
Abstract
Extracellular nucleotides can modulate the immunological response by activating purinergic receptors (P2Rs) on the cell surface of macrophages, dendritic, and other immune cells. In particular, the activation of P2X7R can induce release of cytokines and cell death as well as the uptake of large molecules through the cell membrane by a mechanism still poorly understood. Polyoxotungstate-1 (POM-1) has been proposed as a potent inhibitor of ecto-nucleotidases, enzymes that hydrolyze extracellular nucleotides, regulating the activity of P2Rs. However, the potential impact of POM-1 on P2Rs has not been evaluated. Here, we used fluorescent dye uptake, cytoplasmic free Ca2+ concentration measurement, patch-clamp recordings, scanning electron microscopy, and quantification of inflammatory mediators to investigate the effects of POM-1 on P2Rs of murine macrophages. We observed that POM-1 blocks the P2YR-dependent cytoplasmic Ca2+ increase and has partial effects on the cytoplasmic Ca2+, increasing dependence on P2XRs. POM-1 can inhibit the events related with ATP-dependent inflammasome activation, anionic dye uptake, and also the opening of large conductance channels, which are associated with P2X7R-dependent pannexin-1 activation. On the other hand, this compound has no effects on cationic fluorescent dye uptake, apoptosis, and bleb formation, also dependent on P2X7R. Moreover, POM-1 can be considered an anti-inflammatory compound, because it prevents TNF-α and nitric oxide release from LPS-treated macrophages.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extracellular ATP (eATP) and its derivatives are key mediators in the immune system, acting as pro-inflammatory or anti-inflammatory factors depending on concentration and the receptors expressed in each particular tissue [1]. Nucleotides function as immunomodulators by activation of cell surface P2 receptors (P2Rs) [2]. P2Rs fall into two classes: the ionotropic P2XRs (P2X1–7R) and the G protein-coupled P2YRs (P2Y1, 2, 4, 6, 11, 12, 13, and 14R) [3, 4]. Both types of P2Rs are expressed by macrophages, dendritic, and endothelial cells, as well as by other immune cells [5].
P2X7R is a cation-selective channel permeable to Na+, K+, and Ca2+ and exhibits a number of unusual properties, including the ability to change membrane permeability. After prolonged exposure to millimolar concentrations of ATP, P2X7R-dependent permeabilization pathways with high conductance are opened, allowing the passage of molecules with an upper limit of approximately 900 Da in macrophages [6]. Two main hypotheses have been proposed to explain the mechanism of permeation to larger molecules: (1) dilation of the channel portion of the receptor after prolonged exposure to high agonist concentrations [7] and (2) opening of a protein pore distinct from the receptor itself [8]. It has been proposed that pannexin-1 (pnx-1) channels are the molecules responsible for P2X7R-associated pores [9]. However, recent evidence has shown that uptake process of P2X7R-associated molecules is more complicated than a single permeation pathway and presents different selectivity for cationic and anionic molecules, indicating the possible co-existence of multiple transport mechanisms [10, 11]. Recently, the possibility that the permeation of organic dyes can occur directly through the P2X7R channel without the need of either additional molecules or “pore dilation” has also been proposed [12]. New pharmacological tools are needed to better characterize these different pathways.
P2X7R activation has been widely studied as a trigger of the NLRP3 inflammasome, but eATP through P2X7R can modulate immune functions even in an inflammasome-independent manner [13]. P2X7R can induce cell death, cytokine release, killing of intracellular pathogens, and membrane blebbing [14,15,16,17]. The specific pathways that lead to each P2X7R-dependent response in immune cells are still poorly understood, as is the link between these functional responses and the change in membrane permeability.
The interplay between eATP and P2Rs is influenced by the activity of surface ecto-nucleotidases, which hydrolyze extracellular nucleotides in a stepwise manner to ADP, AMP, and adenosine. Four ecto-nucleotidase families have been described: ecto-nucleoside triphosphate diphosphohydrolases (NTPDases), ecto-5′-nucleotidase (5′-NT), ecto-nucleotide pyrophosphatases/phosphodiesterases (NPPs), and alkaline phosphatases (APs) [18]. NTPDase1/CD39 hydrolyzes extracellular triphosphonuclosides and diphosphonuclosides to monophosphonucleosides and is the dominant ecto-nucleotidase in immune cells [19, 20]. Besides their potential role as modulators of P2Rs, ecto-nucleotidases are also involved in immunomodulation and infection [21, 22]. So, to completely define the roles that nucleotides and nucleosides play in purinergic signaling, it would be useful to prevent the extracellular metabolism of nucleotides by blocking ecto-nucleotidases. However, there are only a few potent inhibitors of these enzymes, and many of the available blockers are expensive, unstable, and/or unspecific, also inhibiting P2Rs [23, 24]. For example, suramin is a well-known and widely used generic P2R antagonist [25] and also an E-NTPDase inhibitor, as are PPADS [26] and BG0 [27].
Polyoxometalates (POMs) are negatively charged inorganic compounds that contain metal ions surrounded by oxygen atoms [28]. Because these compounds have high molecular weights and are not drug-like molecules, they were found to be useful pharmacological tools [23, 29, 30]. Sodium polyoxotungstate (POM-1) was proposed as a potent inhibitor of NTPDase1, 2, and 3 [31], and since then, it has been used in different models. POMs have been shown to exhibit biological activities, including anticancer [32], antibacterial [33], antiprotozoal [34], antiviral [22, 35], and antidiabetic activities [36]. However, their biological mechanisms of action at the molecular level are not well understood.
In particular, the potential effects of POM-1 on purinergic receptors have not been evaluated, even when this compound has been used in P2R studies [37,38,39]. In the present study, we investigated the effects of POM-1 on P2R signaling, with focus on P2X7R and its pro-inflammatory activity in murine macrophages. To evaluate the different phenomena associated with P2Rs, we used fluorescent dye uptake, cytoplasmic free Ca2+ concentration measurement with Fura-2, patch-clamp recordings, scanning electron microscopy, and quantification of inflammatory mediators, such as pro-inflammatory cytokines and nitric oxide (NO). We conclude that POM-1 has inhibitory action on P2YR-dependent cytoplasmic Ca2+ increase and some but not all P2X7R-associated responses. Additionally, POM-1 can be considered an anti-inflammatory compound, because it prevents TNF-α and NO release from LPS-treated macrophages.
Material and methods
Material
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, penicillin, and streptomycin were obtained from Gibco (São Paulo, SP, Brazil); thioglycollate medium was acquired from Difco (Detroit, MI, USA); NaCl and MgCl2 were purchased from Vetec (Rio de Janeiro, RJ, Brazil); KCl and ethanol were obtained from Reagen (Rio de Janeiro, RJ, Brazil); K3Fe(CN)6, CaCl2, EGTA, HEPES, dimethylsulfoxide (DMSO), carboxyfluorescein (CF), ionomycin, probenecid, phosphate-buffered saline (PBS), sulforhodamine B (SR-B), adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), uridine triphosphate (UTP), sodium polyoxotungstate (POM-1), sodium molybdate, malachite green, Triton X-100, ethidium bromide (EB), and LPS from Escherichia coli 0111:B4 were purchased from Sigma-Aldrich (São Paulo, SP, Brazil); Fura-2 AM came from Life Technologies (São Paulo, SP, Brazil), and BAPTA-AM was obtained from Invitrogen (São Paulo, SP, Brazil).
Animals
Swiss-Webster mice were obtained from the animal facilities of the Centro de Criação de Animais de Laboratório (CECAL), FIOCRUZ (Rio de Janeiro, RJ, Brazil). All animals were 8 to 12 weeks old, weighed approximately 16–25 g, and were handled according to the guidelines for animal use in scientific experiments of the Carlos Chagas Filho Institute of Biophysics of Federal University of Rio de Janeiro. The protocols were approved by the Research Ethics Committee on Animal Care of the Health Science Center of Federal University of Rio de Janeiro (protocol number 001200.001568/2013-87).
Macrophage isolation and culture
Thioglycollate-elicited macrophages were obtained from the intraperitoneal cavity of mice, 4 days after thioglycollate injection, as previously described [10]. Briefly, cells were washed in DMEM and plated on 35-mm culture dishes, 12, 24, or 96-well plates or glass coverslips, according to the experiment. The cultures were maintained in DMEM supplemented with 10% fetal bovine serum, 2 g/L sodium bicarbonate, 0.3 mg/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (supplemented medium) at 37 °C in a humidified atmosphere containing 5% CO2. Non-adherent cells were removed after 40 min, and the macrophages were kept for 4 days under the same culture conditions.
Ecto-nucleotidase activity assays
Ecto-nucleotidase activity was determined using the malachite green assay and high-performance liquid chromatography (HPLC). Using the malachite green colorimetric assay, NTPDase activity in macrophages (2 × 105 cells/petri dish) was determined by measuring the amount of liberated inorganic phosphate (Pi) [40]. Prior to testing, the cells were washed with reaction medium containing (in mM) 1 CaCl2, 145 NaCl, 5 KCl, 10 HEPES, and 1 MgCl2 at pH 7.4 (normal saline solution). The reaction was initiated by adding the nucleotide substrate to the culture dish and stopped by the addition of the supernatants of the cultures in a tube containing 1 mL of cold 25% charcoal in 0.1 M HCl. This charcoal suspension was washed at least 20 times with 0.1 M HCl before use to avoid inorganic phosphate (Pi) contamination [41]. The tubes were centrifuged at 1500×g for 10 min. Aliquots (50 μL) of the supernatants containing the released Pi were quantified using the malachite green colorimetric reagent and a solution containing different concentrations of Pi as standard. The malachite green reagent was prepared by mixing 2 g of sodium molybdate, 0.3 g of malachite green, and 0.5 g of Triton X-100 in 1 L of 0.7 M HCl. Controls to determine non-enzymatic Pi release were performed by incubating the cells in the absence of substrate or incubating the substrate in the absence of cells, which were added after the reaction was stopped.
For the HLPC-UV analysis, macrophages (2 × 106 cells/petri dish) cultured as described above were incubated with 3 mM ATP for 30 min with or without 100 μM POM-1 in normal saline solution. The supernatants were collected; the pH was adjusted to 6.5 with 3.5 M NaOH/1 M HCO3, and the samples were centrifuged for 10 min at 15,000×g. The supernatant was analyzed with a Prominence HPLC (Shimadzu, Japan), equipped with a vacuum degasser, a binary pump with a manual injector, and UV detector. Nucleosides were separated at room temperature in a C-18 Kromasil column (250 × 4.6 mm) using 1 mL/min flow rate of 100 mM H3PO4 pH 6.2 adjusted with TEA (mobile phase A) and 5 mM solution of MgSO4 in the mobile phase A (mobile phase B). The elution program was composed of 3% B (0–5 min) followed by a linear gradient of 3 to 100% B (5–38 min) and 100% B (38–90 min). Peaks were detected at 254 nm, identified by their retention times, and confirmed after co-injection of each supernatant spiked with authentic standards. Calibration curves were assessed by the integration of peaks of each standard (prepared as 2 mM stock solutions in PBS) in the range of 1 μM and 1 mM (r 2 > 0.9982).
Intracellular calcium measurements
Macrophages (2 × 105 plated on glass coverslips) were loaded with 5 μM Fura-2 AM at 37 °C in supplemented medium containing 2.5 mM probenecid during 40 min. The cells were then washed twice in normal saline solution and placed in a chamber whose base was formed by the coverslip containing the cells. The cells were maintained at 37 °C or room temperature (depending of the experiment) in normal saline solution. Cytoplasmic calcium concentrations of groups of 20–30 cells were monitored continuously with the use of a fluorescence imaging spectrofluorimeter (Easy Ratio Pro) equipped with a DeltaRAMX Illuminator, an Olympus IX71 microscope, a QuantEM 5125C camera, and the ImagePro Plus V 6.3 software (PTI Photon Technology International, Princeton, NJ, USA) at 37 °C or room temperature. Fura-2 was excited alternately at 340 and 380 nm, and the emission at 510 nm was measured. The ratio measurement is proportional to the cytoplasmic calcium concentration and was determined every 100 ms. Nucleotide application was performed by gently adding 20 μL of the appropriate concentration nucleotide solution with a manual pipette. Maximum and minimum fluorescence values were obtained by adding ionomycin (20 μM) and EGTA (10 mM), respectively.
Dye uptake
Unless otherwise specified, macrophages (1 × 105) were kept at 37 °C for 5 min, in normal saline solution before the experiment. The fluorescent dyes CF (5 mM) or SR-B (200 μM) and ATP (5 mM) were then added, and the cells were kept under the same conditions for an additional period of 10 min. In some experiments, cells were pre-incubated with POM-1 for a period of 30 min, as specified in the figure legends. Dye uptake was then determined either by fluorescence microscopy or by quantitative spectrofluorimetry, as described below.
Fluorescence microscopy was performed using an Axiovert 100 microscope (Karl Zeiss, Oberkochen, Germany) equipped with an HBO lamp, a digital camera (Olympus American Inc., PA, USA), and Image-Pro Plus v6.2 software (Media Cybernetics, Inc., Bethesda, MD, USA).
Quantitative spectrofluorimetry was performed in experiments with CF or SR-B, using an FLX-800 plate reader (BioTek Instruments Inc., Winooski, VT, USA) according to the following protocol. The cells were gently washed five times with PBS, lysed by the addition of 100 μL PBS containing 0.01% BSA and 0.1% Triton X-100, and scraped off the plate, and the content was employed for fluorescence determination using the following excitation and emission wavelength ranges (nm): 420–450 and 528–520 for CF and 516–520 and 620–640 for SR-B. Protein concentrations were determined by the Bradford method.
Electrophysiological measurements
Macrophages (1 × 105) were plated in 35-mm plastic culture dishes for 3 days in the conditions described above. Before the experiment, the culture medium was exchanged for normal saline solution (see below). Experiments were performed at − 40 mV holding potential, maintaining the cells at 37 °C using a heating chamber. Ionic currents were recorded in whole-cell or cell-attached configurations, using an Axonpatch 200B amplifier (Axon Instruments, USA) according to standard patch-clamping techniques [42]. High-resistance seals (1–5 GΩ) were formed after offset potential compensation, using micropipettes filled with a solution containing (in mM) 135 KCl, 5 NaCl, 2 MgCl2, 0.1 EGTA, and 10 HEPES, pH 7.4, in the whole-cell experiments, and normal saline solution in cell-attached experiments. ATP at the indicated concentrations was applied to the cell cultures by pneumatic injection, using a pneumatic pump (NeuroPhore BH-2 system, Harvard Apparatus, USA). Data were collected using pClamp software, version 10.2, and a Digidata 1140 interface (Axon Instruments, USA) and plotted using GraphPad Prism software, version 6.0 (USA).
Induction and analysis of cell death
Apoptosis induction
Macrophages (2 × 105/well plated in 12-well culture plates) were pre-incubated at 37 °C with or without POM-1 for 30 min in DMEM and then with or without 5 mM ATP for an additional 30 min. After this period, cells were gently washed twice and incubated for 6 h at 37 °C with supplemented DMEM. The cells were then centrifuged at 5600×g for 1 min, and the pellet was used for determination of apoptosis, as described in [43, 44]. Briefly, the cells were suspended in 250 μL of a lysing buffer that preserved the nuclei for the determination of DNA content (50 μg/mL EB, 0.1% sodium citrate, 0.1% Triton X-100). The nuclei were then analyzed by flow cytometry (FACSCalibur, Becton Dickinson, Mountain View, USA), and the degree of apoptosis was quantified by counting the number of hypodiploid events. At least 3000 events were collected per sample, and normalized values were calculated by subtracting the spontaneous apoptosis obtained in the controls (absence of any treatment) from all experimental values and calculating the percent of the number of hypodiploid events (percent of apoptosis) induced by 5 mM ATP as 100%.
Pyroptosis induction
Macrophages (5 × 106/well plated in a 24-well plate) were washed two times and then pre-incubated with POM-1 for 30 min in supplemented DMEM at 37 °C. After that, the cells were treated with LPS (100 ng/mL) for 6 h, finally adding ATP (3 mM) for 2 h in the same conditions. Macrophages were also treated only with POM-1 for 8 h, ATP for 2 h, or LPS for 6 h. After this period, the cells were centrifuged at 5600×g for 1 min, and the supernatant was collected to measure the lactate dehydrogenase (LDH) content. The LDH enzymatic activity in the supernatants was determined by using a commercially available colorimetric assay kit according to the maker’s instructions (Doles, Goiania, GO, Brazil). Controls were obtained by using supernatants of untreated cells (0%) and cells treated with 1% saponin for 30 min (100%). Each reading was subtracted from the reading obtained in medium containing POM-1 without cells in order to avoid any possible interference of this compound with the reagents in the LDH detection kit.
Light and scanning electron microscopy
For light microscopy, images of macrophages (1 × 105 per coverslip) were obtained using a differential interference contrast (DIC) system in an Olympus BX 51 microscope with Olympus DP-12 digital camera.
For scanning electron microscopy (SEM), cells adhered in glass coverslips were fixed in solution that consisted of 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2. Cells were fixed for 1 h at room temperature and 12 h at 4 °C. After this period, the samples were washed in 0.1 M cacodylate buffer, post-fixed in 1% OsO4 and 0.8% K3Fe(CN)6, washed in distilled water, dehydrated in a graded ethanol series (20 GL absolute) for 20 min each step, critical point dried in liquid CO2, mounted on metallic stubs, and coated with gold (20–25 nm deposited). The samples were examined using an FEI Quanta 250 scanning electron microscope operating at 25–30 kV, in high vacuum mode.
IL-1β and TNF-α release assays
Macrophages (5 × 106/well plated in a 24-well plate) were washed two times and then pre-incubated with POM-1 for 30 min in supplemented DMEM at 37 °C. After that, the cells were treated with LPS (100 ng/mL) for 6 h, finally adding ATP (3 mM) for 2 h in the same conditions. The supernatant was collected, and the amounts of IL-1β and TNF-α were measured by ELISA kit (PeproTech, USA) according to the manufacturer’s manual. Assays were performed in duplicate for each independent experiment.
Nitric oxide assay
Macrophages (5 × 105/well plated in 96-well plates) were treated or not with LPS (at the concentrations indicated) for 24 h, with or without of 100 μM POM-1. After this time, the culture supernatants were collected for measurement of the amount of nitrite using the Griess colorimetric method [45]. In this method, nitrite reacts with sulfanilamide and N-α-naphthyl-ethylenediamine, giving a coloration that can be evaluated using an ELISA microplate reader with a filter for a wavelength of 550 nm.
Data analysis
Differences between experimental groups were evaluated by the Mann-Whitney non-parametric test. Data were analyzed using GraphPad InsTat software (GraphPad Software Inc., v5.0).
Results
Ecto-nucleotidase activity in macrophage is inhibited by POM-1
To characterize the ecto-nucleotidase activity on the surface of murine macrophages, we incubated the cells with 500 μM ATP or 500 μM AMP at 37 °C and analyzed the time course of nucleotide hydrolysis using the malachite green assay. As shown in Fig. 1a, a high level of ecto-phosphohydrolase activity was observed when ATP was used as substrate, but we did not observe the hydrolysis of AMP. We further evaluated the effect of POM-1 on the ecto-ATPase activity of murine macrophages. Cells were pre-incubated for 10 min with 50 or 100 μM POM-1 before the addition of substrate, and this concentration was maintained during the reaction. As expected, POM-1 displayed a dose-dependent inhibition of the ecto-ATPase activity in macrophages, with the maximum effect (greater than 70% inhibition) being observed at 100 μM after 20 min of reaction (Fig. 1a). We also confirmed the effect of POM-1 on ATP hydrolysis using HPLC. Macrophages were incubated with 3 mM ATP for 30 min in the absence or presence of 100 μM POM-1. As can be seen in Fig. 1b, macrophages hydrolyzed 65% of ATP, generating 13% of ADP and 52% of AMP. However, when cells were treated with POM-1, the ATP hydrolysis was only 30%, reducing the production of ADP to 4.5% and AMP to 24%. The absence of AMP hydrolysis in macrophages was also confirmed by HPLC analysis. We used 3 mM AMP as substrate and did not observe any AMPase activity (data not shown).

Ecto-nucleotidase activity of murine macrophages is inhibited by POM-1. a Mouse peritoneal macrophages were incubated for different periods of time at 37 °C in a reaction medium containing the substrates 500 μM AMP (white squares), ATP (dark circles), and also ATP plus 50 μM POM-1 (white triangles, trace lines) or ATP plus 100 μM POM-1 (dark triangles, dotted lines), and the concentration of Pi in the supernatant was analyzed by the malachite green assay. Data are the means ± SD of five independent experiments. b Mouse peritoneal macrophages were incubated for 30 min in the presence of 3 mM ATP with or without 100 μM POM-1. The quantity of AMP (black bars), ADP (gray bars), and ATP (dark gray bars) present in the supernatant was analyzed using HPLC. The results are expressed as percentage of control (% initial ATP concentration) and are means ± SE of triplicate experiments. *p < 0.05
POM-1 inhibits free intracellular Ca2+ increase induced by nucleotides
We used Fura-2 microfluorimetry to investigate the effect of POM-1 on P2Rs of macrophages. In these cells, ATP, ADP, UTP, and UDP induced a rapid and concentration-dependent intracellular Ca2+ increase, due to Ca2+ release from intracellular stores (triggered by P2YR activation) or influx from the extracellular medium (dependent of P2XR activation). Under our experimental conditions, ADP elicits a Ca2+ response in macrophages only at concentrations greater than or equal to 10 μM, consistent with the expression of P2Y1R (Fig. 2a). The cells were more responsive to UTP (Fig. 2c) and UDP (Fig. 2e) treatments, with a Ca2+ response to doses as low as 500 nM, compatible with the activation of P2Y2R, P2Y4R, and P2Y6 [46]. The presence of 100 μM POM-1 completely inhibited such responses (Fig. 2b, d, f, respectively).

Effect of POM-1 on intracellular calcium response induced by ADP, UTP, and UDP in murine macrophages. Mouse peritoneal macrophages were loaded with Fura-2, and 1, 10, and 100 μM ADP (a, b); 0.5, 1, and 10 μM UTP (c, d); or 0.5, 1, and 10 μM UDP (e, f) were added at times indicated with arrows. Cells were pre-incubated and maintained in the presence of POM-1 (b, d, f) and treated with the nucleotides. Fluorescence is proportional to the intracellular calcium concentration and was measured using arbitrary fluorescence ratio units. Representative data of a single experiment are shown. Experiments were repeated at least four times with similar results
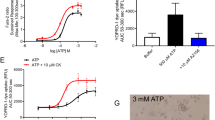
ATP at concentrations 1–100 μM induced a single spike of Ca2+ after each application (Fig. 3a), a response compatible with the activation of P2Y2R, P2Y4R [46], and P2X1–7Rs [47]. Continuous application of 2 mM ATP at 37 °C induced a sustained increase in the free cytosolic Ca2+ concentration, a characteristic of P2X7R activation (Fig. 3c). POM-1 totally inhibited the Ca2+ response induced by 1 μM ATP and partially inhibited the response induced by 10 and 100 μM ATP (Fig. 3b). No inhibition was observed in the response obtained with 2 mM ATP (Fig. 3d). These results show that POM-1 has a partial inhibitory effect on P2X1–6Rs and suggest that it does not affect the sustained increase of cytosolic Ca2+ concentration induced by higher doses of ATP and associated with P2X7R activation.

Effect of POM-1 on intracellular calcium response induced by ATP in murine macrophages. Mouse peritoneal macrophages were loaded with Fura-2, and 1, 10, and 100 μM ATP (a, b) were added at the time indicated with arrows. In another set of experiments, cells were maintained at 37 °C and perfused with 2 mM ATP (c, d), indicated by bars. Some cells were pre-incubated and maintained in the presence of POM-1 (b, d) and treated with the nucleotides in the same conditions. Fluorescence is proportional to the intracellular calcium concentration and was measured using arbitrary fluorescence ratio units. Representative data of a single experiment are shown. Experiments were repeated at least four times with similar results
POM-1 inhibits the ATP-induced uptake of anionic but not cationic dyes
To further investigate the effects of 100 μM POM-1 on P2X7Rs, we studied P2X7R-induced dye uptake in macrophages using a cationic ([sulforhodamine B (SR-B)]) and an anionic ([carboxyfluorescein (CF)]) dye. In the presence of 5 mM ATP at 37 °C, both dyes could be detected in the cytoplasmic compartment of the cells by fluorescence microscopy and by quantification of the total amount of dye inside the cell using a fluorimetric method (Figs. 4 and 5). The presence of POM-1 had no effect on ATP-induced SR-B uptake (Fig. 4), but this compound showed a significant inhibitory effect on ATP-induced CF uptake, reducing uptake by almost 75% compared with the control (Fig. 5). To confirm these results, we used another cationic dye, EB, and obtained the same results (data not shown). It is important to note that no dye uptake was observed in macrophages obtained from P2X7−/− mice, either in the presence or in the absence of POM-1, indicating that this is a P2X7R-dependent and not a POM-1-induced phenomenon (data not shown). These results support the view that the uptake of anionic and cationic dyes involves different pathways in macrophages and show that POM-1 is only able to inhibit the anionic one.

eATP-induced sulforhodamine B uptake in murine macrophages is inhibited by POM-1. Fluorescence microscopy of mouse peritoneal macrophages incubated for 15 min at 37 °C in the absence (a, b) or in the presence (c, d) of 5 mM ATP, with (b, d) or without (a, c) 100 μM POM-1. In all cases, SR-B (200 μM) was added and the samples were washed before transfer to the microscope stage. e Quantitative comparison of uptake of SR-B in the presence or not of POM-1. Cells were incubated at 37 °C with SR-B for 10 min in the presence of 5 mM ATP, with or without POM-1, as indicated, and the amount of dye taken up by the cells was measured by spectrofluorimetry, as described in the “Material and methods” section. The bars represent the means of four independent experiments. Scale bars = 200 μm

POM-1 does not affect eATP-induced carboxifluorescein uptake in murine macrophages. Fluorescence microscopy of mouse peritoneal macrophages incubated for 15 min at 37 °C in the absence (a, b) or in the presence (c, d) of 5 mM ATP, with (b, d) or without (a, c) 100 μM POM-1. In all cases, CF (5 mM) was added and the samples were washed before transfer to the microscope stage. e Quantitative comparison of uptake of CF in the presence or not of POM-1. Cells were incubated at 37 °C with CF for 10 min in the presence of 5 mM ATP, with or without POM-1, as indicated, and the amount of dye taken up by the cells was measured by spectrofluorimetry as described in the “Material and methods” section. The bars represent the means of four independent experiments. Scale bars = 200 μm
POM-1 inhibits the large ATP-induced channels
P2X7R activation by brief exposure to micromolar concentration of eATP opens a selective cation channel with an 8–9 pS conductance that allows the influx of Na+ and Ca2+ [6]. We performed whole-cell current recordings and observed that 100 μM ATP triggers the typical inward currents (Fig. 6a). When the macrophages were pre-treated with 100 μM POM-1, the same concentration of eATP only induced a smaller current, indicating that POM-1 has partial inhibition of the P2X7R cation channel (Fig. 6b).

Effect of POM-1 on eATP-induced ionic currents. Currents evoked by 100 μM ATP in murine macrophages, under voltage clamp at − 40 mV, using whole-cell mode, in the absence (a) or presence of 100 μM POM-1 (b). Single-channel currents activated by 4 mM ATP, under voltage clamp at − 40 mV, using cell-attached mode, in the absence (c) or presence of 100 μM POM-1 (d). Representative data of a single experiment are shown. Experiments were repeated at least four times with similar results
We previously identified a P2X7R-associated non-selective channel that is activated by eATP and can be recorded using cell-attached configuration [10]. We therefore investigated whether this large conductance channel could be inhibited by the presence of POM-1. We observed that, whereas the channel opening was detected in the macrophages treated with 5 mM ATP (Fig. 6c), no large conductance channels were observed in the cells treated with the same ATP concentration in the presence of 100 μM POM-1 (Fig. 6d). These results indicate that POM-1 inhibits the large conductance P2X7-associated channel and partially the small conductance cationic channel.
POM-1 inhibits ATP-induced P2X7-associated pyroptosis but not apoptosis
The prolonged activation of P2X7R with millimolar concentrations of eATP triggers cell death, by apoptosis [48] and pyroptosis [49]. Thus, we performed two different experimental protocols that allowed distinguishing between both types of eATP-induced cell death. For apoptotic death induction, macrophages were incubated with 5 mM ATP for 20 min, in the presence or not of 100 μM POM-1 (induction period), washed, and kept in culture medium, without eATP for 6 h more (effector period). Apoptosis was quantified by measuring the percent of hypodiploid nuclei using flow cytometry, while cell lysis was quantified by LDH release at the end of the effector period. As can be seen in Fig. 7a, the POM-1 treatment did not block the ATP-induced apoptosis.

eATP-induced cell death in the presence or not of POM-1. Murine macrophages were exposed to zero or 5 mM ATP, with or without addition of 100 μM POM-1 for 20 min, washed, and maintained in culture for 6 h. After that, macrophages were incubated with apoptosis buffer containing EB and the nuclei were analyzed by flow cytometry. The percentage of apoptosis was calculated according to the number of hypodiploid events, considering the result obtained with 5 mM ATP as 100% (a). Cells were pre-incubated or not with 100 μM POM-1 for 30 min and treated with 100 ng/mL LPS for 6 h and with 3 mM ATP for 2 h. Macrophages were also treated only with POM-1 for 8 h, ATP for 2 h, or LPS for 6 h. LDH activity was measured in the supernatants. Control lyses were obtained by using supernatants of untreated cells (0%), and cells treated with 1% saponin for 30 min (100%) (b). Data are the means ± SE of at least four independent experiments. *p < 0.01; **p < 0.0001
We evaluated the effects of POM-1 on pyroptosis by treating the macrophages with 100 ng/mL LPS for 6 h, followed by 2 h with 3 mM ATP, in the presence or not of 100 μM POM-1. The measurement of the LDH release showed that POM-1 significantly inhibits the pyroptotic death induced by LPS plus ATP and also the necrotic cell death caused by the incubation with ATP alone for 2 h (Fig. 7b). These results again show a selective inhibition of P2X7R-activated phenomena by POM-1. The treatment only with POM-1 did not cause any difference in macrophage viability.
Bleb formation in response to ATP is not blocked by POM-1
The activation of P2X7R induces extensive changes in cell morphology, cytoskeletal architecture, and membrane blebbing, a phenomenon defined as the formation of large membrane-bound vesicles, which protrude from the cell surface and are roughly hemispherical and devoid of organelles [50, 51]. To investigate the effect of POM-1 on P2X7R-dependent blebbing phenomena, we treated macrophages with 5 mM ATP in the presence or not of POM-1 and observed the morphological changes after 30 min using DIC microscopy and SEM. Using DIC microscopy, we observed that ATP-treated cells exhibit a substantial change in cell morphology and the formation of membrane blebs (Fig. 8a). The macrophages appeared rounded, with retracted filopodia, and 85% of them showed membrane blebs. In contrast, cells treated with control buffer or POM-1 had numerous filopodia, appeared more elongated, and did not have membrane blebs. The pre-incubation with POM-1 did not prevent the morphological changes induced by eATP (Fig. 8a, b).

POM-1 does not affect membrane blebbing induced by eATP in macrophages. Murine macrophages were plated on glass coverslips and treated with saline solution (control) or 5 mM ATP for 30 min, with or without 100 μM POM-1, washed, and visualized using light microscopy or processed for SEM. a Images obtained using differential interference contrast (DIC). Scale bars 7 μm. b Cells from five random fields were counted and scored for blebbing. The graph represents the mean percentages of macrophages that were positive for blebbing + ES, from four independent experiments. c Scanning electron microscopy of macrophages with the different treatments. Scale bars 5 μm
To better characterize the eATP response in macrophages and to observe if there is any morphological change in membrane surface of the cells as a consequence of POM-1 treatment, we performed SEM. As we can see in Fig. 8c, only the macrophages treated with eATP showed bleb formation, even in the presence of POM-1. In the absence of eATP, untreated cells presented similar morphological aspect to that of cells treated with POM-1, showing that POM-1 per se does not cause membrane surface change in macrophages.
TNF-α, IL-1β, and NO release in LPS-primed macrophages is blocked by POM-1
eATP, acting via P2X7R, induces production and release of IL-1β by activation of the NLRP3/caspase-1 complex [13]. We examined the effects of POM-1 on IL-1β and TNF-α secretion by LPS-primed macrophages in response to 3 mM ATP. Macrophages treated with 100 μM POM-1 displayed a significant decrease in ATP-induced IL-1β release compared to cells without treatment (Fig. 9a). In our conditions, LPS with or without eATP can lead to TNF-α release, which was also observed by others [52]. Both types of stimulation were inhibited by pre-treatment with POM-1, blocking TNF-α production by macrophages (Fig. 9b).

POM-1 blocks TNF-α, IL-1β, and NO release in LPS-primed macrophage. Peritoneal mouse macrophages were incubated with LPS (100 ng/mL) for 6 h, finally adding ATP (3 mM) for 2 h. In the cases indicated, 100 μM POM-1 was present in the culture medium all the time. The supernatants were collected, and the cytokine measured by an ELISA detection kit for IL-1β (a) or TNF-α (b). For the NO measurements, macrophages were treated with LPS (at the indicated concentrations) for 24 h, in the presence or not of 100 μM POM-1. The supernatant was collected, and the NO released was measured by the Griess method. Data are the means ± SE of five independent experiments. *p < 0.01; **p < 0.001
We next tested whether POM-1 inhibits LPS-induced production of NO, a process that may involve the release of ATP and the activation of P2 receptors [53, 54]. For that, macrophages were incubated for 24 h with different concentration of LPS in the absence or presence of 100 μM POM-1, and the release of NO was quantified indirectly by measuring nitrite, the product of the reaction of NO with oxygen. POM-1 inhibited the release of NO at all the concentrations of LPS tested (Fig. 9c).
Taken together, these results indicate that POM-1 has a potent anti-inflammatory effect on macrophages.
Discussion
NTPDase1 (CD39) is the dominant ecto-nucleotidase expressed in murine peritoneal macrophages elicited with thioglycollate, as shown by Lévesque and collaborators using RT-PCR and western blot, and who did not detect the presence of CD73 in this cell type [55]. Zanin and co-workers showed that CD73 is expressed in resident, LPS-stimulated and IL-4-stimulated macrophages, but did not detect any AMPase activity or expression in thioglycollate-elicited macrophages [56]. Consistent with these previous data, we found that thioglycollate-elicited murine macrophages have ecto-ATPase activity compatible with CD39, but we did not detect AMP hydrolysis, consistent with the lack of at CD73 activity (Fig. 1). Moreover, the ATP hydrolysis was inhibited by 100 μM POM-1, as expected [57].
The CD39 activity has potent implications for inflammation, due to the reduction of the eATP levels. So, POM-1, as a CD39 inhibitor, is expected to increase the inflammatory response of macrophages. However, our data show that POM-1 has anti-inflammatory effects on these cells. This suggests that the effects of POM-1 on macrophage inflammatory response could be due to the inhibition of P2 receptors.
Extracellular ATP, ADP, UDP, and UTP can induce cytosolic Ca2+ elevation in macrophages through the activation of Gq-coupled P2Y and P2X receptors [58]. In our conditions, extracellular application of ATP, ADP, UTP, and UDP induced elevations of free intracellular Ca2+ concentrations at doses consistent with the activation of P2Y1, 2, 4, and 6, in agreement with previously published results [59]. POM-1 inhibited the cytosolic Ca2+ elevation induced by all these P2Y receptors (Fig. 2). eATP can also induce the influx of extracellular Ca2+ through P2X1–7 channels, most of them previously described in peritoneal macrophages [59]. Here, we showed that POM-1 can partially inhibit the spike of Ca2+ response triggered by P2X1–6, but not the sustained Ca2+ increase evoked by the P2X7R activation (Fig. 3).
We previously described that P2X7R activation in macrophages is associated with two different transport mechanisms through the plasma membrane: a diffusional pathway, which allows the entry of anions, and another pathway for non-diffusional uptake of cations [10]. In HEK-293 cells transfected with P2X7R, we observed the uptake of cationic dyes and sustained increase in cytosolic Ca2+ levels, as well as the ionic inward currents typically associated with the activation of this receptor. In the same previous study, we showed that P2X7R-transfected HEK cells do not present uptake of anionic dyes or opening of large conductance P2X7R-associated channel [10]. In the present study, POM-1 inhibited the uptake of anionic fluorescent dyes (Fig. 5) and the activity of the large conductance ATP-dependent channels (Fig. 6), but not cationic dye uptake or changes in intracellular Ca2+ concentration in thioglycollate-elicited murine macrophages stimulated with 2 mM ATP (Figs. 3 and 4). These results add additional support to the existence of these two distinct P2X7R-dependent pathways for molecule uptake and suggest that POM-1 is a new pharmacological tool to study them. We previously also showed that ATP-induced cationic dye uptake in macrophages does not require intracellular Ca2+ signals [60]. However, the link between the cytosolic Ca2+ elevations, the solute transport mechanisms induced by extracellular nucleotides, and the cellular responses evoked by them in macrophages is not yet clear.
Priming of macrophages with LPS induces intracellular accumulation of pro-IL-1β and pro-IL-18, which will be released after eATP stimulation. P2X7R activation promotes assembly of inflammasome, caspase-1-dependent cleavage, and release of biologically active cytokines [61]. In addition, caspase-1 activation induces pyroptotic cell death [62] and activates NF-κB [63], leading to the secretion of TNF-α [64] and NO production [65]. It is important to mention that caspase-1 induces NF-κB activation independently of its enzymatic role in cytokine maturation [63]. Our results show that POM-1 inhibits the sequence of events trigged by eATP via P2X7R activation in LPS-primed macrophages, since this drug can prevent LDH (Fig. 7b), IL-1β, TNF-α, and NO release (Fig. 9). Interestingly, POM-1 is also able to inhibit the release of TNF-α and NO even without eATP treatment, suggesting that this compound can block the canonical LPS-induced NF-κB activation mediated by TLR4-MyD88 signaling pathway. However, in these conditions, one must also consider the possibility that the LPS stimulation triggers ATP release by the macrophages, as previously shown [53, 54].
Peritoneal macrophages from Entpd1 −/− mice are more susceptible to ATP-induced necrotic cell death and release more IL-1β when stimulated with LPS plus ATP, compared with macrophages from wild-type mice [55]. This finding indicates that the effect of POM-1 in decreasing P2X7-dependent cell death and cytokine release is not caused by inhibition of CD39 activity.
On the other hand, stimulation of P2X7R also activates the caspase-8/9/3 pathway and triggers apoptotic cell death [66]. Hanley and collaborators showed that activation of P2X7R induces blebbing, uptake of cationic dyes, caspase-3/7 activation, and cell death independent of caspase-1 and pannexin-1 [67]. P2X7R-dependent blebbing involves both Ca2+-dependent and Ca2+-independent mechanisms [52, 68,69,70] and is dissociated from IL-1β secretion or capase-1 activation [52]. In accordance with that second pathway activated by P2X7R, we did not observe POM-1 inhibition of cationic uptake (Fig. 4a), apoptosis (Fig. 7a), or bleb formation (Fig. 8) in macrophages treated with ATP. Also, our results are in concordance with previous data, which showed the existence of two different pathways for P2X7R-induced cell death, with separate intracellular signaling events for apoptosis and LDH release [49]. Based on the results obtained so far, a hypothetical model for P2X7R intracellular pathway and POM-1 inhibition is presented in Fig. 10.

Hypothetic model for of POM-1 inhibition of P2X7R intracellular pathways. P2X7R activation by eATP leads to the entry of extracellular Na+ and Ca2+. Prolonged exposure to millimolar concentrations of eATP triggers the opening of different transport mechanisms, which allow the passage of larger molecules, such as the anionic carboxiflourescein (CF) and the cationic sulforhodamine-B (SR-B) dye. Different intracellular cascades are also triggered. We propose two major separated pathways dependent on P2X7R. One sequence of events includes the activation of caspase-3, caspase-8, and caspase-9, resulting in the induction of membrane blebbing and apoptosis. This cascade is associated with uptake of large cationic molecules and cannot be inhibited by POM-1. The other intracellular pathway triggered by P2X7R involves the inflammasome assembly, with caspase-1 activation and induction of release of cytokines (TNF-α, IL-1β), nitric oxide (NO), and pyroptosis. This cascade is associated with the uptake of both large cationic and anionic molecules and can be inhibited by POM-1. Because the TNF-α and NO release triggered by LPS are also inhibited by POM-1, an effect of this compound on the TLR4 canonical pathway should be considered
The role of pannexin-1 in the signaling cascades that mediate the different responses to P2X7R activation in macrophages remains unknown. One difficulty in the effort to better understand this question is the fact that often researchers use the ATP-dependent cationic dye uptake as a measure of pannexin-1 involvement in a particular phenomenon [9, 67, 71]. However, we [10] and others [11] showed that the P2X7-dependent dye uptake has at least two associated pathways, with differences between cations and anions. The participation of pannexin-1 in each transport mechanism stays unclear.
The activation of pannexin-1 channels has been associated with activation of NLRP3 inflammasome/capspase-1 and the induction of cell death and cytokine release [9, 71]. However, Qu and colleagues showed that the activation of caspase-1 and secretion of IL-1β in response to ATP are normal in pannexin-1-deficient macrophages [72].
Non-canonical inflammasome activates another class of inflammatory caspases, including caspase-11, to induce pyroptosis and promote IL-1β by triggering the canonical NLRP3 inflammasome [73]. Recently, Yang and collaborators presented data indicating that pannexin-1 and P2X7R are required for susceptibility to endotoxic shock in mice induced via the non-canonical inflammasome pathway [74]. However, how P2X7R regulates the caspase-11 activation and what the role of pannexin-1 is in this process is not known yet.
POM-1 can inhibit the events related with ATP-dependent inflammasome activation, anionic dye uptake, and also the opening of large conductance channels, which are associated with P2X7R-dependent pannexin-1 activation. Considering that, it seems reasonable to think that these phenomena occur in the same signaling pathway dependent on P2X7R, different from the pathway that includes triggering of apoptosis, blebbing, and cationic dye uptake, which cannot be inhibited by POM-1 (Fig. 10).
References
Junger WG (2011) Immune cell regulation by autocrine purinergic signaling. Nat Rev Immunol 11:201–212. https://doi.org/10.1038/nri2938
Burnstock G, Boeynaems JM (2014) Purinergic signalling and immune cells. Purinergic Signal 10:529–564. https://doi.org/10.1007/s11302-014-9427-2
Burnstock G (2014) Purinergic signalling: from discovery to current developments. Exp Physiol 99:16–34. https://doi.org/10.1113/expphysiol.2013.071951
Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442:527–532. https://doi.org/10.1038/nature04886
Di Virgilio F, Vuerich M (2015) Purinergic signaling in the immune system. Auton Neurosci 191:117–123. https://doi.org/10.1016/j.autneu.2015.04.011
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067. https://doi.org/10.1152/physrev.00015.2002
Virginio C, MacKenzie A, North RA, Surprenant A (1999) Kinetics of cell lysis, dye uptake and permeability changes in cells expressing the rat P2X7 receptor. J Physiol Lond 519:335–346. https://doi.org/10.1111/j.1469-7793.1999.0335m.x
Coutinho-Silva R, Persechini PM (1997) P2Z purinoceptor-associated pores induced by extracellular ATP in macrophages and J774 cells. Am J Phys 273:C1793–C1800
Pelegrin P, Surprenant A (2006) Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J 25:5071–5082. https://doi.org/10.1038/sj.emboj.7601378
Schachter J, Motta AP, de Souza Zamorano A, da Silva-Souza HA, Guimarães MZ, Persechini PM (2008) ATP-induced P2X7-associated uptake of large molecules involves distinct mechanisms for cations and anions in macrophages. J Cell Sci 121:3261–3270. https://doi.org/10.1242/jcs.029991
Cankurtaran-Sayar S, Sayar K, Ugur M (2009) P2X7 receptor activates multiple selective dye-permeation pathways in RAW 264.7 and human embryonic kidney 293 cells. Molecular Pharmacol 76:1323–1332. https://doi.org/10.1124/mol.109.059923
Li M, Toombes GE, Silberberg SD, Swartz KJ (2015) Physical basis of apparent pore dilation of ATP-activated P2X receptor channels. Nat Neurosci 18:1577–1583. https://doi.org/10.1038/nn.4120
Csóka B, Németh ZH, Törő G, Idzko M, Zech A, Koscsó B, Spolarics Z, Antonioli L, Cseri K, Erdélyi K, Pacher P, Haskó G (2015) Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. FASEB J 29:3626–3637. https://doi.org/10.1096/fj.15-272450
Di Virgilio F, Chiozzi P, Ferrari D, Falzoni S, Sanz JM, Morelli A, Torboli M, Bolognesi G, Baricordi OR (2001) Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 97:587–600. https://doi.org/10.1182/blood.V97.3.587
Coutinho-Silva R, Stahl L, Raymond MN, Jungas T, Verbeke P, Burnstock G, Darville T, Ojcius DM (2003) Inhibition of chlamydial infectious activity due to P2X7R-dependent phospholipase D activation. Immunity 19:403–412. https://doi.org/10.1016/S1074-7613(03)00235-8
Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F (2006) The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176:3877–3883. https://doi.org/10.4049/jimmunol.176.7.3877
Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ (2004) The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J Leukoc Biol 75:1173–1182. https://doi.org/10.1189/jlb.1203648
Zimmermann H, Zebisch M, Sträter N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 5:437–502. https://doi.org/10.1007/s11302-012-9309-4
Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC (2007) CD39 and control of cellular immune responses. Purinergic Signal 3:171–180. https://doi.org/10.1007/s11302-006-9050-y
Morandini AC, Savio LE, Coutinho-Silva R (2014) The role of P2X7 receptor in infectious inflammatory diseases and the influence of ectonucleotidases. Biom J 37:169–177. https://doi.org/10.4103/2319-4170.127803
Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Hasko G (2016) Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer 2:95–109. https://doi.org/10.1016/j.trecan.2016.01.003
Schachter J, Delgado KV, Barreto-de-Souza V, Bou-Habib DC, Persechini PM, Meyer-Fernandes JR (2015) Inhibition of ecto-ATPase activities impairs HIV-1 infection of macrophages. Immunobiology 220(5):589–596. https://doi.org/10.1016/j.imbio.2014.12.004
Baqi Y (2015) Ecto-nucleotidase inhibitors: recent developments in drug discovery. Mini Rev Med Chem 15:21–33. https://doi.org/10.2174/1389557515666150219115141
Munkonda MN, Kauffenstein G, Kukulski F, Lévesque SA, Legendre C, Pelletier J, Lavoie EG, Lecka J, Sévigny J (2007) Inhibition of human and mouse plasma membrane bound NTPDases by P2 receptor antagonists. Biochem Pharmacol 74:1524–1534. https://doi.org/10.1016/j.bcp.2007.07.033
Dunn PM, Blakeley AG (1988) Suramin: a reversible P2-purinoceptor antagonist in the mouse vas deferens. Br J Pharmacol 93:243–245
Chen BC, Lee CM, Lin WW (1996) Inhibition of ecto-ATPase by PPADS, suramin and reactive blue in endothelial cells, C6 glioma cells and RAW 264.7 macrophages. Br J Pharmacol 119:1628–1634
Gendron FP, Benrezzak O, Krugh BW, Kong Q, Weisman GA, Beaudoin AR (2002) Purine signaling and potential new therapeutic approach: possible outcomes of NTPDase inhibition. Curr Drug Targets 3:229–245. https://doi.org/10.2174/1389450023347713
Lee SY, Fiene A, Li W, Hanck T, Brylev KA, Fedorov VE, Lecka J, Haider A, Pietzsch HJ, Zimmermann H, Sévigny J, Kortz U, Stephan H, Müller CE (2015) Polyoxometalates-potent and selective ecto-nucleotidase inhibitors. Biochem Pharmacol 15:171–181. https://doi.org/10.1016/j.bcp.2014.11.002
Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Müller CE, Eltzschig HK (2007) CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation 116:1784–1794. https://doi.org/10.1161/CIRCULATIONAHA.107.690180
Grenz A, Zhang H, Hermes M, Eckle T, Klingel K, Huang DY, Müller CE, Robson SC, Osswald H, Eltzschig HK (2007) Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J 21:2863–2873. https://doi.org/10.1096/fj.06-7947com
Müller CE, Iqbal J, Baqi Y, Zimmermann H, Röllich A, Stephan H (2006) Polyoxometalates-a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorg Med Chem Lett 16:5943–5947. https://doi.org/10.1016/j.bmcl.2006.09.003
Zhai F, Li D, Zhang C, Wang X, Li R (2008) Synthesis and characterization of polyoxometalates loaded starch nanocomplex and its antitumoral activity. Eur J Med Chem 43:1911–1917. https://doi.org/10.1016/j.ejmech.2007.11.032
Seko A, Yamase T, Yamashita K (2009) Polyoxometalates as effective inhibitors for sialyl- and sulfotransferases. J Inorg Biochem 103:1061–1066. https://doi.org/10.1016/j.jinorgbio.2009.05.002
Turner TL, Nguyen VH, McLauchlan CC, Dymon Z, Dorsey BM, Hooker JD, Jones MA (2012) Inhibitory effects of decavanadate on several enzymes and Leishmania tarentolae in vitro. J Inorg Biochem 108:96–104. https://doi.org/10.1016/j.jinorgbio.2011.09.009
Qi Y, Xiang Y, Wang J, Qi Y, Li J, Niu J, Zhong J (2013) Inhibition of hepatitis C virus infection by polyoxometalates. Antivir Res 100:392–398. https://doi.org/10.1016/j.antiviral.2013.08.025
Nomiya K, Torii H, Hasegawa T, Nemoto Y, Nomura K, Hashino K, Uchida M, Kato Y, Shimizu K, Oda M (2001) Insulin mimetic effect of a tungstate cluster. Effect of oral administration of homopolyoxotungstates and vanadium-substituted polyoxotungstates on blood glucose level of STZ mice. J Inorg Biochem 86:657–667. https://doi.org/10.1016/S0162-0134(01)00233-1
Amobi NI, Guillebaud J, Smith IC (2012) Perspective on the role of P2X-purinoceptor activation in human vas deferens contractility. Exp Physiol 97:583–602. https://doi.org/10.1113/expphysiol.2011.063206
Pinheiro AR, Paramos-de-Carvalho D, Certal M, Costa C, Magalhães-Cardoso MT, Ferreirinha F, Costa MA, Correia-de-Sá P (2013) Bradykinin-induced Ca2+ signaling in human subcutaneous fibroblasts involves ATP release via hemichannels leading to P2Y12 receptors activation. Cell Commun Signal 18:70. https://doi.org/10.1186/1478-811X-11-70
Wan P, Liu X, Xiong Y, Ren Y, Chen J, Lu N, Guo Y, Bai A (2016) Extracellular ATP mediates inflammatory responses in colitis via P2X7 receptor signaling. Sci Rep 6:19108. https://doi.org/10.1038/srep19108
Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 100:95–97. https://doi.org/10.1016/0003-2697(79)90115-5
Russo-Abrahão T, Cosentino-Gomes D, Gomes MT, Alviano DS, Alviano CS, Lopes AH, Meyer-Fernandes JR (2011) Biochemical properties of Candida parapsilosis ecto-5′-nucleotidase and the possible role of adenosine in macrophage interaction. FEMS Microbiol Lett 317:34–42. https://doi.org/10.1111/j.1574-6968.2011.02216.x
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Eur. J Physiol 391:85–100
Riccardi C, Nicoletti I (2006) Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc 1:1458–1461. https://doi.org/10.1038/nprot.2006.238
da Silva-Souza HA, Lira MN, Costa-Junior HM, da Cruz CM, Vasconcellos JS, Mendes AN, Pimenta-Reis G, Alvarez CL, Faccioli LH, Serezani CH, Schachter J, Persechini PM (2014) Inhibitors of the 5-lipoxygenase arachidonic acid pathway induce ATP release and ATP-dependent organic cation transport in macrophages. Biochim Biophys Acta 1838:1967–1977. https://doi.org/10.1016/j.bbamem.2014.04.006
Granger DL, Taintor RR, Boockvar KS, Hibbs JB Jr (1996) Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol 268:142–151
von Kügelgen I, Hoffmann K (2016) Pharmacology and structure of P2Y receptors. Neuropharmacology 104:50–61. https://doi.org/10.1016/j.neuropharm.2015.10.030
Jacobson KA, Müller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104:31–49. https://doi.org/10.1016/j.neuropharm.2015.12.001
Costa-Junior HM, Mendes AN, Davis GHNG, Monteiro-da-Cruz C, Ventura AL, Serezani CH, Faccioli LH, Freire-de-Lima CG, Bisaggio RC, Persechini PM (2009) ATP-induced apoptosis involves a Ca2+-independent phospholipase A2 and 5-lipoxygenase in macrophages. Prostaglandins Other Lipid Mediat 88:51–61. https://doi.org/10.1016/j.prostaglandins.2008.09.004
Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, Nagaoka I (2014) Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS One 9:e85765. https://doi.org/10.1371/journal.pone.0085765
Keller H, Rentsch P, Hagmann J (2002) Differences in cortical actin structure and dynamics document that different types of blebs are formed by distinct mechanisms. Exp Cell Res 277:161–172. https://doi.org/10.1006/excr.2002.5552
Verhoef PA, Estacion M, Schilling W, Dubyak GR (2003) P2X7 receptor-dependent blebbing and the activation of Rho-effector kinases, caspases, and IL-1 release. J Immunol 170:5728–5738. https://doi.org/10.4049/jimmunol.170.11.5728
Pan Y, Shen B, Gao Q, Zhu J, Dong J, Zhang L, Zhang Y (2016) Caspase-1 inhibition attenuates activation of BV2 microglia induced by LPS-treated RAW 264.7 macrophages. J Biomed Res 30:225–233. https://doi.org/10.7555/JBR.30.20150141
Sperlagh B, Hasko G, Nemeth Z, Vizi ES (1998) ATP released by LPS increases nitric oxide production in raw 264.7 macrophage cell line via P2Z/P2X7 receptors. Neurochem Int 33:209–215. https://doi.org/10.1016/S0197-0186(98)00025-4
Sakaki H, Tsukimoto M, Harada H, Moriyama Y, Kojima S (2013) Autocrine regulation of macrophage activation via exocytosis of ATP and activation of P2Y11 receptor. PLoS One 8:e59778. https://doi.org/10.1371/journal.pone.0059778
Lévesque SA, Kukulski F, Enjyoji K, Robson SC, Sévigny J (2010) NTPDase1 governs P2X7-dependent functions in murine macrophages. Eur J Immunol 40:1473–1485. https://doi.org/10.1002/eji.200939741
Zanin RF, Braganhol E, Bergamin LS, Campesato LF, Filho AZ, Moreira JC, Morrone FB, Sévigny J, Schetinger MR, de Souza Wyse AT, Battastini AM (2012) Differential macrophage activation alters the expression profile of NTPDase and ecto-5′-nucleotidase. PLoS One 7:e31205. https://doi.org/10.1371/journal.pone.0031205
Wall MJ, Wigmore G, Lopatár J, Frenguelli BG, Dale N (2008) The novel NTPDase inhibitor sodium polyoxotungstate (POM-1) inhibits ATP breakdown but also blocks central synaptic transmission, an action independent of NTPDase inhibition. Neuropharmacology:1251–1258. https://doi.org/10.1016/j.neuropharm.2008.08.005
Van Kolen K, Slegers H (2006) Integration of P2Y receptor-activated signal transduction pathways in G protein-dependent signalling networks. Purinergic Signal 2:451–469. https://doi.org/10.1007/s11302-006-9008-0
Coutinho-Silva R, Ojcius DM, Górecki DC, Persechini PM, Bisaggio RC, Mendes AN, Marks J, Burnstock G, Dunn PM (2005) Multiple P2X and P2Y receptor subtypes in mouse J774, spleen and peritoneal macrophages. Biochem Pharmacol 69:641–655. https://doi.org/10.1016/j.bcp.2004.11.012
da Cruz CM, Ventura AL, Schachter J, Costa-Junior HM, da Silva Souza HA, Gomes FR, Coutinho-Silva R, Ojcius DM, Persechini PM (2006) Activation of ERK1/2 by extracellular nucleotides in macrophages is mediated by multiple P2 receptors independently of P2X7-associated pore or channel formation. Br J Pharmacol 147:324–334. https://doi.org/10.1038/sj.bjp.0706559
Mehta VB, Hart J, Wewers MD (2001) ATP-stimulated release of interleukin (IL)-1 and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem 276:3820–3826. https://doi.org/10.1074/jbc.M006814200
Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunol Rev 243:206–214. https://doi.org/10.1111/j.1600-065X.2011.01044.x
Lamkanfi M, Kalai M, Saelens X, Declercq W, Vandenabeele P (2004) Caspase-1 activates nuclear factor of the kappa-enhancer in B cells independently of its enzymatic activity. J Biol Chem 279:24785–24793. https://doi.org/10.1074/jbc.M400985200
Pan Y, Shen B, Gao Q, Zhu J, Dong J, Zhang L, Zhang Y (2016) Caspase-1 inhibition attenuates activation of BV2 microglia induced by LPS-treated RAW264.7 macrophages. J Biomed Res 30:225–233. https://doi.org/10.7555/JBR.30.20150141
Lee J, Choi J, Kim S (2015) Effective suppression of pro-inflammatory molecules by DHCA via IKK-NF-κB pathway, in vitro and in vivo. Br J Pharmacol 172:3353–3369. https://doi.org/10.1111/bph.13137
Kong Q, Wang M, Liao Z, Camden JM, Yu S, Simonyi A, Sun GY, Gonzalez FA, Erb L, Seye CI, Weisman GA (2005) (2005) P2X7 nucleotide receptors mediate caspase-8/9/3-dependent apoptosis in rat primary cortical neurons. Purinergic Signal 1:337–347. https://doi.org/10.1007/s11302-005-7145-5
Hanley PJ, Kronlage M, Kirschning C, del Rey A, Di Virgilio F, Leipziger J, Chessell IP, Sargin S, Filippov MA, Lindemann O, Mohr S, Königs V, Schillers H, Bähler M, Schwab A (2012) Transient P2X7 receptor activation triggers macrophage death independent of Toll-like receptors 2 and 4, caspase-1, and pannexin-1 proteins. J Biol Chem 287:10650–10663. https://doi.org/10.1074/jbc.M111.332676
Mackenzie AB, Young MT, Adinolfi E, Surprenant A (2005) Pseudoapoptosis induced by brief activation of ATP-gated P2X7 receptors. J Biol Chem 280:33968–33976. https://doi.org/10.1074/jbc.M502705200
Roger S, Pelegrin P, Surprenant A (2008) Facilitation of P2X7 receptor currents and membrane blebbing via constitutive and dynamic calmodulin binding. J Neurosci 28:6393–6401. https://doi.org/10.1523/JNEUROSCI.0696-08.2008
Noronha-Matos JB, Coimbra J, Sá-e-Sousa A, Rocha R, Marinhas J, Freitas R, Guerra-Gomes S, Ferreirinha F, Costa MA, Correia-de-Sá P (2014) P2X7-induced zeiosis promotes osteogenic differentiation and mineralization of postmenopausal bone marrow-derived mesenchymal stem cells. FASEB J 28:5208–5222. https://doi.org/10.1096/fj.14-257923
Pelegrin P, Surprenant A (2009) The P2X(7) receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal 5:129–137. https://doi.org/10.1007/s11302-009-9141-7
Qu Y, Misaghi S, Newton K, Gilmour LL, Louie S, Cupp JE, Dubyak GR, Hackos D, Dixit VM (2011) Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol 186:6553–6561. https://doi.org/10.4049/jimmunol.1100478
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121. https://doi.org/10.1038/nature10558
Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G (2015) Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43:923–932. https://doi.org/10.1016/j.immuni.2015.10.009
Acknowledgements
We thank the National Institute of Science and Technology for Structural Biology and Bioimaging—Microscopy Division (CENABIO-UFRJ) for allowing use of the scanning electron microscope.
Funding
This research received financial support from CNPq, FAPERJ and FINEP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Gabriela Pimenta-dos-Reis declares that she has no conflict of interest.
Eduardo José Lopes Torres declares that he has no conflict of interest.
Paula Gabriela Quintana declares that she has no conflict of interest.
Lincon Onorio Vidal declares that he has no conflict of interest.
Bárbara Andréa Fortes dos Santos declares that she has no conflict of interest.
Chuan-Sheng Lin declares that he has no conflict of interest.
Norton Heise declares that he has no conflict of interest.
Pedro Muanis Persechini declares that he has no conflict of interest.
Julieta Schachter declares that she has no conflict of interest.
Ethical approval
All animals were used according to the guidelines for animal use in scientific experiments of the Carlos Chagas Filho Institute of Biophysics of Federal University of Rio de Janeiro. The protocols were approved by the Research Ethics Committee on Animal Care of the Health Science Center of Federal University of Rio de Janeiro (protocol number 001200.001568/2013-87).
Rights and permissions
About this article

Cite this article
Pimenta-dos-Reis, G., Torres, E.J.L., Quintana, P.G. et al. POM-1 inhibits P2 receptors and exhibits anti-inflammatory effects in macrophages. Purinergic Signalling 13, 611–627 (2017). https://doi.org/10.1007/s11302-017-9588-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-017-9588-x