
Abstract
Activity of the A3 adenosine receptor (AR) allosteric modulators LUF6000 (2-cyclohexyl-N-(3,4-dichlorophenyl)-1H-imidazo [4,5-c]quinolin-4-amine) and LUF6096 (N-{2-[(3,4-dichlorophenyl)amino]quinolin-4-yl}cyclohexanecarbox-amide) was compared at four A3AR species homologs used in preclinical drug development. In guanosine 5′-[γ-[35S]thio]triphosphate ([35S]GTPγS) binding assays with cell membranes isolated from human embryonic kidney cells stably expressing recombinant A3ARs, both modulators substantially enhanced agonist efficacy at human, dog, and rabbit A3ARs but provided only weak activity at mouse A3ARs. For human, dog, and rabbit, both modulators increased the maximal efficacy of the A3AR agonist 2-chloro-N 6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide as well as adenosine > 2-fold, while slightly reducing potency in human and dog. Based on results from N 6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide ([125I]I-AB-MECA) binding assays, we hypothesize that potency reduction is explained by an allosterically induced slowing in orthosteric ligand binding kinetics that reduces the rate of formation of ligand-receptor complexes. Mutation of four amino acid residues of the human A3AR to the murine sequence identified the extracellular loop 1 (EL1) region as being important in selectively controlling the allosteric actions of LUF6096 on [125I]I-AB-MECA binding kinetics. Homology modeling suggested interaction between species-variable EL1 and agonist-contacting EL2. These results indicate that A3AR allostery is species-dependent and provide mechanistic insights into this therapeutically promising class of agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Selective A3 adenosine receptor (AR) agonists are currently being developed as pharmacological therapy for a number of inflammatory diseases including rheumatoid arthritis, psoriasis, and cancer [1]. However, the full therapeutic benefit of these drugs could be limited by dose-limiting side effects [2]. Clinical development of A3AR ligands is further challenged by substantial differences in A3AR pharmacology among species [3,4,5,6]. This complication hinders assessment of therapeutic efficacy of A3AR ligands in non-primate models of human disease.
Allosteric modulators of G protein-coupled receptors (GPCRs) are ligands that alter the affinity or intrinsic activity of orthosteric ligands by interacting with a distinctly different binding site on the receptor [7,8,9,10]. From a therapeutic perspective, allosteric modulators have the advantage of regulating activity of an endogenous ligand in a spatially and temporally specific manner [7,8,9,10]. Allosteric ligands typically mediate their effects by inducing conformational changes in the receptor that alter orthosteric agonist binding affinity and/or intrinsic activity thereby causing positive or negative allosteric modulation. These effects can vary depending on the nature of the orthosteric ligand (“probe dependence”) and have the capability to selectively modulate specific intracellular signaling pathways (“signaling bias”).
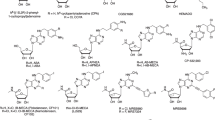
Several classes of compounds have been reported to be allosteric modulators for the human A3AR, including the imidazoquinolinamines and the 2,4-disubstituted quinolines [11,12,13,14,15,16,17,18]. Derivatives of these two chemical classes function as positive allosteric modulators (PAMs) that increase the intrinsic activity of orthosteric agonists, while tending to reduce potency. 2-Cyclohexyl-N-(3,4-dichlorophenyl)-1H-imidazo [4,5-c]quinolin-4-amine (LUF6000) (Fig. 1) emerged from structure-activity relationship (SAR) studies of the imidazoquinolinamine template as having a pronounced effect to increase agonist efficacy with less propensity to decrease potency (Fig. 1; [16]) and to exhibit selectivity versus the other three AR subtypes [15, 16]. Modification of LUF6000 involving scission of the imidazole ring led to the development of the 2,4-disubstituted quinoline LUF6096 (Fig. 1), which similarly retained desirable allosteric effects and A3AR selectivity [17]. Both of these compounds have begun to be tested for therapeutic efficacy in animal models of disease [19, 20]. These compounds were developed for activity at the human A3AR, however, and it remains to be determined whether they exhibit similar modulating activity at the A3AR from other species.

Chemical structures of A3AR allosteric modulator compounds used in the present investigation
The goal of this investigation was to characterize the modulatory activity of LUF6000 and LUF6096 for the A3AR from a panel of animal species commonly used in drug development research. Functional modulation was assessed using a guanosine 5′[γ-[35S]thio]triphosphate ([35S]GTPγS) binding assay that detects receptor-induced G protein activation with membranes prepared from human embryonic kidney cells (HEK 293) expressing recombinant A3ARs from the various species. Orthosteric effects were explored in radioligand binding assays with the agonist radioligand N 6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide ([125I]I-AB-MECA). Based on the results of initial studies, we focused on examining the potential role of the first extracellular loop (EL1) of the A3AR in mediating activity of the modulators using a mutagenesis and molecular modeling approach.
Materials and methods
Materials
Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). [35S]GTPγS (specific activity 1250 Ci/mmol) and carrier-free Na125I (2200 Ci/ml) were from PerkinElmer (Boston, MA). LUF6000 and LUF6096 were synthesized as reported [16, 17]. Plasmid purification kits were purchased from Qiagen (Valencia, Ca). Adenosine deaminase (ADA) was obtained from Roche Applied Sciences (Indianapolis, IN) and glass fiber filters (GF/B and GF/C) were obtained from Whatman (Sanford, ME). All remaining drugs and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Creation of stable HEK 293 cell lines expressing A3ARs
Full-length dog, rabbit, and mouse A3AR cDNAs subcloned into the mammalian expression vector pCDNA3.1 were obtained, as previously described [21,22,23]. The human A3AR cDNA in pCDNA3.1 was purchased from the cDNA Resource Center (www.cdna.org). Site-directed mutations (G75Q/I76V/T77K/I78M) were introduced into the human A3AR cDNA using the QuikChange Site-Directed Mutagenesis kit (Stratagene), according to the manufacturer’s instructions. The following primers were used to create the mutations: 5′-GCC TTT GGC CAT TGT TGT CAG CCT GCA GGT CAA AAT GCA CTT CTA CAG CTG CCT TTT TAT GA-3′ and 5′-TCA TAA AAA GGC AGC TGT AGA AGT GCA TTT TGA CCT GCA GGC TGA CAA CAA TGG CCA AAG GC-3′. Following amplification in Escherichia coli, plasmids were purified using Qiagen (Valencia, CA) purification kits and sequenced through the entire coding region to establish the presence of the desired mutations and the absence of unintended mutations potentially generated during the amplification steps. The plasmids were transfected into human HEK 293 cells using Lipofectamine 2000 reagent (Invitrogen) and selected with 2 mg/ml of G418. Cell lines derived from individual clones were maintained in cell culture media (Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and antibiotics) containing 0.6 mg/ml G418. The level of expression of the receptors in each of the cell lines was equivalent based on saturation radioligand binding analyses (not shown).
Membrane preparations
Cell membranes were prepared as described previously [16, 17, 20]. Briefly, HEK 293 cells stably expressing recombinant wild-type or mutant human A3ARs were washed in phosphate-buffered saline followed by homogenization in hypotonic lysis buffer containing Tris-HCl buffer (50 mM, pH 7.4) containing 1 mM EDTA and 5 mM MgCl2 and then centrifuged at 27,000×g for 30 min at 4 °C. Cell pellets were washed twice in the same buffer, after which the resultant pellets were re-suspended in 50 mM Tris-HCl buffer (pH 7.4) containing 10% sucrose and stored at − 80 °C. Because the modulators failed to increase [35S]GTPγS binding in preliminary studies with the mouse A3AR using crude P1 preparations, for some studies, plasma membranes were enriched by preparing P2 pellets. Cells were homogenized in lysis buffer containing 10% sucrose and centrifuged at 500×g for 10 min to remove nuclear and other cellular debris. The pellet was re-suspended in sucrose buffer and centrifuged again at 500×g. The pooled supernatants were diluted at least threefold, pelleted, and washed twice by centrifugation at 27,000×g for 30 min in 50 mM Tris-HCl buffer (pH 7.4). The resultant pellets were re-suspended and frozen in sucrose buffer.
[35S]GTPγS binding assays
[35S]GTPγS binding assays were conducted as described [15, 20]. In brief, 200 μl of buffer containing 50 mM Tris-HCl (pH 7.4), 1 mM EGTA, 10 mM MgCl2, 1 μM GDP, 1 mM dithiothreitol, 100 mM NaCl, 0.2 nM [35S]GTPγS, 300 nM 8-[4-[4-(4-chlorophenyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine (PSB-603) to block A2BARs expressed endogenously in HEK 293 cells; 0.005% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS); and 0.5% bovine serum albumin was added to polypropylene incubation tubes. Adenosine deaminase (3 U/ml) was also included in all assays except those examining the effects of adenosine. Then, reactions were started by addition of the membrane suspension (5 μg protein) to the tubes and carried out in quadruplicate for 2 h at room temperature. In studies with the modulators, the membranes were pre-incubated with the compounds for 30 min prior to initiating the assay. The reactions were stopped by rapid filtration through Whatman GF/B filters presoaked in 50 mM Tris-HCl buffer (pH 7.4) containing 5 mM MgCl2 and 0.02% CHAPS. The filters were washed three times with 3 ml of wash buffer (10 mM Tris-HCl and 1 mM MgCl2, pH 7.4, 4 °C), and radioactivity trapped in the filters was measured by liquid scintillation counting. Non-specific binding of [35S]GTPγS was measured in the presence of 10 μM unlabeled GTPγS.
Binding assays with [125I]I-AB-MECA
Cell membranes (50 μg) were incubated in 100 μl binding buffer (50 mM Tris-HCl [pH 7.4], 1 mM EDTA, 10 mM MgCl2, and 3 units/ml adenosine deaminase) containing ~ 0.3 nM [125I]I-AB-MECA and indicated concentrations of the A3AR allosteric modulator compounds. The reactions were incubated at room temperature for the times indicated after which bound and free radioligands were separated by rapid filtration through GF/C glass fiber filters. Radioactivity trapped in the filters was measured using a gamma counter. For dissociation studies, [125I]I-AB-MECA (~ 0.3 nM) was incubated with HEK 293 cell membranes (50 μg) expressing ARs for 2 h at room temperature in binding buffer (100 μl), after which the assay was initiated by the addition of 100 μM adenosine-5′-N-ethylcarboxamide (NECA, a nonselective agonist) along with the enhancer compounds or equivalent vehicle. The time-course of dissociation of specific [125I]I-AB-MECA binding was measured by rapid filtration at the indicated time intervals. For association/equilibration binding assays, membranes (50 μg) were incubated with [125I]I-AB-MECA (~ 0.3 nM) and the modulator compounds for the indicated times. For all assays, non-specific binding was determined by incubation in the presence of 100 μM NECA. [125I]I-AB-MECA was prepared by radioiodination of AB-MECA using the chloramine-T method [21, 24, 25].
Molecular modeling
A3ARs homology models
Homology models of the human A3AR, mouse A3AR, and mEL1-hA3AR were built based on the human A2AAR crystal structure of highest available resolution (PDB ID: 4EIY; [26]) and by using the alignment and homology modeling tools implemented in the MOE suite [27]. In particular, A3AR homology models were built using the automated Homology Modeling protocol. The AMBER99 force field was used for protein modeling and the Protonate 3D methodology was used for protonation state assignment [28]. The final models were refined through energy minimization until a root mean square (RMS) gradient of 0.1 kcal/mol/Å was achieved. The stereochemical quality of the models was checked using several tools (Ramachandran plot; backbone bond lengths, angles, and dihedral plots; clash contacts report; rotamer strain energy report) implemented in the MOE suite.
Loop refinement
For the final A3AR models, EL1 and EL2 were subjected to refinement by means of the Prime [29] package portion of the Schrödinger Suite [30] to better explore their conformational space. The two loops were cooperatively refined in pairs using an implicit membrane whose orientation and thickness were derived from the spatial arrangement of the human A2AAR structure stored in the Orientations of Proteins in Membranes (OPM) database [31]. For the human A3AR model, EL1 was refined from Ser73 to His79 and EL2 from Gly148 to Cys166. Corresponding residues were refined for the other A3AR models.
Molecular docking of Cl-IB-MECA at the A3AR
The Cl-IB-MECA structure was built using the builder tool implemented in the MOE suite [27] and subjected to energy minimization using the MMFF94x force field, until a RMS gradient of 0.05 kcal/mol Å was achieved. A homology model of the human A3AR was built based on an agonist-bound human A2AAR crystal structure (PDB ID: 3QAK; [32]) following the same procedure described above. Molecular docking of the ligand at the human A3AR model was performed by means of the Glide [33] package portion of the Schrödinger Suite [30]. The docking site was defined using key residues in the binding pocket of the human A3AR model, namely Phe (EL2), Asn (6.55), Trp (6.48), and His (7.43), and a 20 Å × 20 Å × 20 Å box was centered on those residues. Docking of the ligand was performed in the rigid binding site using the extra precision (XP) protocol. The top scoring docking conformations were subjected to visual inspection and comparison with the nucleoside crystallographic pose at the human A2AAR [32] to select the final binding conformation. Due to lack of knowledge of the binding pocket, we did not attempt docking analyses with the allosteric modulators at this time.
Data analysis
EC50 and E max values were calculated by fitting the data to: E = (E max × x) / (EC50 + x), in which x is the concentration of the test compound. In the kinetic binding assays, dissociation rate constants were determined by fitting the specific binding data to a two-phase exponential decay model: Y = Y 0 × f 1 × e (−k1 × t) + Y 0 × f 2 × e (−k2 × t), where Y o denotes specific binding at time 0, f 1 and f 2 denote the fraction of receptors in the two phases, k 1 and k 2 denote the dissociation rate constants, and t denotes the elapsed time. For association assays, association binding rates were determined by fitting the specific binding data to a one-phase exponential association model: Y = Y max × (1 − e (−k × t), where Y max denotes maximal binding and k denotes the observed association rate constant. The association half-time was calculated as 0.693/k. All values were statistically compared using an unpaired Student’s t test or a one-way ANOVA followed by Student’s t test with the Bonferroni correction, as appropriate. All values are presented as the mean ± SEM. A p value < 0.05 was considered to be statistically significant.
Results
[35S]GTPγS binding
In previous functional studies with the human A3AR [15, 17], both LUF6000 and LUF6096 have been shown to increase the maximal efficacy of the A3AR agonist 2-chloro-N 6-(3-iodobenzyl)-adenosine-5′-N-methylcarboxamide (Cl-IB-MECA). As shown in Fig. 2, this was confirmed in the present study using the [35S]GTPγS binding assay. In the presence of 10 μM of either LUF6000 or LUF6096, the E max of Cl-IB-MECA was increased ~ 2–3-fold, while its EC50 was increased 5–6-fold. Neither of the compounds directly stimulated [35S]GTPγS binding, supporting an allosteric mechanism. Thus, with the human A3AR, both compounds function as PAMs with respect to efficacy enhancement but also slightly reduce agonist potency. Similar findings were observed in assays with the dog A3AR (Fig. 2), whereby the compounds increased the efficacy of Cl-IB-MECA ~ 2–3-fold while decreasing its potency (5–6-fold). In assays with the rabbit A3AR, however, the efficacy of Cl-IB-MECA was increased without a reduction in potency, and in assays with the mouse A3AR, no enhancement was detected (Fig. 2). A nearly identical profile was observed among the species with LUF6096 (10 μM) on efficacy and potency in assays using adenosine as the orthosteric agonist (Table 1). To compare the potency of the enhancers among the different species, concentration-response curves with LUF6096 were conducted in the presence of a maximal concentration (10 μM) of Cl-IB-MECA. As shown in Fig. 3, the calculated EC50 values for LUF6096 for the responding species (human, dog, rabbit) differed only within a factor of 3.

Modulatory effect of 10 μM LUF6000 or LUF6096 on Cl-IB-MECA-induced [35S]GTPγS binding in assays with HEK 293 cell membranes expressing human (a), dog (b), rabbit (c), or mouse (d) A3ARs. Incubations were started by addition of the membrane suspension (5 μg protein) to reactions containing ~ 0.1 nM [35S]GTPγS and Cl-IB-MECA, as described in “Materials and methods,” and were carried out in quadruplicate for 2 h at room temperature. The membrane solutions were pre-incubated with the modulators for 30 min before beginning the assays. EC50 and E max values are reported in the tables. Data are the mean ± SEM of four to eight experiments performed in quadruplicate. Asterisk: P < 0.05 versus control by one-way ANOVA followed by post hoc analysis by a Student’s t test for unpaired data with the Bonferroni correction

Modulatory effect of increasing concentrations of LUF6096 on [35S]GTPγS binding in response to a maximal concentration (10 μM) of Cl-IB-MECA in assays using HEK 293 cell membranes expressing human, canine, rabbit, or mouse A3ARs. Assays were conducted as described in Fig. 2 legend. The membrane preparations were pre-incubated with LUF6096 for 30 min before beginning the assays. EC50 values and E max values calculated from the data for each species of A3AR are reported in the table. Data are the mean ± SEM of four to eight experiments performed in quadruplicate
Due to lack of an effect of the modulators on mouse A3ARs, we repeated the [35S]GTPγS binding assays using enriched P2 membrane preparations (see “Materials and methods”). As shown in Fig. 4, in these experiments, we observed a small efficacy enhancing effect of both modulator compounds. In the presence of 10 μM of either LUF6000 or LUF6096, the E max of Cl-IB-MECA was increased 20–30%, with no change in the EC50. Similarly, LUF6096 produced a detectable efficacy enhancing effect (46%) in assays using adenosine as the orthosteric agonist (Fig. 4). The EC50 of LUF6096 to produce efficacy enhancement, determined from concentration-response curves in the presence of 10 μM Cl-IB-MECA, was calculated to be 1.33 ± 0.83 μM (n = 5), suggesting reduced (~ 25-fold) potency at mouse A3ARs compared to the other species. Overall, these observations indicate that the modulatory effects of LUF6000 and LUF6096 vary among the species tested with respect to efficacy enhancement, potency, and the propensity to reduce agonist potency.

Modulatory effect of 10 μM LUF6000 or LUF6096 on a Cl-IB-MECA-induced or b adenosine-induced [35S]GTPγS binding in assays with enriched P2 HEK 293 cell membranes expressing mouse A3ARs. Assays were conducted as described in Fig. 2 legend. EC50 and E max values are reported in the tables. Data are the mean ± SEM of three to seven experiments performed in quadruplicate. Asterisk: P < 0.05 versus control by one-way ANOVA followed by post hoc analysis by a Student’s t test for unpaired data with the Bonferroni correction
[125I]I-AB-MECA binding
We compared among the species the effects of LUF6000 and LUF6096 on binding of the orthosteric agonist [125I]I-AB-MECA. We began with a dissociation binding assay, because in previous studies with the human A3AR both modulators produce prominent slowing of the rate of [125I]I-AB-MECA dissociation [17]. For these studies, membranes from HEK 293 cells expressing A3ARs from the various species were incubated with [125I]I-AB-MECA for 2 h to reach equilibrium, after which displacement was initiated by the addition of NECA (100 μM) either alone or in combination with either modulator (10 μM). When the A3AR is expressed in HEK 293 cells, [125I]I-AB-MECA binds to two affinity states, which relates to G protein-coupling status [21, 34]. Thus, the dissociation of [125I]I-AB-MECA is biphasic, whereby the slow component corresponds to the high-affinity, G protein-coupled state of the A3AR and the fast component corresponds with binding to the low-affinity, uncoupled state of the receptor [21, 34].
The results of the [125I]I-AB-MECA dissociation kinetic experiments are shown in Fig. 5, and the calculated dissociation rate constants along with the fraction of receptors for the slow phase (high-affinity state) are reported in Table 2. At a concentration of 10 μM, both LUF6000 and LUF6096 reduced the rate of dissociation of [125I]I-AB-MECA from human and dog A3ARs. The modulators did not influence the rate of dissociation of [125I]I-AB-MECA from rabbit or mouse A3ARs. When an effect on the dissociation rate was observed, the modulators produced a significant decrease in the slow [125I]I-AB-MECA dissociation rate. The modulators also tended to decrease the fast [125I]I-AB-MECA dissociation rate, although this did not reach the level of significance, likely due to difficulty in obtaining accurate measurements of this rapid phase. Thus, the allosteric effect of the modulators to alter agonist dissociation is also species-dependent.

Effect of LUF6000 or LUF6096 on dissociation of [125I]I-AB-MECA binding to HEK 293 cell membranes expressing human (a), dog (b), rabbit (c), or mouse (d) A3ARs. Membranes (50 μg) were incubated with [125I]I-AB-MECA (~ 0.3 nM) for 2 h at room temperature after which dissociation was initiated by adding 100 μM NECA mixed with either vehicle or modulator compound. The fraction of specific binding at various times after the addition of NECA is shown. The data were fitted to a two-phase exponential decay model. Data are the mean ± SEM of five to eight experiments performed in triplicate. See Table 1 that reports dissociation rate constants
We next performed a preliminary association binding assay with the human A3AR. Membranes from HEK 293 cells were incubated with [125I]I-AB-MECA (0.3 nM) for up to 18 h in the presence or absence of 10 μM LUF6096. As shown in Fig. 6, inclusion of LUF6096 markedly delayed the rate at which [125I]I-AB-MECA binding reached equilibrium. In control assays, binding reached equilibrium in ~ 2 h, whereas in the presence of LUF6096, binding reached equilibrium at a time well beyond 5 h. After fitting the data to a single-site association binding model, the association half-time of [125I]I-AB-MECA was calculated to be increased by LUF6096 more than fivefold from ~ 31 min in the control assays to ~ 185 min in the presence of LUF6096. Maximal [125I]I-AB-MECA binding in the presence of LUF6096 was not reduced.

Effect of LUF6096 on association kinetic binding of [125I]I-AB-MECA to HEK 293 cell membranes expressing the human A3AR. Membranes (50 μg) were incubated with [125I]I-AB-MECA (~ 0.3 nM) for the times indicated at room temperature with either vehicle or 10 μM LUF6096. Non-specific binding was determined in the presence of 100 μM NECA. The fraction of specific binding obtained at the 18-h time-point in the presence of vehicle is shown. The binding data were fitted to a one-phase exponential association model. Data are the mean ± SEM of four to six experiments performed in triplicate
Based on these findings, we utilized a hybrid association/equilibrium binding experiment to efficiently compare activity of the modulators on [125I]I-AB-MECA binding among the A3ARs from the different species. For these assays, transfected HEK 293 cells expressing A3ARs were incubated with 0.3 nM [125I]I-AB-MECA in the presence of increasing concentrations of either LUF6000 or LUF6096 and the extent of specific binding was compared at 3 h with binding obtained at 18 h. Assessment at the 18-h time-point will detect an effect of the modulators on [125I]I-AB-MECA binding under equilibrium conditions, whereas assessment at the 3-h time-point will detect whether the modulators might have an impact on the rate at which [125I]I-AB-MECA binding achieves equilibrium. As shown in Fig. 7, both of the modulators produced a concentration-dependent reduction in [125I]I-AB-MECA binding to the A3AR from all of the species tested at the 3-h time-point, while having no effect on binding at the 18-h time-point. The magnitude of this effect (i.e., the extent of reduction in [125I]I-AB-MECA binding at 3 h) among the species was human = dog > rabbit = mouse. These results demonstrate a general effect of the modulators to allosterically impact [125I]I-AB-MECA binding kinetics without affecting the extent of binding at equilibrium.

Effect of LUF6000 and LUF6096 on binding of the orthosteric radioligand [125I]I-AB-MECA in assays with HEK 293 cell membranes expressing human, dog, rabbit, or mouse A3ARs. Membranes (50 μg) were incubated with [125I]I-AB-MECA (~ 0.3 nM) for either 3 h (a and c) or 18 h (b and d) at room temperature in the presence of increasing concentrations of LUF6000 (a and b) or LUF6096 (c and d). Non-specific binding was determined in the presence of 100 μM NECA. The fraction of specific binding obtained in the absence of the modulator compounds is shown. Data are the mean ± SEM of four to six experiments performed in triplicate
Participation of the first extracellular loop
The amino acid sequences of those species whereby the modulators produced prominent efficacy enhancement (human, dog, rabbit) were compared to those that were minimally responsive (rat, mouse) to potentially identify regions of the A3AR that might participate in mediating the actions of the modulator compounds. We included the rat A3AR as a poor responding species in this analysis, since we found that both LUF6000 and LUF6096 produced no detectable efficacy enhancing activity in [35S]GTPγS binding assays (P1 membranes) with the rat A3AR (not shown). As depicted in Fig. 8, this analysis revealed that the first extracellular loop (EL1) region, composed of five amino acids of which four were conserved in human, dog, and rabbit (75G-I-T-I-H/Q/G79), was markedly different in mouse and rat and notably contained charged residues (E or K). Based on this analysis, assays were conducted with a stably transfected HEK 293 cell line expressing a mutated form of the human A3AR, named the mEL1-hA3AR, in which the EL1 region was changed to the mouse sequence (76Q-V-K-M-H80). A clonal cell line was selected that expressed the mEL1-hA3AR at levels similar to that of the wild-type human A3AR-expressing HEK 293 cell line. As determined in saturation equilibrium radioligand binding assays, mutation of EL1 of the human A3AR to the mouse sequence did not influence the binding affinity of [125I]I-AB-MECA to either the high- (G protein-coupled) or low- (uncoupled) affinity states and also did not influence the fraction of receptors in the high-affinity state (Table 3).

“Snake” diagram of the human A3AR sequence. Amino acid residues that are conserved in species found to be responsive to both modulator compounds (human, dog, rabbit) but are not conserved in weakly responding species (rat, mouse) are highlighted in red. According to this criterion for comparison, marked variability was identified in the first extracellular loop region. Detailed amino acid sequence comparison of this region is shown in the lower left-hand section of the illustration. The dendrogram in the lower right-hand corner reveals that A3AR sequences from responsive and less responsive species cluster together. This diagram was prepared using the GPCRdb database (http://www.gpcrdb.org).
In [35S]GTPγS binding assays, LUF6096 increased the efficacy of Cl-IB-MECA (10 μM) with HEK 293 cell membranes expressing the mEL1-hA3AR to a similar extent as compared to assays with the wild-type human A3AR (Fig. 9). However, the EC50 of LUF6096 was reduced 17-fold, determined from [35S]GTPγS binding assays in which concentration-response curves with LUF6096 were conducted in the presence of 10 μM Cl-IB-MECA. As shown in Fig. 9, LUF6096 did not reduce the extent of [125I]I-AB-MECA binding to the mEL1-hA3AR at the 18-h time-point in the hybrid association/equilibrium binding assay described earlier. At the 3-h time-point, however, LUF6096 produced a greater reduction in [125I]I-AB-MECA binding to the mEL1-hA3AR compared to the wild-type human A3AR (Fig. 9). The potency (EC50) of LUF6096 to reduce binding to the mEL1-hA at the 3-h time-point was comparable to the wild-type receptor (wild-type = 1.91 ± 0.35 μM; mEL1-hA3AR = 2.54 ± 0.5 μM; Fig. 9). LUF6096 also produced a greater effect to slow [125I]I-AB-MECA dissociation from the mEL1-hA3AR (Fig. 9). These results indicate that changing the EL1 region of the human A3AR to the mouse sequence results in a greater effect of LUF6096 to (i) reduce the potency of Cl-IB-MECA and (ii) slow [125I]I-AB-MECA binding kinetics.

Modulatory effect of LUF6096 in assays with HEK 293 cell membranes expressing the mEL1-hA3AR mutant. a Effect of LUF6096 (10 μM) on [35S]GTPγS binding while stimulating with a maximal concentration of Cl-IB-MECA (10 μM). b Effect of increasing concentrations of LUF6096 on [35S]GTPγS binding in response to Cl-IB-MECA (10 μM). Data in this panel are presented as a percentage of maximal [35S]GTPγS binding. c Effect of increasing concentrations of LUF6096 on binding of [125I]I-AB-MECA in the hybrid association/equilibrium binding assay involving comparison of specific binding at 3 and 18 h. d Effect of LUF6096 on [125I]I-AB-MECA dissociation kinetics. All of the assays were conducted as described in “Materials and methods” and in previous figure legends. Data are the mean ± SEM of four to eight experiments performed in triplicate ([125I]I-AB-MECA binding assays) or quadruplicate ([35S]GTPγS binding assays). Asterisk: P < 0.05 versus the wild-type human A3AR by an unpaired Student’s t test
Molecular modeling
In an attempt to analyze the structural details of the EL1 region, we built homology models of the human A3AR, mouse A3AR, and mEL1-hA3AR and performed a conformational search of the extracellular loop areas. The models were based on the reported crystal structure of the human A2AAR [26]. As part of the modeling, EL1 and EL2 were cooperatively refined due to their close proximity and the potential for interaction. Loop modeling is still a challenging task due to marked flexibility, making it difficult to obtain unequivocal information. Figure 10 shows the top scoring conformations obtained for the three different receptors.

Comparison of human A3AR (magenta), mouse A3AR (orange), and mEL1-hA3AR (yellow) homology models, after loop refinement. Ribbon region corresponding to the mutated residues in EL1 is colored green. Docking pose of Cl-IB-MECA is shown in cyan
The following information regarding EL1 and EL2 was obtained from the analyses. The general structure of EL1 was predicted to be similar between the human and mouse receptors because of its short length (five residues) despite marked dissimilarity in the amino acid sequence in this region. In some cases, it formed a β-sheet with part of EL2, as observed in several human A2AAR crystal structures [26, 32]. The EL2 residues predicted from the model to make contact with the EL1 sequence are different between the species: 162T-F-L-S-C-Q167 for the human A3AR and 163S-T-L-L-C-H168 for the mouse A3AR. Therefore, replacement of the EL1 region of the human A3AR with the mouse sequence affects the interaction pattern between the two loops in comparison with the native receptor, which could result in modification of EL2 structure and function of the mEL1-hA3AR. Notably, EL1 does not make direct contact with Cl-IB-MECA in the binding cavity while EL2 does, as shown from the Cl-IB-MECA docking pose reported in Fig. 10. In fact, some EL2 residues have a role in shaping the binding cavity and stabilizing the ligand. For example, a phenylalanine that is conserved among ARs (F168 at the human A3AR) forms a π-π stacking interaction with the adenine core of the ligand in agreement with the binding mode of AR agonists in the A2AAR crystallographic structures [32]. EL2 appears to be very flexible given the wide range of possible conformations it assumed during the search. At the human A2AAR, EL2 is involved in three disulfide bridges that constrain its structure, while in the A3AR, only one disulfide is present. Therefore, the conformation of EL2 of the A3AR could be quite different from that of the A2AAR.
Discussion
This study compared the modulatory activity of LUF6000 and LUF6096 at the A3AR from four different mammalian species (human, dog, rabbit, and mouse) expressed in HEK 293 cells. These ligands, which are currently under investigation in preclinical animal models [19, 20], emerged from SAR studies of the imidazoquinolinamine and 2,4-disubstituted quinoline classes of allosteric modulators of the human A3AR that enhance the intrinsic activity of orthosteric agonists [11,12,13,14,15,16,17,18]. We demonstrate that the pharmacology of the modulators is unique for each of the species investigated. Most notably, both modulators showed weak efficacy enhancing activity on mouse A3ARs.
The observation that both LUF6000 and LUF6096 exert low efficacy-enhancing activity versus rodent A3ARs was not unexpected. Previous studies have documented that orthosteric pharmacology of rodent A3ARs differs substantially from other mammalian species, explained by a greater degree of sequence divergence that influences the orthosteric ligand binding pocket and receptor activation mechanisms [3, 4, 6]. The A3AR is known to be capable of stimulating G protein-independent translocation of β-arrestin2 upon ligand binding that contributes to rapid desensitization of the receptor and may also lead to activation of additional intracellular signaling pathways [14]. Since LUF6000 has been reported to modulate β-arrestin translocation mediated by the human A3AR [14], it would be interesting to compare activity of the modulator compounds on this signaling response in other species.
Other notable species differences include a variable effect of the modulators to decrease agonist potency in the [35S]GTPγS binding assay and to influence [125I]I-AB-MECA binding properties. Interestingly, both of the modulators decreased agonist (Cl-IB-MECA and adenosine) potency in the [35S]GTPγS binding assays in the same species (human and dog) that they substantially slowed [125I]I-AB-MECA binding kinetics. These findings raise the possibility that potency reduction by the modulators might be explained by a slowed rate of receptor occupancy. The effect of the modulators to alter [125I]I-AB-MECA binding kinetics appears to be complex, whereby the on and off rates are influenced in a manner that results in slowed ligand-receptor complex formation but no change in binding at equilibrium. In general, the properties of the two different modulators were comparable. Both produced a similar degree of efficacy enhancement as well as effects on [125I]I-AB-MECA binding, and both displayed a similar activity profile among the different species of receptors. While the two different modulators are structurally dissimilar, the data suggest that they both likely interact with the same allosteric site on the receptor.
The mutagenesis studies suggest that the EL1 region of the A3AR participates in mediating the allosteric effect of LUF6096 to alter orthosteric ligand binding kinetics. It is possible that the EL1 region provides structural support that allows access of agonists to the orthosteric ligand binding site. Based on findings from the molecular modeling analyses of the extracellular loop regions, this may occur through interactions with EL2. These analyses predicted that EL2, which is much larger, can assume different conformations that are influenced by EL1, as shown by the formation of a β-sheet between the two loops in some cases. It is interesting that the EL1 region of the M4 muscarinic receptor has been implicated to serve a similar function. Nawaratne and colleagues [35] identified that a single mutation of an isoleucine93(2.65) of EL1 of the M4 muscarinic receptor to a threonine residue selectively reduced the modulatory activity of the allosteric ligand LY2033298 to increase orthosteric agonist binding while leaving efficacy enhancing activity unchanged. Another mutagenesis study of the human A3AR by our group determined that specific amino acid residues in transmembrane regions 1, (asparagine30(1.50)), 2 (aspartic acid58(2.50)), 3 (aspartic acid107(3.49)), 5 (phenylalanine182(5.43)), and 7 (asparagine274(7.45)) abolished the effect of the imidazoquinolinamine A3AR allosteric modulator DU124183 to decrease the rate of dissociation of [125I]I-AB-MECA, implicating these regions as additional sites required for allosteric interactions [12]. All of these amino acid residues are conserved among the human, dog, rabbit, mouse, and rat sequences. In this earlier study, the effect of the mutations on functional enhancing activity was not addressed.
In conclusion, species variation in the actions of GPCR allosteric ligands has been underappreciated. Unlike the orthosteric binding site, allosteric domains of GPCRs are not subject to evolutionary pressures to accommodate endogenous ligands and can therefore exhibit greater variation between receptor subtypes and between species. The possibility for species variability lies not only with the ability of modulator compounds to bind to an allosteric site but also with the magnitude of allosteric interactions that influence signaling responses. This study provides evidence that allosteric pharmacology of the A3AR displays prominent species variability. This information is important for future development of this class of compounds for therapeutic use [8, 20] and should be considered when validating A3AR modulation by PAMs in animal models. It will be useful to focus efforts in future investigations to develop pan-species A3AR PAMs with minimal negative allosteric properties.
Abbreviations
- AR:
-
Adenosine receptor
- CHAPS:
-
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Cl-IB-MECA:
-
2-Chloro-N 6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide
- GPCR:
-
G protein-coupled receptor
- [35S]GTPγS:
-
Guanosine 5′-[γ-[35S]thio]triphosphate
- HEK 293:
-
Human embryonic kidney 293
- [125I]I-AB-MECA:
-
N 6-(4-amino-3-[125I]iodobenzyl)adenosine-5′-N-methylcarboxamide
- LUF6000:
-
2-Cyclohexyl-N-(3,4-dichlorophenyl)-1H-imidazo [4,5-c]quinolin-4-amine
- LUF6096:
-
N-{2-[(3,4-dichlorophenyl)amino]quinolin-4-yl}cyclohexanecarbox-amide
- NECA:
-
Adenosine-5′-N-ethylcarboxamide
- PAM:
-
Positive allosteric modulator
References
Jacobson KA, Gao ZG (2006) Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 5(3):247–264. https://doi.org/10.1038/nrd1983
van Troostenburg AR, Clark EV, Carey WD, Warrington SJ, Kerns WD, Cohn I, Silverman MH, Bar-Yehuda S, Fong KL, Fishman P (2004) Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int J Clin Pharmacol Ther 42(10):534–542
Linden J (1994) Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions. Trends Pharmacol Sci 15(8):298–306
Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA (2008) Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett 18(9):2813–2819. https://doi.org/10.1016/j.bmcl.2008.04.001
Paoletta S, Tosh DK, Finley A, Gizewski ET, Moss SM, Gao ZG, Auchampach JA, Salvemini D, Jacobson KA (2013) Rational design of sulfonated A3 adenosine receptor-selective nucleosides as pharmacological tools to study chronic neuropathic pain. J Med Chem 56(14):5949–5963. https://doi.org/10.1021/jm4007966
Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA (2012) Structure-guided design of A3 adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem 55(10):4847–4860. https://doi.org/10.1021/jm300396n
Hill SJ, May LT, Kellam B, Woolard J (2014) Allosteric interactions at adenosine A1 and A3 receptors: new insights into the role of small molecules and receptor dimerization. Br J Pharmacol 171(5):1102–1113. https://doi.org/10.1111/bph.12345
Jacobson KA, Gao ZG, Göblyös A, IJzerman AP (2011) Allosteric modulation of purine and pyrimidine receptors. Adv Pharmacol 61:187–220. https://doi.org/10.1016/B978-0-12-385526-8.00007-2
Langmead CJ, Christopoulos A (2014) Functional and structural perspectives on allosteric modulation of GPCRs. Curr Opin Cell Biol 27:94–101. https://doi.org/10.1016/j.ceb.2013.11.007
May LT, Leach K, Sexton PM, Christopoulos A (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. https://doi.org/10.1146/annurev.pharmtox.47.120505.105159
Gao ZG, Kim SG, Soltysiak KA, Melman N, AP I, Jacobson KA (2002) Selective allosteric enhancement of agonist binding and function at human A3 adenosine receptors by a series of imidazoquinoline derivatives. Mol Pharmacol 62(1):81–89
Gao ZG, Kim SK, Gross AS, Chen A, Blaustein JB, Jacobson KA (2003) Identification of essential residues involved in the allosteric modulation of the human A3 adenosine receptor. Mol Pharmacol 63(5):1021–1031
Gao ZG, Van Muijlwijk-Koezen JE, Chen A, Muller CE, IJzerman AP, Jacobson KA (2001) Allosteric modulation of A3 adenosine receptors by a series of 3-(2-pyridinyl)isoquinoline derivatives. Mol Pharmacol 60(5):1057–1063
Gao ZG, Verzijl D, Zweemer A, Ye K, Göblyös A, IJzerman AP, Jacobson KA (2011) Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem Pharmacol 82(6):658–668. https://doi.org/10.1016/j.bcp.2011.06.017
Gao ZG, Ye K, Göblyös A, IJzerman AP, Jacobson KA (2008) Flexible modulation of agonist efficacy at the human A3 adenosine receptor by the imidazoquinoline allosteric enhancer LUF6000. BMC Pharmacol 8:20. https://doi.org/10.1186/1471-2210-8-20
Göblyös A, Gao ZG, Brussee J, Connestari R, Santiago SN, Ye K, IJzerman AP, Jacobson KA (2006) Structure-activity relationships of new 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric enhancers of the A3 adenosine receptor. J Med Chem 49(11):3354–3361. https://doi.org/10.1021/jm060086s
Heitman LH, Göblyös A, Zweemer AM, Bakker R, Mulder-Krieger T, van Veldhoven JP, de Vries H, Brussee J, IJzerman AP (2009) A series of 2,4-disubstituted quinolines as a new class of allosteric enhancers of the adenosine A3 receptor. J Med Chem 52(4):926–931. https://doi.org/10.1021/jm8014052
Kim Y, de Castro S, Gao ZG, IJzerman AP, Jacobson KA (2009) Novel 2- and 4-substituted 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric modulators of the A3 adenosine receptor. J Med Chem 52(7):2098–2108. https://doi.org/10.1021/jm801659w
Cohen S, Barer F, Bar-Yehuda S, IJ AP, Jacobson KA, Fishman P (2014) A3 adenosine receptor allosteric modulator induces an anti-inflammatory effect: in vivo studies and molecular mechanism of action. Mediat Inflamm 2014:708746. https://doi.org/10.1155/2014/708746
Du L, Gao ZG, Nithipatikom K, IJzerman AP, Veldhoven JP, Jacobson KA, Gross GJ, Auchampach JA (2012) Protection from myocardial ischemia/reperfusion injury by a positive allosteric modulator of the A3 adenosine receptor. J Pharmacol Exp Ther 340(1):210–217. https://doi.org/10.1124/jpet.111.187559
Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J (1997) Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol Pharmacol 52(5):846–860
Kreckler LM, Wan TC, Ge ZD, Auchampach JA (2006) Adenosine inhibits tumor necrosis factor-a release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor. J Pharmacol Exp Ther 317(1):172–180. https://doi.org/10.1124/jpet.105.096016
Takano H, Bolli R, Black RG Jr, Kodani E, Tang XL, Yang Z, Bhattacharya S, Auchampach JA (2001) A1 or A3 adenosine receptors induce late preconditioning against infarction in conscious rabbits by different mechanisms. Circ Res 88(5):520–528
van der Hoeven D, Wan TC, Auchampach JA (2008) Activation of the A3 adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Mol Pharmacol 74(3):685–696. https://doi.org/10.1124/mol.108.048066
Chandrasekera PC, Wan TC, Gizewski ET, Auchampach JA, Lasley RD (2013) Adenosine A1 receptors heterodimerize with b1- and b2-adrenergic receptors creating novel receptor complexes with altered G protein coupling and signaling. Cell Signal 25(4):736–742. https://doi.org/10.1016/j.cellsig.2012.12.022
Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, IJzerman AP, Cherezov V, Stevens RC (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337(6091):232–236. https://doi.org/10.1126/science.1219218
Molecular Operating Environment (MOE), version 2012.10, Chemical Computing Group Inc., 1255 University St., Suite 1600, Montreal, QC, H3B 3X3 (Canada)
The UnitProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UnitProt). Nucleic Acids Res 40:D71–D75
Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55(2):351–367. https://doi.org/10.1002/prot.10613
Schrödinger Suite (2012). Schrödinger, LLC, New York, NY
Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22(5):623–625. https://doi.org/10.1093/bioinformatics/btk023
Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332(6027):322–327. https://doi.org/10.1126/science.1202793
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749. https://doi.org/10.1021/jm0306430
Murphree LJ, Marshall MA, Rieger JM, MacDonald TL, Linden J (2002) Human A2A adenosine receptors: high-affinity agonist binding to receptor-G protein complexes containing Gb4. Mol Pharmacol 61(2):455–462
Nawaratne V, Leach K, Felder CC, Sexton PM, Christopoulos A (2010) Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: identification of ligand-specific and global activation mechanisms. J Biol Chem 285(25):19012–19021. https://doi.org/10.1074/jbc.M110.125096
Acknowledgements
This research was supported in part by the National Institutes of Health (R01HL133589, R01HL111392, and the NIDDK Intramural Research Program Z01 DK01117-26).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Lili Dua declares that she has no conflict of interest.
Zhan-Guo Gao declares that he has no conflict of interest.
Silvia Paoletta declares that she has no conflict of interest.
Tina C. Wan declares that she has no conflict of interest.
Elizabeth T. Gizewski declares that she has no conflict of interest.
Samantha Barbour declares that she has no conflict of interest.
Jacobus P. D. van Veldhoven declares that he has no conflict of interest.
Adriaan P. IJzerman declares that he has no conflict of interest.
Kenneth A. Jacobson declares that he has no conflict of interest.
John A. Auchampach declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Du, L., Gao, ZG., Paoletta, S. et al. Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signalling 14, 59–71 (2018). https://doi.org/10.1007/s11302-017-9592-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-017-9592-1