predict the influence of particular compounds on our perception of tomato flavour or on consumer preference. Although each technique has its own pros and cons, a sample processing method starting from frozen material such as paste B would be the most adequate from an-omics perspective, as the same sample can be used for the different types of analysis. Concerning the capture method, headspace solid phase microextraction yields the highest number of volatile compounds even from a small amount of sample. The combination of these two techniques would probably be the most adequate for a multi-omics approach.
References
Allwood, J. W., Cheung, W., Xu, Y., et al. (2014). Metabolomics in melon: A new opportunity for aroma analysis. Phytochemistry, 99, 61–72.
Aubert, C., Baumann, S., & Arguel, H. (2005). Optimization of the analysis of flavor volatile compounds by liquid-liquid microextraction (LLME). Application to the aroma analysis of melons, peaches, grapes, strawberries, and tomatoes. Journal of Agricultural and Food Chemistry, 53, 8881–8895.
Baldwin, E. A., Goodner, K., & Plotto, A. (2008). Interaction of volatiles, sugars, and acids on perception of tomato aroma and flavor descriptors. Journal of Food Science, 73, S294–S307.
Baldwin, E. A., Goodner, K., Plotto, A., & Einstein, M. (2004). Effect of volatiles and their concentration on perception of tomato descriptors. Journal of Food Science, 69, S310–S318.
Baldwin, E. A., Nisperos-Carriedo, M. O., & Moshonas, M. G. (1991). Quantitative analysis of flavor and other volatiles and for certain constituents of 2 tomato cultivars during ripening. Journal of the American Society for Horticultural Science, 116, 265–269.
Baldwin, E. A., Scott, J. W., Einstein, M. A., et al. (1998). Relationship between sensory and instrumental analysis for tomato flavor. Journal of the American Society for Horticultural Science, 123, 906–915.
Beltran, J., Serrano, E., Lopez, F. J., Peruga, A., Valcarcel, M., & Rosello, S. (2006). Comparison of two quantitative GC-MS methods for analysis of tomato aroma based on purge-and-trap and on solid-phase microextraction. Analytical and Bioanalytical Chemistry, 385, 1255–1264.
Buttery, R. G. (1993). Quantitative and sensory aspects of flavor of tomato and other vegetables and fruits. In T. E. Acree & R. Teranishi (Eds.), Flavor science: Sensible principles and techniques (pp. 259–286). Washington: American Chemical Society.
Buttery, R. G., Teranishi, R., & Ling, L. C. (1987). Fresh tomato aroma volatiles: A quantitative study. Journal of Agricultural and Food Chemistry, 35, 540–544.
Buttery, R. G., Teranishi, R., Ling, L. C., Flath, R. A., & Stern, D. J. (1988). Quantitative studies on origins of fresh tomato aroma volatiles. Journal of Agricultural and Food Chemistry, 36, 1247–1250.
Cameron, A. C., & Yang, S. F. (1982). A simple method for the determination of resistance to gas diffusion in plant organs. Plant Physiology, 70, 21–23.
Carbonell-Barrachina, A. A., Agustí, A., & Ruiz, J. J. (2006). Analysis of flavor volatile compounds by dynamic headspace in traditional and hybrid cultivars of Spanish tomatoes. European Food Research and Technology, 222, 536–542.
Causse, M., Saliba-Colombani, V., Lecomte, L., Duffe, P., Rousselle, P., & Buret, M. (2002). QTL analysis of fruit quality in fresh market tomato: A few chromosome regions control the variation of sensory and instrumental traits. Journal of Experimental Botany, 53, 2089–2098.
Cebolla-Cornejo, J., Roselló, S., Valcárcel, M., Serrano, E., Beltrán, J., & Nuez, F. (2011). Evaluation of genotype and environment effects on taste and aroma flavor components of Spanish fresh tomato varieties. Journal of Agricultural and Food Chemistry, 59, 2440–2450.
Chen, G. P., Hackett, R., Walker, D., Taylor, A., Lin, Z., & Grierson, D. (2004). Identification of a specific isoform of tomato lipoxygenase (TomloxC) involved in the generation of fatty acid-derived flavor compounds. Plant Physiology, 136, 2641–2651.
Díaz de León-Sánchez, F., Pelayo-Zaldívar, C., Rivera-Cabrera, F., et al. (2009). Effect of refrigerated storage on aroma and alcohol dehydrogenase activity in tomato fruit. Postharvest Biology and Technology, 54, 93–100.
Farneti, B., Alarcon, A. A., Cristescu, S. M., et al. (2013). Aroma volatile release kinetics of tomato genotypes measured by PTR-MS following artificial chewing. Food Research International, 54, 1579–1588.
Gamero, A., Wesselink, W., & de Jong, C. (2013). Comparison of the sensitivity of different aroma extraction techniques in combination with gas chromatography-mass spectrometry to detect minor aroma compounds in wine. Journal of Chromatography A, 1272, 1–7.
Goff, S. A., & Klee, H. J. (2006). Plant volatile compounds: Sensory cues for health and nutritional value? Science, 311, 815–819.
Goulet, C., Mageroy, M. H., Lam, N. B., Floystad, A., Tieman, D. M., & Klee, H. J. (2012). Role of an esterase in flavour volatile variation within the tomato clade. Proceedings of the National Academy of Sciences, USA, 109, 19009–19014.
Granell, A., & Rambla, J. L. (2013). Biosynthesis of volatile compounds. In G. Seymour, G. A. Tucker, M. Poole, & J. J. Giovannoni (Eds.), The molecular biology and biochemistry of fruit ripening (pp. 135–161). Oxford, UK: Wiley-Blackwell. doi:10.1002/9781118593714.
Jaeger, S. R., McRae, J. F., Bava, C. M., et al. (2013). A Mendelian trait for olfactory sensitivity affects odor experience and food selection. Current Biology, 23, 1601–1605.
Mageroy, M. H., Tieman, D. M., Floystad, A., Taylor, M. G., & Klee, H. J. (2012). A Solanum lycopersicum catechol-O-methyltransferase involved in synthesis of the flavor molecule guaiacol. Plant Journal, 69, 1043–1051.
Mathieu, S., Dal Cin, V., Fei, Z., et al. (2009). Flavour compounds in tomato fruits: Identification of loci and potential pathways affecting volatile composition. Journal of Experimental Botany, 60, 325–337.
Matsui, K., Sugimoto, K., Mano, J., Ozawa, R. & Takabayashi, J. (2012). Differential metabolisms of green leaf volatiles in injured and intact parts of a wounded leaf meet distinct ecophysiological requirements. Plos One, 7, e36433. doi:10.1371/journal.pone.0036433.
Maul, F., Sargent, S. A., Balaban, M. O., Baldwin, E. A., Huber, D. J., & Sims, C. A. (1998). Aroma volatile profiles from ripe tomatoes are influenced by physiological maturity at harvest: An application for electronic nose technology. Journal of the American Society for Horticultural Science, 123, 1094–1101.
Maul, F., Sargent, S. A., Sims, C. A., Baldwin, E. A., Balaban, M. O., & Huber, D. J. (2000). Tomato flavor and aroma quality as affected by storage temperature. Journal of Food Science, 65, 1228–1237.
McDonald, R. E., McCollum, T. G., & Baldwin, E. A. (1996). Prestorage heat treatments influence free sterols and flavor volatiles stored at chilling temperature. Journal of the American Society for Horticultural Science, 121, 531–536.
McRae, J. F., Jaeger, S. R., Bava, C. M., et al. (2013). Identification of regions associated with variation in sensitivity to food-related odors in the human genome. Current Biology, 23, 1596–1600.
Niinemets, U., Kannaste, A. & Copolovici, L. (2013). Quantitative patterns between plant volatile emissions induced by biotic stresses and the degree of damage. Frontiers in Plant Science, 4, 262. doi:10.3389/fpls.2013.00262.
Nongonierma, A., Cayot, P., Le Quere, J. L., Springett, M., & Voilley, A. (2006). Mechanisms of extraction of aroma compounds from foods, using adsorbents. Effect of various parameters. Food Reviews International, 22, 51–94.
Ortiz-Serrano, P., & Gil, J. V. (2010). Quantitative comparison of free and bound volatiles of two commercial tomato cultivars (Solanum lycopersicum L.) during ripening. Journal of Agricultural and Food Chemistry, 58, 1106–1114.
Petró-Turza, M. (1987). Flavor of tomato and tomato products. Food Reviews International, 2, 309–351.
Rambla, J. L., Tikunov, Y. M., Monforte, A. J., Bovy, A. G., & Granell, A. (2014). The expanded tomato fruit volatile landscape. Journal of Experimental Botany, 65, 4613–4623.
Renard, C. M. G. C., Ginies, C., Gouble, B., Bureau, S., & Causse, M. (2013). Home conservation strategies for tomato (Solanum lycopersicum): Storage temperature vs. duration—is there a compromise for better aroma preservation? Food Chemistry, 139, 825–836.
Ruiz, J. J., Alonso, A., García-Martínez, S., Valero, M., Blasco, P., & Ruiz-Bevia, F. (2005). Quantitative analysis of flavour volatiles detects differences among closely related traditional cultivars of tomato. Journal of the Science of Food and Agriculture, 85, 54–60.
Schilmiller, A., Shi, F., Kim, J., et al. (2010). Mass spectrometry screening reveals widespread diversity in trichome specialized metabolites of tomato chromosomal substitution lines. Plant Journal, 62, 391–403.
Selli, S., Kelebek, H., Ayseli, M. T., & Tokbas, H. (2014). Characterization of the most aroma-active compounds in cherry tomato by application of the aroma extract dilution analysis. Food Chemistry, 165, 540–546.
Shen, J. Y., Tieman, D., Jones, J. B., et al. (2014). A 13-lipoxygenase, TomloxC, is essential for synthesis of C5 flavour volatiles in tomato. Journal of Experimental Botany, 65, 419–428.
Simkin, A. J., Schwartz, S. H., Auldridge, M., Taylor, M. G., & Klee, H. J. (2004). The tomato carotenoid cleavage dioxygenase 1 genes contribute to the formation of the flavor volatiles beta-ionone, pseudoionone, and geranylacetone. Plant Journal, 40, 882–892.
Tadmor, Y., Fridman, E., Gur, A., et al. (2002). Identification of malodorous, a wild species allele affecting tomato aroma that was selected against during domestication. Journal of Agricultural and Food Chemistry, 50, 2005–2009.
Tandon, K. S., Baldwin, E. A., Scott, J. W., & Shewfelt, R. L. (2003). Linking sensory descriptors to volatile and nonvolatile components of fresh tomato flavor. Journal of Food Science, 68, 2366–2371.
Tieman, D., Bliss, P., & McIntyre, L. M. (2012). The chemical interactions underlying tomato flavour preference. Current Biology, 22, 1035–1039.
Tieman, D. M., Loucas, H. M., Kim, J. Y., Clark, D. G., & Klee, H. J. (2007). Tomato phenylacetaldehyde reductases catalyze the last step in the synthesis of the aroma volatile 2-phenylethanol. Phytochemistry, 68, 2660–2669.
Tieman, D., Taylor, M., Schauer, N., Fernie, A. R., Hanson, A. D., & Klee, H. J. (2006a). Tomato aromatic amino acid decarboxylases participate in synthesis of the flavour volatiles 2-phenylethanol and 2-phenylacetaldehyde. Proceedings of the National Academy of Sciences, USA, 103, 8287–8292.
Tieman, D. M., Zeigler, M., Schmelz, E. A., et al. (2006b). Identification of loci affecting flavour volatile emissions in tomato fruits. Journal of Experimental Botany, 57, 887–896.
Tieman, D., Zeigler, M., Schmelz, E., et al. (2010). Functional analysis of a tomato salicylic acid methyl transferase and its role in synthesis of the flavor volatile methyl salicylate. Plant Journal, 62, 113–123.
Tikunov, Y. M., de Vos, R. C. H., Gonzalez-Paramas, A. M. G., Hall, R. D., & Bovy, A. G. (2010). A role for differential glycoconjugation in the emission of phenylpropanoid Volatiles from tomato fruit discovered using a metabolic data fusion approach. Physiology, 152, 55–70.
Tikunov, Y., Lommen, A., De Vos, C. H. R., et al. (2005). A novel approach for nontargeted data analysis for metabolomics. Large-scale profiling of tomato fruit volatiles. Plant Physiology, 139, 1125–1137.
Tikunov, Y. M., Molthoff, J., de Vos, R. C., et al. (2013). Non-smoky glycosyltransferase 1 prevents the release of smoky aroma from tomato fruit. Plant Cell, 25, 3067–3078.
Vogel, J. T., Tieman, D. M., Sims, C. A., Odabasi, A. Z., Clark, D. G., & Klee, H. J. (2010). Carotenoid content impacts flavour acceptability in tomato (Solanum lycopersicum). Journal of the Science of Food and Agriculture, 90, 2233–2240.
Zanor, M. I., Rambla, J. L., Chaib, J., et al. (2009). Metabolic characterization of loci affecting sensory attributes in tomato allows an assessment of the influence of the levels of primary metabolites and volatile organic contents. Journal of Experimental Botany, 60, 2139–2154.
Acknowledgments
We thank Rafael Fernández for providing excellent tomato fruits for this study. Funding to AG was provided through CALITOM and ESPSOL from FECYT and EUSOL (EU FP7 program) and Quality Fruit FA 1106.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jose Luis Rambla, Cristina Alfaro, Aurora Medina, Manuel Zarzo, Jaime Primo and Antonio Granell declared that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11306_2015_824_MOESM1_ESM.pptx
Supplementary material 1 Table S1. Relative abundance of volatile compounds after different capturing methods. Footnote. Values represent the contribution of each individual compound related to the sum of the areas of all the peaks in the chromatogram, expressed as a percentage. RT, Retention Time (min); Kovats RI, Kovats Retention Index; m/z, specific ion used for compound quantitation; nd, not detected. (PPTX 103 kb)
11306_2015_824_MOESM2_ESM.pptx
Supplementary material 2 Figure S1. Representative Total Ion Chromatograms (TIC) obtained by each of the capturing methods used. Chromatograms are scaled to the highest peak (100 %). Numbers on the top right of each chromatogram indicate the absolute height (counts) as registered by the detector for the highest peak. In order to facilitate visual alignment of chromatograms, the peak corresponding to (Z)-3-hexenal + hexanal has been marked with an asterisk. (PPTX 80 kb)
11306_2015_824_MOESM3_ESM.xlsx
Supplementary material 3 Figure S2. Representative Total Ion Chromatograms (TIC) obtained by each of the sample processing methods used. Chromatograms are scaled to the highest peak (100 %). Numbers on the top right of each chromatogram indicate the absolute height (counts) as registered by the detector for the highest peak (XLSX 24 kb)
Rights and permissions
About this article

Cite this article
Rambla, J.L., Alfaro, C., Medina, A. et al. Tomato fruit volatile profiles are highly dependent on sample processing and capturing methods. Metabolomics 11, 1708–1720 (2015). https://doi.org/10.1007/s11306-015-0824-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-015-0824-5