
Abstract
Peripheral purinergic signaling plays an important role in nociception. Increasing evidence suggests that metabotropic P2Y receptors are also involved, but little is known about the underlying mechanism. Herein, we report that selective P2Y receptor agonist uridine 5′-triphosphate (UTP) can exert an enhancing effect on the functional activity of acid-sensing ion channels (ASICs), key sensors for extracellular protons, in rat dorsal root ganglia (DRG) neurons. First, UTP dose-dependently increased the amplitude of ASIC currents. UTP also shifted the concentration–response curve for proton upwards, with a 56.6 ± 6.4 % increase of the maximal current response to proton. Second, UTP potentiation of proton-gated currents can be mimicked by adenosine 5′-triphosphate (ATP), but not by P2Y1 receptor agonist ADP. Potentiation of UTP was blocked by P2Y receptor antagonist suramin and by inhibition of intracellular G protein, phospholipase C (PLC), protein kinase C (PKC), or protein interacting with C-kinase 1 (PICK1) signaling. Third, UTP altered acidosis-evoked membrane excitability of DRG neurons and caused a significant increase in the amplitude of the depolarization and the number of spikes induced by acid stimuli. Finally, UTP dose-dependently exacerbated nociceptive responses to injection of acetic acid in rats. These results suggest that UTP enhanced ASIC-mediated currents and nociceptive responses, which reveal a novel peripheral mechanism underlying UTP-sensitive P2Y2 receptor involvement in hyperalgesia by sensitizing ASICs in primary sensory neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Following inflammation, tissue injury or ischemia, nucleotides like adenosine 5′-triphosphate (ATP) and uridine 5′-triphosphate (UTP) are released from damaged cells and sensory neurons [1]. These molecules can produce pain and hyperalgesia via ionotropic P2X receptors and metabotropic P2Y receptors located in primary afferent neurons [2]. In sensory ganglia, P2 receptors are also expressed by glial cells and contribute to pain transmission [3]. Although the contribution of P2X receptors to nociception has been understood, increasing evidence suggests that P2Y receptors play an important role in pain [3, 4]. P2Y receptors are composed of eight subtypes, which are found in primary afferent neurons [5]. In particular, P2Y1 and P2Y2 receptors have been shown to be the major P2Y subtypes expressed in small-diameter sensory neurons with thin primary afferent fibers including nociceptors [5, 6]. In the periphery, activation of P2Y2 receptors by UTP sensitizes transient receptor potential vanilloid receptor 1 (TRPV1) and contributes to TRPV1-mediated thermal hypersensitivity [7, 8]. Activation of P2Y2 receptors also potentiates mechanosensitive currents in peptidergic nociceptive dorsal root ganglia (DRG) neurons and sensitizes cutaneous C-fiber nociceptors to mechanical stimuli [9]. In contrast, inhibition of P2Y2 receptors by antisense oligodeoxynucleotides alleviates the mechanical hyperalgesia in trigeminal neuropathic pain [10]. Mice lacking P2Y2 receptors fail to develop thermal hyperalgesia during CFA-evoked inflammation [11]. Thus, P2Y2 receptors play a key role in thermal and mechanical nociception.
Proton is also released under multiple pathological conditions such as inflammation and ischemia [12]. It is known that the local extracellular pH levels drop to 5.4 in acute inflammation and 6.3 or lower in severe ischemia [13, 14]. The released protons can cause pain through activating proton-gated ion channels on peripheral sensory afferents. Studies show that acid-sensing ion channels (ASICs), rather than TRPV1, mediate pain sensation induced by peripheral protons [15, 16]. To date, seven subunits of ASICs encoded by four genes have been identified [17]. All other ASICs, except ASIC4, are expressed in both DRG cell bodies and sensory terminals, where they contribute to pain signaling [18, 19]. Among the ASIC subunits, ASIC3 is specifically localized in nociceptive fibers and considered as the most essential pH sensor for pain [15, 20]. Increasing evidence has shown that ASICs play an important role in various pain conditions such as inflammatory pain, postoperative pain, and migraine [21–23].
Recently, Seo et al. report that acidic pH facilitates ATP-mediated mechanical allodynia via the activation of P2X receptors and ASICs, indicating a crosstalk between P2X receptors and ASICs [24]. Herein, we further report that activation of UTP-sensitive P2Y receptor on nociceptive DRG neurons sensitizes ASICs and contributes to acidosis-evoked pain.
Material and methods
Isolation of DRG neurons
The experimental protocol was approved by the animal research ethics committee of Hubei University of Science and Technology. All procedures conformed to international guidelines on the ethical use of animals, and every effort was made to minimize the number of animals used and their sufferings. Five- to 6-week old Sprague–Dawley male rats were anesthetized with ethyl ether and then decapitated. The DRGs were taken out and transferred immediately into Dulbecco’s modified Eagle’s medium (DMEM, Sigma) at pH 7.4. After the removal of the surrounding connective tissues, the DRGs were minced with fine spring scissors and the ganglion fragments were placed in a flask containing 5 ml of DMEM, in which 0.5 mg/ml of trypsin (type II-S, Sigma), 1.0 mg/ml of collagenase (type I-A, Sigma), and 0.1 mg/ml of DNase (type IV, Sigma) had been dissolved, and incubated at 35 °C in a shaking water bath for 25–30 min. Soybean trypsin inhibitor (type II-S, Sigma) at 1.25 mg/ml was then added to stop trypsin digestion. Dissociated neurons were placed into a 35-mm petri dish and kept for at least another 60 min before electrophysiological recordings. The neurons selected for electrophysiological experiment were 15–35 μm in diameter.
Electrophysiological recordings
Whole-cell patch clamp and voltage clamp recordings were carried out at room temperature (22–25 °C) using a MultiClamp 700B amplifier and Digidata 1440A A/D converter (Axon Instruments, CA, USA). Recording pipettes were pulled using a Sutter P-97 puller (Sutter Instruments, CA, USA). The micropipettes were filled with internal solution containing the following (in mM): KCl 140, MgCl2 2.5, HEPES 10, EGTA 11, and ATP 5; its pH was adjusted to 7.2 with KOH and osmolarity was adjusted to 310 mOsm/L with sucrose. Cells were bathed in an external solution containing the following (in mM): NaCl 150, KCl 5, CaCl2 2.5, MgCl2 2, HEPES 10, and d-glucose 10; its osmolarity was adjusted to 330 mOsm/L with sucrose and its pH to 7.4. The resistance of the recording pipette was in the range of 3–6 MΩ. A small patch of membrane underneath the tip of the pipette was aspirated to form a gigaseal and then a negative pressure was applied to rupture it, thus establishing a whole-cell configuration. The adjustment of capacitance compensation and series resistance compensation was done before recording the membrane currents. The membrane voltage was maintained at −60 mV in all voltage clamp experiments unless otherwise specified. Current clamp recordings were obtained by switching to current clamp mode after a stable whole-cell configuration was formed in voltage clamp mode. Only cells with a stable resting membrane potential (more negative than −50 mV) were used in the study. Signals were sampled at 10 to 50 kHz and filtered at 2 to 10 kHz, and the data were stored in a compatible PC computer for offline–online analysis using the pCLAMP 10 acquisition software (Axon Instruments, CA, USA).
Drug application
Drugs were purchased from Sigma and used in the experiments, including the following: hydrochloric acid, UTP, ATP, ADP, suramin, MRS2179, capsazepine, and tetrodotoxin (TTX). All drugs were dissolved daily in the external solution just before use and were held in a linear array of fused silica tubes (o.d./i.d. = 500 μm/200 μm) connected to a series of independent reservoirs. The application pipette tips were positioned ∼30 μm away from the recorded neurons. The application of each drug was driven by gravity and controlled by the corresponding valve, and rapid solution exchange could be achieved within about 100 ms by shifting the tubes horizontally with a PC-controlled micromanipulator. Cells were constantly bathed in normal external solution flowing from one tube connected to a larger reservoir between drug applications. In some experiments where guanosine 5′[beta-thio]diphosphate (GDP-β-S (Sigma), U-73122(Sigma), GF109203X (RBI), and FSC-231 (Gether Lab, University of Copenhagen, DK-2200 Copenhagen, Denmark) were applied for intracellular dialysis, they were dissolved in the internal solution before use. To functionally characterize ASIC activity, we used capsazepine (10 μM) to block TRPV1 in this study [25].
Nociceptive behavior induced by acetic acid in rats
Rats were placed in a 30 × 30 × 30 cm Plexiglas chamber and were allowed to habituate for at least 30 min before nociceptive behavior experiments. Separate groups of rats were pretreated with 20 μl capsazepine (100 μM) together with vehicle, different dosages of UTP, and MRS2179 in the ipsilateral hind paw before injection of acetic acid. After 5 min, the other experimenters subcutaneously administered acetic acid solution (0.6 %, 20 μl) into the dorsal face of the hind paw using a 30-gauge needle connected to a 100-μL Hamilton syringe. Nociceptive behavior (that is, number of flinches) was counted over a 5-min period starting immediately after the injection [15, 26].
Data analysis
Data were statistically compared using Student’s t test or analysis of variance (ANOVA), followed by Bonferroni post hoc test. Statistical analysis of concentration–response data was performed using nonlinear curve-fitting program ALLFIT. Data are expressed as mean ± SEM.
Results
UTP enhanced ASIC currents in rat dorsal root ganglia neurons
Whole cell recordings were conducted in small and medium diameters (15 to 35 μm) acutely isolated DRG neurons of adult rats in this study. To functionally characterize ASIC currents, we measured proton-gated currents in the presence of capsazepine (10 μM) to block proton-induced TRPV1 activation in the whole-cell patch clamp configuration [25]. ASIC currents were induced by extracellular protons. We found that a rapid reduction of extracellular pH from 7.4 to 6.0 for 5 s evoked an inward current (IpH6.0) in most native DRG neurons (85.3 %, 145/170). Given a 3-min washout period to allow full recovery, the cells were able to respond to the same degree as in the initial application of acid stimuli. As shown in Fig. 1a, most (70.3 %, 102/145) of these proton-evoked currents can be characterized by a large transient current followed by fast inactivation and then a small sustained current with no or very slow inactivation [27]. Our previous and current studies show that these characterized currents could be completely blocked by broad-spectrum ASIC channel blocker amiloride, also by ASIC3 channels blocker APETx2 (Fig. 1a) [28]. These currents were thus considered to be ASIC3-like currents and were mainly investigated in this study.

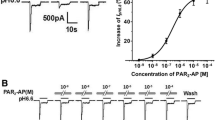
UTP enhanced ASIC currents in rat DRG neurons. a Representative current traces were evoked by extracellular application of a pH 6.0 solution for 5 s in the presence of capsazepine (10 μM) to block proton-induced TRPV1 activation. The proton-induced current could be completely blocked by100 μM of amiloride (Amil), a broad-spectrum ASIC channel blocker. It was also blocked by 3 μM of APETx2, an ASIC3 blocker. b The sequential current traces illustrate the potentiation of proton-gated currents by different concentrations of UTP (10−9–10−4 M) and recovery after washout of UTP. Representative currents were recorded for more than 80 min in a DRG neuron tested. UTP was pre-applied to external solution for 1 min. c The graph shows that UTP increased the peak amplitude of proton-gated currents in a dose-dependent manner with an EC50 of 1.2 × 10−6 M. UTP was re-treated for 1 min. DRG neurons with membrane potential clamped at −60 mV. Each point represents the mean ± SEM of eight to ten neurons
In the majority of neurons sensitive to acid stimuli (76.5 %, 78/102), we observed that the proton-evoked currents were enhanced by the pre-application of the selective P2Y agonist UTP (10−5 M) for 1 min. The ASIC currents recovered to initial response after a washout of UTP. Figure 1b shows that the amplitude of IpH6.0 increased as concentration of re-treated UTP increased from 10−9 to 10−4 M in a representative DRG neuron. Figure 1c shows the dose–response curve for UTP in the potentiation of proton-gated currents. UTP had a maximum effect (64.7 ± 7.8 %, n = 9) at a concentration of 10−4 M. The half-maximal response (EC50) value of the dose–response curve for UTP was 1.2 ± 0.3 μM. The results indicated that UTP dose-dependently enhanced the ASIC current in rat DRG neurons.
We then investigated whether the potentiation of UTP was dependent upon pH. Figure 2a shows the enhancing effects of UTP (10−5 M) pretreatment for 1 min on currents evoked by three different pHs. Figure 2b shows the concentration–response curve to protons in the presence and absence of UTP (10−5 M). First, pretreatment of UTP shifted the concentration–response curve to protons upwards, as indicated by an increase of 56.6 ± 6.4 % in the maximal current response to protons when UTP was pre-applied. However, the slopes or Hill coefficients of those two curves were essentially similar (n = 1.39 ± 0.20 in the absence of UTP vs n = 1.31 ± 0.17 in the presence of UTP; P > 0.1, Bonferroni post hoc test). Second, the pH0.5 values of both curves had no statistical difference (pH0.5 of 6.09 ± 0.05 without UTP pretreatment vs pH0.5 of 6.13 ± 0.05 with UTP pretreatment; P > 0.1, Bonferroni post hoc test). Third, the threshold pH values of both curves had no significant difference in the presence and absence of UTP.

Concentration–response relationship for protons with or without the pre-application of UTP. a Sequential currents evoked by different pHs in the absence or presence of UTP (10−5 M for 1 min). b The concentration–response curves for protons with or without UTP pre-application. Each point represents the mean ± SEM of eight to ten neurons. All current values were normalized to the current response induced by pH 4.5 applied alone (marked with asterisk). The curves shown are a best fit of the data to the logistic equation I = Imax / [1 + (pH0.5/C)n], where C is the concentration of protons, I is the normalized current response value, pH0.5 is the proton concentration that produced half the maximal current response to protons, and n is the Hill coefficient. The curves for protons without and with UTP pre-application were drawn according to the equation described above
Intracellular mechanisms underling the potentiation of proton-gated currents by UTP
Metabotropic P2Y receptors belong to G-protein-couple receptors and are expressed in DRG neurons [5, 29]. To further explore intracellular signal transduction mechanisms underlying potentiation of ASIC currents by P2Y agonist UTP, we performed the same experiment as described above while intracellularly injecting the some signal blockers. Firstly, recordings were performed with 500 μM of nonhydrolyzable GDP analog GDP-β-S in the solution of the recording electrode, the enhancing effect of ASIC currents by 10−5 M UTP-induced P2Y receptor activation was significantly attenuated (UTP 58.7 ± 5.5 %, GDP-β-S + UTP 9.8 ± 6.2 %; P < 0.01, Bonferroni post hoc test, n = 9; Fig. 3a (A1, A2), b). Secondly, intracellular application of 10 μM phospholipase C (PLC) blocker U73122 also significantly blocked UTP potentiation of ASIC currents (U73122 + UTP: 13.6 ± 4.6 %; P < 0.01, n = 9; Fig. 3a (A3), b). Thirdly, pretreatment of 10−5 M UTP for 1 min only caused an increase of 13.2 ± 6.0 % on ASIC currents after 2 μM of protein kinase C (PKC) inhibitor GF109203X were applied internally to DRG neurons (P < 0.01, n = 9; Fig. 3a (A4), b). Finally, UTP potentiation of ASIC currents was also significantly attenuated by intracellular injection of 50 μM protein interacting with C-kinase 1 (PICK1) inhibitor FSC-231 (FSC-231 + UTP 9.2 ± 9.8 %; P < 0.01, n = 9; Fig. 3a (A5), b). Intracellular dialysis of these blockers did not significantly alter the amplitude of IpH6.0 (data not shown). These data collectively indicated that involvement of GPCR, PLC, PKC, and PICK1 signal pathways in the intracellular mechanisms of UTP potentiation of ASIC currents in rat DRG neurons.

Intracellular mechanisms underlying the potentiation of ASIC currents by UTP. a The current traces in A1 show that UTP enhanced proton-gated currents with recording pipette filled with the normal internal solution in a DRG neuron tested. The current traces in A2–5 show that UTP potentiation of proton-gated currents was blocked by intracellular dialysis of nonhydrolyzable GDP analog GDP-β-S (500 μM), PLC inhibitor U-73122 (10 μM), PKC inhibitor GF109203X (2 μM), or PICK1 (protein interacting with C-kinase 1) inhibitor FSC-231 (50 μM) by the recording pipettes. Proton-gated currents were elicited by application of pH 6.0 for 5-s durations. UTP (10−5 M) was pre-applied to external solution for 1 min. b The bar graph shows the percentage increases in the IpH6.0 induced by UTP (10−5 M) with recording pipettes filled with the normal internal solution, GDP-β-S, U-73122, GF109203X, or FSC-231 containing internal solution. **P < 0.01, post hoc Bonferroni test, compared with normal internal solution. n = 9 in each column
P2Y2 receptor subtypes were involved in UTP potentiation of ASIC currents
The main candidates for Gq-coupled P2Y receptors expressed in DRG neurons are P2Y1 and P2Y2 [5, 6]. Although the contribution of P2Y1 is less likely because it is not sensitive to UTP, experiments were conducted with the other P2Y agonists and selective antagonists to further clarify which of the P2Y receptor subtypes is/are involved. Firstly, we compared the effects of ATP, ADP, or UTP on IpH6.0. As shown in Fig. 4a, b, after 10−5 M of ATP or ADP was pre-applied to DRG neurons instead of UTP, ATP caused an enhancing effect equivalent to those observed for UTP (ATP 65.1 ± 10.3 %; UTP 62.8 ± 6.7 %), whereas ADP had no effect on IpH6.0 (4.6 ± 2.7 %). When 10−5 M of ATP and UTP were applied together, IpH6.0 increased at 63.8 ± 8.7 %. These results suggested that UTP potentiation of IpH6.0 can be mimicked by ATP, but not by ADP, an agonist for P2Y1 receptor subtype. We then observed the effect of P2Y receptor antagonists on UTP-induced potentiation of ASIC currents. Suramin has been shown to block all nucleotide-sensitive P2Y receptors with the exception of the P2Y4 receptor subtype [30]. We found that 100 μM suramin significantly blocked the potentiation of UTP (Fig. 4c). The amplitude of IpH6.0 increased at 57.4 ± 5.5 % after pretreatment with UTP (10−5 M) alone for 1 min in nine DRG neurons. In contrast, UTP produced only an increase of 9.6 ± 4.1 % on ASIC currents in 100 μM suramin pretreated nine neurons (P < 0.05, Bonferroni post hoc test, n = 8; Fig. 4d). Thus, we ruled out the involvement of P2Y4 receptor subtype in UTP potentiation of ASIC currents. In addition, ATP potentiation of IpH6.0 was also blocked by suramin (100 μM), and ATP increased IpH6.0 from 65.1 ± 10.3 to 8.7 ± 5.2 % in the absence and presence of suramin (P < 0.05, Bonferroni post hoc test, n = 8). MRS2179, a P2Y1-selective antagonist, was also pre-applied to DRG neurons. In the presence and absence of 10−4 M of MRS2179, application of 10−5 M UTP for 1 min produced a similar enhancing effect on ASIC currents in DRG neurons recorded (UTP 57.4 ± 5.5 %, MRS2179 + UTP 62.1 ± 14.1 %; P > 0.1, n = 9; Fig. 4c, d). These results indicated that the UTP enhancement of ASIC currents did not involve P2Y1 receptor subtype. Thus, potentiation of ASIC currents by UTP may be mediated by activation of the metabotropic P2Y2 receptor subtype.

P2Y2 receptor subtypes were involved in UTP potentiation of ASIC currents. The current traces in a and bar graphs in b show that IpH6.0 was enhanced by 10−5 M of UTP or ATP, but not by ADP, an agonist for P2Y1 receptor subtype. The current traces in c and bar graphs in d show that UTP potentiation of ASIC currents was blocked by P2Y receptor antagonist suramin (100 μM), but not by P2Y1 selective antagonist MRS2179 (10−4 M). Statistical tests were performed using Bonferroni post hoc test, and significance is shown as follows: **P < 0.01, compared with normal white column; ##P < 0.01, compared with UTP alone column; n = 9 in each column
UTP increased proton-induced membrane excitability of rat DRG neurons
Activation of ASICs by protons causes an inward current, which depolarizes the resting membrane potential to the threshold and initiates action potentials (APs) [31]. Further experiments were performed to record rat DRG neuron excitability under a current clamp model in the presence of capsazepine (10 μM) to block proton-induced TRPV1 activation. As shown in Fig. 5a, a pH drop from 7.4 to 6.0 for 5 s evoked a whole-cell inward current with voltage clamp recording in a DRG neuron, which could also trigger bursts of APs (spikes) under current clamp conditions in the cell tested. Similar to UTP potentiation of proton-gated currents under voltage clamp conditions, pretreatment of 10−5 M UTP for 1 min also increased acidosis-evoked spikes (Fig. 5a). In the nine DRG neurons tested, pretreatment of UTP increased the mean number of spikes induced by acidosis from 6.3 ± 0.7 of control condition to 10.5 ± 1.3 (P < 0.05, paired t test, n = 9; Fig. 5b). After a washout of UTP, the acidosis-evoked spikes recovered to the control condition.

UTP increased proton-evoked membrane excitability of rat DRG neurons. a Original current and spike recordings from the same DRG neuron. Left panel: In the presence of the TRPV1 inhibitor capsazepine (10 μM), a pH 6.0 acid stimulus induced an inward current with voltage clamp recording. Right panel: The pH 6.0 acid stimulus produced cell spikes with current clamp recording in the same neuron. The administration with UTP (10−5 M) for 1 min increased the acidosis-induced spiking activity. c Original current and membrane potential recordings from the same DRG neuron. Left panel: Voltage clamp recording of current induced by a pH 6.0 acid stimulus. Holding potential was −60 mV. Right panel: Current clamp recording (I = 0 pA) of the membrane depolarization evoked by the pH 6.0 acid stimulus from the same neuron as the left panel. The treatment of UTP (10−5 M) for 1 min also increased the acidosis-induced membrane depolarization. No action potential was triggered by the membrane depolarization in the neuron tested in the presence of capsazepine (10 μM) and TTX (1 μM) to block proton-induced TRPV1 activation and Na+ channel-mediated action potentials, respectively. b, d Bar graphs show the effects of UTP on the number of spikes and membrane potential depolarization produced by pH 6.0. The acidosis-evoked depolarization and spikes recovered to control condition after washout of UTP. *P < 0.05, paired t test, compared with pH alone, n = 9 in each column
We then observed the membrane potential of rat DRG neurons in the presence of capsazepine (10 μM) and TTX (1 μM) to block proton-induced TRPV1 activation and Na + channel-mediated action potentials, respectively. As shown in Fig. 5c, a pH 6.0 acid stimulus induced an inward current with voltage clamp recording and a depolarization of the resting membrane potential under current clamp recording (I = 0 pA) conditions in a DRG neuron. In this particular cell, pretreatment of 10−5 M UTP increased the depolarization evoked by the acidic stimuli (Fig. 5c). In the nine DRG neurons treated with 10−5 M UTP, the magnitude of membrane potential depolarization (ΔVm) induced by acidosis exposure increased from 18.5 ± 1.9 mV of control condition to 24.2 ± 1.5 mV (paired t test, P < 0.05, n = 9; Fig. 5d). The ΔVm recovered to control condition after a washout of UTP. In addition, UTP alone did not cause depolarization and spikes. These results indicated that UTP reversibly increased proton-induced membrane excitability of rat DRG neurons.
UTP exacerbated acid-induced nocifensive behaviors in rats
The above experiments provided electrophysiological evidence that P2Y agonist UTP potentiated ASIC activity in vitro, so we further ascertain whether UTP facilitated ASIC-mediated pain in vivo. It has been shown that intraplantar injection of acetic acid elicits an intense flinch/shaking response mediated by ASICs [15, 26]. As shown in Fig. 6, intraplantar injection of acetic acid solution (0.6 %, 20 μL) in the presence of the TRPV1 inhibitor capsazepine (100 μM) caused an intense flinch/shaking response in rats, and pre-administration of UTP in the ipsilateral hind paw dose-dependently exacerbated the acid-induced nocifensive behaviors. The acetic acid-induced flinch response was significantly greater in rats treated with medium and high doses (1 and 10 μg) of UTP than that observed in rats injected with acetic acid alone (Bonferroni post hoc test, p < 0.05 and p < 0.01, n = 10). However, the low dose (0.01 and 0.1 μg) of UTP had no effect on the acid-induced nocifensive behaviors (Bonferroni post hoc test, p > 0.05, n = 10). In addition, the UTP-mediated exacerbating effect on acid-induced nocifensive behaviors was significantly blocked by co-treatment of 20 μg P2Y antagonist suramin, but not by co-treatment of 10 μg P2Y1 antagonist MRS2179 (Fig. 6). These results indicated that peripheral P2Y agonist UTP contributed to acid-induced nocifensive behaviors in rats.

UTP exacerbated nociceptive responses to intraplantar injection of acetic acid in rats. Intraplantar injection of acetic acid evoked a flinch/shaking response in the presence of 100 μM capsazepine. The pretreatment of UTP dose-dependently (0.01–10 μg) increased flinching behavior induced by acetic acid. The effect of UTP (10 μg) was blocked by co-treatment of 20 μg P2Y antagonist suramin, but not by co-treatment of 10 μg P2Y1 antagonist MRS2179. Flinching shaking of paw was recorded as the number of flinches during the first 5 min after injection of acetic acid. *P < 0.05, **P < 0.01, Bonferroni post hoc test, compared with vehicle white column; ##P < 0.01, n = 10 rats in each column
Discussion
The current study demonstrated that P2Y agonist UTP can exert an enhancing effect on the functional activity of ASICs. UTP increased the amplitude of ASIC currents and acidosis-evoked membrane excitability in dissociated rat DRG neurons. Potentiation of ASIC currents by UTP was mediated via PLC, PKC, and PICK1 signal pathways following activation of G-protein-coupled P2Y receptors. Peripherally, administration of UTP dose-dependently exacerbated nociceptive responses to intraplantar injection of acetic acid in rats.
Extracellular protons have been found to generate currents in a subset of primary afferents since 1980 [32]. The current study showed that a rapid drop in the extracellular pH from 7.4 to 6.0 produced an inward current in native DRG neurons. These acid currents are ASIC currents in the presence of capsazepine to block the activation of TRPV1 channels, since they could be completely blocked by broad-spectrum ASIC channel blocker amiloride. Most of these acid currents can be characterized by a large transient current followed by fast inactivation and then a small sustained current with no or very slow inactivation [27]. Our previous and current studies have identified that the characterized acidosis-evoked currents are mainly mediated by ASIC3, since they could be completely blocked by ASIC3 channels blocker APETx2 [28]. We thus considered them to be ASIC3-like currents, although precise ASIC subunits need to be identified. It has been shown that all seven ASIC subunits besides ASIC4 are present in DRG neurons [18, 19]. Moreover, ASIC3 have emerged as critical pH sensors predominantly expressed in small-diameter DRG neurons and nociceptors [15, 20].
The current study demonstrated that UTP, a selective P2Y agonist, enhanced the ASIC currents in rat DRG neurons in a dose-dependent manner. UTP sensitized ASICs by shifting the proton concentration–response curve upward and increasing the maximum response of ASICs to protons. The present study also showed that blockade of G-protein signaling with intracellular dialysis of GDP-β-S completely prevented UTP-mediated potentiation of ASIC currents, indicating that G-proteins were involved in the intracellular mechanisms of this potentiation. Metabotropic P2Y receptors belong to the G-protein-coupled receptor family, and almost all P2Y receptor subtypes are expressed in DRG [5, 29]. However, the levels of expression of P2Y family members vary widely: P2Y2∼P2Y1 >> P2Y6 >> P2Y4 [11]. The main candidates for Gq-coupled P2Y receptors expressed in DRG neurons are P2Y1 and P2Y2 receptors [5, 6]. UTP potentiation of ASIC currents can be mimicked by ATP, but not by ADP, an agonist for P2Y1 receptor subtype. P2Y1-selective antagonist MRS2179 did not alter the potentiation of UTP. Thus, P2Y1 receptor subtype was not involved in UTP potentiation of ASIC currents. UTP is an agonist for P2Y2 and P2Y4 receptors [29]. P2Y4, a Gq-coupled UTP receptor, is expressed in sensory ganglia [33, 34], but functional analysis shows low levels of P2Y4 receptor expression in DRG neurons [8, 35]. Suramin has been shown to block all nucleotide-sensitive P2Y receptors with the exception of the P2Y4 receptor subtype [30]. UTP potentiation of ASIC currents was blocked by suramin, indicating that P2Y4 receptor subtype was also not involved in UTP potentiation of ASIC currents. In DRG neurons, P2Y receptor involved in the potentiation of TRPV1 and mechanosensitive currents by UTP is reported to be P2Y2 receptor subtype [7, 8]. UTP enhancement of ASIC currents was most likely mediated by activation of the metabotropic P2Y2 receptor subtype.
P2Y2 receptors are coupled to Gq proteins and can thus in principal activate PLC and PKC pathways [36]. Lack of the enhancing effect in cells treated with PLC inhibitor U-73122 indicating a PLC-dependent pathway was involved in the ASIC sensitization by activation of UTP-sensitive P2Y receptors. It has been shown that inhibition of P2X3 currents by activation of UTP-sensitive P2Y2 receptors also requires PLC activation [37]. Further, we found that UTP potentiation of ASIC currents was blocked by intracellular dialysis of PKC inhibitor GF109203X. The observation suggested that activation of PLC and PKC signaling pathways by P2Y agonist UTP contributed to sensitization of ASICs in DRG neurons. Similarly, sensitization of TRPV1 by activation of P2Y2 receptors also involves PLC and PKC signaling pathways [7, 8]. Our previous studies show that PKC activator PMA produces a similar enhancing effect on ASIC currents in DRG neurons [38]. PKC pathway stimulation has been reported to induce an increase of ASIC3-like currents recorded from cultured rat DRG neurons [39]. Morever, this regulation by PKC requires PICK-1, a protein interacting with C kinase [39]. The current study showed that potentiation of ASIC currents by UTP may depend upon an interaction with PICK1, since the potentiation was also reversed by intracellular dialysis of FSC-231, a small molecule inhibitor of PICK1. However, ASIC3 does not seem to interact with PICK1, which was essential for the potentiation of ASIC3-like currents by UTP. One possible explanation was that the present ASIC3-like currents were mediated by ASIC3/2 heteromers, but not by ASIC3 homomers, since heteromeric ASIC3/2b channels, but not homomeric ASIC3 channels, are positively regulated by PKC via the partner protein PICK-1 [39].
Activation of ASICs by extracellar protons mainly induces sodium influx, resulting in membrane depolarization and bursts of action potentials [31]. The current clamp experiments showed that UTP increased the amplitude of the depolarization and the number of action potentials induced by extracellular acid stimuli. The increased acid-evoked neuronal excitability appeared to correlate with UTP potentiation of ASIC currents in voltage clamp experiments. ATP and UTP have been found to control the excitability of sensory neurons via P2Y1 and P2Y2 receptors [8]. In many cases, acutely isolated sensory neurons are good models to study the physiological events that occur at the sensory ending of the skin. Behavioral studies showed that UTP dose-dependently exacerbated acid-induced nocifensive behaviors in rats. Obviously, the electrophysiological data in vitro corroborated the behavioral studies in vivo and vice versa. The current results strongly indicated that P2Y agonist UTP could potentiate ASIC currents via an intracellular cascade and then increase the number of action potentials in primary sensory neurons, resulting in amplification of proton-induced pain.
In general, inflammation, tissue injury, ischemia, and even intense exercises are closely associated with both an increase in extracellular ATP and an elevated proton concentration (i.e., a decrease in tissue pH) [40–43]. ATP may cause pain and hyperalgesia not only by the activation of ionotropic P2X receptors, but also via metabotropic P2Y receptors located in primary afferent neurons (Lazarowski et al., 2003). The released protons also elicit pain and hyperalgesia through activating ASICs on peripheral sensory afferents. Once both ATP and protons are released together, a functional crosstalk may occur between P2 purinergic receptors and ASICs. It has been shown that activation of ASICs by protons facilitates ATP-induced mechanical allodynia mediated by P2X receptors [24]. P2Y2 receptors confer responsiveness to UTP and ATP to a similar extent. Moreover, UTP has been reported to be released from ruptured cells [44, 45]. The current results demonstrated that the activation of P2Y receptors by released ATP or UTP could sensitize ASICs, which can further exacerbate proton-evoked pain.
Taken together, our results indicated that P2Y receptor activation of nociceptive DRG neurons by UTP enhanced ASICs function and contributed to acidosis-evoked pain, which revealed a novel peripheral mechanism underlying involvement in P2Y receptor-mediated hyperalgesia by sensitizing ASICs in primary sensory neurons.
Abbreviations
- ASIC:
-
Acid-sensing ion channels
- ATP:
-
Adenosine 5′-triphosphate
- DRG:
-
Dorsal root ganglion
- IpH :
-
Proton-gated current
- PICK1:
-
Protein interacting with C-kinase 1
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- TRPV1:
-
Transient receptor potential vanilloid receptor-1
- UTP:
-
Uridine 5′-triphosphate
References
Cook SP, McCleskey EW (2002) Cell damage excites nociceptors through release of cytosolic ATP. Pain 95:41–47
Lazarowski ER, Boucher RC, Harden TK (2003) Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol Pharmacol 64:785–795
Gerevich Z, Illes P (2004) P2Y receptors and pain transmission. Purinergic Signal 1:3–10
Magni G, Ceruti S (2013) P2Y purinergic receptors: new targets for analgesic and antimigraine drugs. Biochem Pharmacol 85:466–477
Kobayashi K, Fukuoka T, Yamanaka H, Dai Y, Obata K, Tokunaga A et al (2006) Neurons and glial cells differentially express P2Y receptor mRNAs in the rat dorsal root ganglion and spinal cord. J Comp Neurol 498:443–454
Molliver DC, Cook SP, Carlsten JA, Wright DE, McCleskey EW (2002) ATP and UTP excite sensory neurons and induce CREB phosphorylation through the metabotropic receptor, P2Y2. Eur J Neurosci 16:1850–1860
Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H et al (2003) Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosc: Off J Soc Neurosci 23:6058–6062
Yousuf A, Klinger F, Schicker K, Boehm S (2011) Nucleotides control the excitability of sensory neurons via two P2Y receptors and a bifurcated signaling cascade. Pain 152:1899–1908
Lechner SG, Lewin GR (2009) Peripheral sensitisation of nociceptors via G-protein-dependent potentiation of mechanotransduction currents. J Physiol 587:3493–3503
Li N, Lu ZY, Yu LH, Burnstock G, Deng XM, Ma B (2014) Inhibition of G protein-coupled P2Y2 receptor induced analgesia in a rat model of trigeminal neuropathic pain. Mol Pain 10:21
Malin SA, Davis BM, Koerber HR, Reynolds IJ, Albers KM, Molliver DC (2008) Thermal nociception and TRPV1 function are attenuated in mice lacking the nucleotide receptor P2Y2. Pain 138:484–496
Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139:267–284
Kweon HJ, Suh BC (2013) Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep 46:295–304
Steen KH, Reeh PW, Anton F, Handwerker HO (1992) Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci: Off J Soc Neurosci 12:86–95
Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V et al (2008) ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J 27:3047–3055
Krishtal O (2003) The ASICs: signaling molecules? Modulators? Trends Neurosci 26:477–483
Deval E, Lingueglia E (2015) Acid-sensing ion channels and nociception in the peripheral and central nervous systems. Neuropharmacology 94:49–57
Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM (2002) Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A 99:2326–2331
Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ et al (2002) Heteromultimers of DEG/ENaC subunits form H + −gated channels in mouse sensory neurons. Proc Natl Acad Sci U S A 99:2338–2343
Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE et al (2001) The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32:1071–1083
Deval E, Noel J, Gasull X, Delaunay A, Alloui A, Friend V et al (2011) Acid-sensing ion channels in postoperative pain. J Neurosci: Off J Soc Neurosci 31:6059–6066
Dussor G (2015) ASICs as therapeutic targets for migraine. Neuropharmacology 94:64–71
Wemmie JA, Taugher RJ, Kreple CJ (2013) Acid-sensing ion channels in pain and disease. Nat Rev Neurosci 14:461–471
Seo HS, Roh DH, Kwon SG, Yoon SY, Kang SY, Moon JY et al (2011) Acidic pH facilitates peripheral alphabetameATP-mediated nociception in rats: differential roles of P2X, P2Y, ASIC and TRPV1 receptors in ATP-induced mechanical allodynia and thermal hyperalgesia. Neuropharmacology 60:580–586
Poirot O, Berta T, Decosterd I, Kellenberger S (2006) Distinct ASIC currents are expressed in rat putative nociceptors and are modulated by nerve injury. J Physiol 576:215–234
Omori M, Yokoyama M, Matsuoka Y, Kobayashi H, Mizobuchi S, Itano Y et al (2008) Effects of selective spinal nerve ligation on acetic acid-induced nociceptive responses and ASIC3 immunoreactivity in the rat dorsal root ganglion. Brain Res 1219:26–31
Wang X, Li WG, Yu Y, Xiao X, Cheng J, Zeng WZ et al (2013) Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J Neurosci: Off J Soc Neurosci 33:4265–4279
Qiu F, Qiu CY, Cai H, Liu TT, Qu ZW, Yang Z et al (2014) Oxytocin inhibits the activity of acid-sensing ion channels through the vasopressin, V1A receptor in primary sensory neurons. Br J Pharmacol 171:3065–3076
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C et al (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
von Kugelgen I (2006) Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther 110:415–432
Kellenberger S, Schild L (2015) International union of basic and clinical pharmacology. XCI. Structure, function, and pharmacology of acid-sensing ion channels and the epithelial Na + channel. Pharmacol Rev 67:1–35
Krishtal OA, Pidoplichko VI (1980) A receptor for protons in the nerve cell membrane. Neuroscience 5:2325–2327
Ruan HZ, Birder LA, de Groat WC, Tai C, Roppolo J, Buffington CA et al (2005) Localization of P2X and P2Y receptors in dorsal root ganglia of the cat. J Histochem Cytochem: Off J Histochem Soc 53:1273–1282
Ruan HZ, Burnstock G (2003) Localisation of P2Y1 and P2Y4 receptors in dorsal root, nodose and trigeminal ganglia of the rat. Histochem Cell Biol 120:415–426
Sanada M, Yasuda H, Omatsu-Kanbe M, Sango K, Isono T, Matsuura H et al (2002) Increase in intracellular Ca(2+) and calcitonin gene-related peptide release through metabotropic P2Y receptors in rat dorsal root ganglion neurons. Neuroscience 111:413–422
von Kugelgen I, Wetter A (2000) Molecular pharmacology of P2Y-receptors. Naunyn Schmiedeberg’s Arch Pharmacol 362:310–323
Mo G, Peleshok JC, Cao CQ, Ribeiro-da-Silva A, Seguela P (2013) Control of P2X3 channel function by metabotropic P2Y2 utp receptors in primary sensory neurons. Mol Pharmacol 83:640–647
Qiu CY, Liu YQ, Qiu F, Wu J, Zhou QY, Hu WP (2012) Prokineticin 2 potentiates acid-sensing ion channel activity in rat dorsal root ganglion neurons. J Neuroinflammation 9:108
Deval E, Salinas M, Baron A, Lingueglia E, Lazdunski M (2004) ASIC2b-dependent regulation of ASIC3, an essential acid-sensing ion channel subunit in sensory neurons via the partner protein PICK-1. J Biol Chem 279:19531–19539
Issberner U, Reeh PW, Steen KH (1996) Pain due to tissue acidosis: a mechanism for inflammatory and ischemic myalgia? Neurosci Lett 208:191–194
Sutherland SP, Cook SP, McCleskey EW (2000) Chemical mediators of pain due to tissue damage and ischemia. Prog Brain Res 129:21–38
Hagberg H (1985) Intracellular pH during ischemia in skeletal muscle: relationship to membrane potential, extracellular pH, tissue lactic acid and ATP. Pflugers Arch - Eur J Physiol 404:342–347
Sinoway LI, Li J (2005) A perspective on the muscle reflex: implications for congestive heart failure. J Appl Physiol 99:5–22
Anderson CM, Parkinson FE (1997) Potential signalling roles for UTP and UDP: sources, regulation and release of uracil nucleotides. Trends Pharmacol Sci 18:387–392
Lazarowski ER, Boucher RC, Harden TK (2000) Constitutive release of ATP and evidence for major contribution of ecto-nucleotide pyrophosphatase and nucleoside diphosphokinase to extracellular nucleotide concentrations. J Biol Chem 275:31061–31068
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 31471062 and 81171039) and the Natural Science Foundation of Hubei Province of China (No. 2015CFA145).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The author(s) declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Ren, C., Gan, X., Wu, J. et al. Enhancement of acid-sensing ion channel activity by metabotropic P2Y UTP receptors in primary sensory neurons. Purinergic Signalling 12, 69–78 (2016). https://doi.org/10.1007/s11302-015-9479-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-015-9479-y