
Abstract
Fragmentation acting over geological times confers wide, biogeographical scale and genetic diversity patterns to species, through demographic and natural selection processes. To test the effects of historical fragmentation on the genetic diversity and differentiation of a widespread forest tree, Pinus nigra Arnold, the European black pine, and to resolve its demographic history, we described and modelled its spatial genetic structure and gene genealogy. We then tested which Pleistocene event, whether recent or ancient, could explain its widespread but patchy geographic distribution. We used a set of different genetic markers, both neutral and potentially adaptive, and either bi-parentally or paternally only inherited, and we sampled natural populations across the entire species range. We analysed the data using both frequentist population genetic and Bayesian inference methods to calibrate realistic, demographic timed scenarios. We also considered how habitat suitability might have affected demography by correlating climate variables at different recent Pleistocene ages with genetic diversity estimates. Species with geographically fragmented distribution areas are expected to display significant among-population genetic differentiation and low within-population genetic diversity. Contrary to these expectations, we show that the current diversity of Pinus nigra and its weak genetic spatial structure result from the Late Pleistocene or Early Holocene fragmentation of one ancestral population into six distinct genetic lineages. Gene flow among the different lineages is strong across forests and many current populations are admixed between lineages. We propose to modify the currently accepted international nomenclature made of five sub-species and name these six lineages using regionally accepted sub-species-level names.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Species with geographically fragmented distribution areas are expected to display strong among-population genetic differentiation and low within-population genetic diversity, similarly to biogeographic islands (Mac Arthur and Wilson 1967). This is because gene flow will necessarily be low among long-separated fragments while isolated fragments, if small enough, will lose diversity because of genetic drift and inbreeding (Young et al. 1996). Such patterns are often found in the wild, both in animals and in plants and both when distribution areas are large or small (e.g. Riginos and Liggins 2013; Young et al. 1996). However, examples also abound, where, despite fragmentation, among-population genetic differentiation remains low or modest: e.g. the European mountain pine Pinus mugo (Heuertz et al. 2010) or the North American white-footed mouse Peromyscus leucopus (Mossman and Waser 2001), while within-population genetic diversity is kept at high levels: e.g. the short-leaved cedar Cedrus brevifolia (Eliades et al. 2011) and see Andrén (1994) for a review.
The climate cycles of the Pleistocene have reshuffled species geographical distributions and genetic diversity patterns (Hewitt 1999; Petit et al. 2003). The imprint left on their current genetic diversity pattern is variable and depends greatly on their life history traits and their ecological preference. Species with ecological niches occurring with isolated or patchy distributions at mid-elevation on mountains today, during the Holocene, were probably widespread at lower elevation repeatedly during the many glacial periods of the Pleistocene (Feurdean et al. 2012), offering several opportunities for gene flow to occur unrestrictedly. In such species, current day patchiness and isolation may not indicate strong genetic differentiation and, a fortiori, the onset of speciation events (see Futuyma (2010) for factors constraining genetic divergence).
Pinus nigra Arnold, the European black pine, belongs to the section Pinus and the subgenus Pinus of the Pinaceae family (Eckert and Hall 2006). It has a wide, discontinuous distribution area, which spreads from isolated occurrences in North Africa to the Northern Mediterranean and eastwards to the Black Sea and Crimea (Gaussen et al. 1964; Barbéro et al. 1998; Isajev et al. 2004). The European black pine can also grow and adapt to several different soil types and topographic conditions supporting a wide variety of climates across its geographic range. It grows at altitudes ranging from sea level to 2000 m, most commonly between 800 and 1500 m above sea level. The European black pine is one of the most economically important native conifers in southern and central Europe and one of the most used species in European reforestation programs since as early as the nineteenth century and throughout the twentieth century (Isajev et al. 2004). Several geographic sub-species are described but its taxonomy is still considered as unresolved (Rubio-Moraga et al. 2012).
Pinus nigra has a large, but highly fragmented geographic distribution area (Fig. 1). Yet, what is known of its genetic diversity is that of a typical temperate forest tree species, with high within- and low among-population genetic diversity (Fady and Conord 2010). The genetic data of the European black pine contradict what is expected from a habitat fragmentation perspective, where patchy distributions equate to high ecological and evolutionary divergence (Young et al. 1996). One of the favoured explanation for this a priori contradiction is a historically high gene flow (dispersal events over millennia of colonization) over long distances (Kremer et al. 2012). In forest trees, long generation time and late sexual maturity increase the effect of gene flow (Austerlitz et al. 2000).

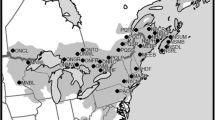
Native geographic distribution area of Pinus nigra (in blue, from Isajev et al. 2004) and location of sampled populations (stars) with their code names. The colour of the star determined the sub-species name according to the nomenclature of the Catalogue of Life (http://www.catalogueoflife.org/)
The population genetic pattern of the Pinus nigra biogeographical islands of today (particularly at the southern and western edges of the species) should thus be the result of relatively recent fragmentation. However, widely contradicting divergence timings have been proposed for explaining the genetic diversity and structure of the European black pine, from very recent Holocene or Quaternary divergence (Rafii and Dodd 2007) to pre-Pleistocene divergence (Naydenov et al. 2016, 2017).
The goal of this study is to test the effects of historical fragmentation on the genetic diversity and differentiation of the European black pine and to resolve its biogeographic history. For this, we describe and model its spatial genetic structure and we test whether its large and patchy distribution results from recent Holocene or earlier Pleistocene events using population genetic data and realistic demographic, timed scenarios. We use a set of different genetic markers, both neutral and putatively adaptive, and both bi-parentally and uni-parentally (cytoplasmic) inherited. Although population genetic studies traditionally use selectively neutral genetic makers, loci influenced by selection are also particularly helpful for assessing relative differences in levels of gene flow, especially in high gene flow species (Guichoux et al. 2013).
We sampled natural populations across the entire range of the species. We discuss why our results differ from, or concur with, the few previous molecular studies carried out at a similar scale on this species (Nikolić and Tucić 1983; Naydenov et al. 2016, 2017; Rafii and Dodd 2007). We also considered how habitat suitability might have affected demography by correlating climate variables at different Pleistocene ages with genetic diversity estimates under the assumption that harsher climate conditions and thus declining habitat suitability would produce demographic contractions with observable signatures in the genetic data (Conord et al. 2012).
Materials and methods
Sampling and DNA extraction
Needles were collected from 19 different populations originating from the Mediterranean Basin and southern Europe where black pine is naturally occurring. Although our populations cannot be claimed to account for all diversity found at landscape scale within the range of the species, they cover the entire distribution of the species, all of its described sub-species and are well suited to identify all evolutionary lineages that can explain present-day genetic diversity and structure. Table 1 indicates the origin of the material, either directly from needles collected in the wild or from individuals planted in common gardens. In both cases, collections in natural populations were carried out to avoid inbreeding, by collecting identical age-class individuals located at least 30 m from each other. Populations were represented by 12 individuals each, with the exception of HRV-01 for which 16 individuals were collected. The 19 populations selected cover the full extent of the geographic distribution of Pinus nigra as well as its taxonomic diversity (Table 1). DNA was extracted from needles using the DNeasy 96 Plant Kit (QIAGEN, Germany) at the INRA molecular biology laboratory of Avignon, France. Microsatellite data analyses, both chloroplast (cpSSRs) and nuclear (nSSRs), were carried out on the 19 selected populations, while gene sequencing was performed on 18 populations.
Genotyping and sequencing
Four paternally inherited cpSSRs, previously identified as polymorphic in black pine, were selected for this study (Appendix S1, Table S1.1 in the Supporting Information; Vendramin et al. 1996). The 14 bi-parentally inherited nSSRs used were those characterized for black pine by Giovannelli et al. (2017; Table S1.2 in Appendix S1). The 14 putatively adaptive nuclear genes selected for this study were those that could be transferred from P. taeda (Mosca et al. 2012) in a preliminary test (Table S1.3 in Appendix S1). As their putative metabolic functions are mostly identified, we call these genes “candidate genes” in the rest of the manuscript. Finally, we also used four organelle regions that can reveal sub-species level differences in widely distributed and taxonomically complex species (Kress and Erickson 2007, Table S1.4 in Appendix S1). These four regions were matK, rbcL, trnH-psbA (chloroplast DNA regions, paternally inherited in the Pinaceae) and nad5–4 (mitochondrial DNA region, maternally inherited in the Pinaceae).
All laboratory procedures are described in Appendix S1.
Data analysis
Genetic diversity and differentiation estimates: cpSSR and nSSR
We used MICRO-CHECKER (Oosterhout et al. 2004) to estimate the presence of null alleles among the 14 selected nSSRs performing 1000 randomizations. We then used ML-NullFreq (Kalinowski and Taper 2006) to calculate the frequencies of the null alleles. This two-step method minimizes the false negative rate of null allele detection (Dąbrowski et al. 2014). When the presence of a null allele was detected, genotypes were corrected using MICRO-CHECKER.
SPAGeDi1.4 (Hardy and Vekemans 2002) was used to compute the rarefied allelic richness (AR) and the expected heterozygosity corrected for sample size (HE). In addition, we inspected the deviations from Hardy–Weinberg genotypic proportions by computing the fixation index (FIS) and tested deviation from zero using 10,000 permutations of alleles within populations in SPAGeDi. Finally, the rarefied number of private alleles (PR) was computed using ADZE (Szpiech et al. 2008).
Genetic differentiation between populations was estimated using GST (Nei 1973), the “standardized” measures G’ST (Hedrick 2005) and Jost’s D (Jost 2008) following the recommendations for correction of sampling bias of Meirmans and Hedrick (2011). Genepop v4.2.1 was used to test for genotypic disequilibrium among the 14 nSSR loci using likelihood ratio statistics (Rousset 2008) and default Markov chain parameters.
Genetic diversity and differentiation estimates: Genes
PHASE v.2.1.1 included in DnaSP v5 (Librado and Rozas 2009) was used to infer haplotypes. Linkage disequilibrium between pair of SNPs within each gene and within each population was inferred from the exact test of linkage disequilibrium (Raymond and Rousset 1995) available in Arlequin v3.5.2.2 (Excoffier and Lischer 2010). As for SSR, we used SPAGeDi to compute the rarefied allelic richness (AR), the expected heterozygosity corrected for sample size (HE), the fixation index (FIS) and its significance. ADZE was used to compute the rarefied number of private alleles (PR). Genetic differentiation (FST) was calculated for each population using Arlequin.
Phylogeographic patterns
SPAGeDi (Hardy and Vekemans 2002) was used to calculate and compare FST and RST for nSSRs and FST and NST for cpSSR haplotypes and genes. RST and NST are analogues of FST which take into account the similarities between alleles (Goldstein and Pollock 1997; Pons and Petit 1996). The tests were performed globally and within 19 (18 for genes) classes of geographic distances equaling the number of populations analysed. For adaptive genes, the analysis was performed considering each gene separately, whereas for nSSR and cpSSR the analysis considered all loci together. The presence of a spatial genetic structure was tested by permuting locations (10,000 permutations). The presence of phylogeographic signals was tested by permuting (10,000) microsatellites allele sizes for nSSRs and by permuting rows and columns of distance matrices between alleles for cpSSRs and candidate genes (Hardy et al. 2003; Pons and Petit 1996).
Range-wide structure
To test for the presence of population genetic structure without a priori on the geographic origin of the individuals, we performed a Bayesian clustering approach using the software STRUCTURE v2.3 (Pritchard et al. 2000; Falush et al. 2003). An admixture model for which individuals may have mixed ancestry and a correlated allele frequency model in which frequencies in the different populations are likely to be similar (due to migration or shared ancestry) were considered. The analysis was first performed independently for nSSRs, cpSSRs and candidate genes, then including all markers together, following Falush et al. (2003). The two types of analyses did not differ significantly in the number of lineages identified (Giovannelli 2017). The analysis using all markers together was much more significant and we do not present the results of the analysis carried out for each marker independently. Five independent runs for each K value ranging from 1 to 20 were performed after a burn-in period of 5 × 105 steps followed by 1 × 106 Markov chain Monte Carlo replicates. The most likely number of clusters (K) and the rate of change of L(K) between successive K values were estimated following Evanno et al. (2005) using the web application StructureHarvester (Earl and von Holdt 2012). Results from five runs for the most likely K were averaged using the Clumpak program (Kopelman et al. 2015).
In addition to STRUCTURE, we used the PopTree software (Takezaki et al. 2014) to build a neighbour-joining tree based on Nei’s standard genetic distance corrected for sampled size (DST). We used the same data set as before but removed two populations (ESP-02 and Crimea-02) which had many missing data. To test the confidence of the phylogenetic tree, we performed 1000 bootstrap tests where genetic loci were resampled with replacement in each replication.
Demographic inference
DIYABC v2.1.0 (Cornuet et al. 2014) was used to infer the species’ demographic history within an approximate Bayesian computation (ABC) framework. Among the 19 sampled populations of P. nigra, only a subset was used for the demographic inference. The selection of the populations was performed based on the results of STRUCTURE, which identified six evolutionary lineages and several admixed populations suggesting the presence of past or continuous gene flow. Since the latter is not modelled by DIYABC, we focused on the past history reconstruction of both divergence and admixture events. To simplify the historical reconstruction, we first focused on the divergence between the six lineages (Fig. 2).

Demographic scenarios tested to infer past range-wide divergence events between six populations of Pinus nigra. The y-axis is time (T) in generation. Given the large number of possible scenarios when six genetic lineages are present, we reduced our investigation by focusing on how populations were grouped after the STRUCTURE analysis. The most likely number of clusters using nSSRs, cpSSRs and genes was six. We thus choose, as the simplest scenario, a rake shape tree (scenario 1) with six branches. We used as alternative scenario that which reproduces a genetic structure at a higher phylogenetic level as suggested by the delta K analysis which shows a peak at K = 2. This structure leads to two groups corresponding to the populations of the west (DZA-01, ITA-01) and of the east (SCG-01, HRV-01, CRIMEA-01, CYP-01)
For this first analysis, six P. nigra populations were selected, which best represented the identified lineages (ancestry proportion higher than 0.85): DZA-01 (P. n. salzmannii, Algeria), ITA-01 (P. n. laricio, Italy), SCG-02 (P.n. nigra, Serbia), HRV-01 (P. n. dalmatica, Croatia), CRIMEA-01 (P.n. pallasiana, Crimea) and CYP-01 (P. n. pallasiana, Cyprus) (Table 1). Sample size per population was kept above 10 and varied between 10 and 12, after removing individuals with too many missing data. Two scenarios of divergence were tested. The first one corresponds to the divergence of the six clusters from a common ancestor at time T1. The second scenario reproduces the genetic structure at a higher hierarchical level, as suggested by the results of the delta K in the STRUCTURE analysis: two ancestral lineages, which diverged from a common ancestor at time T1, and each of them diverging at independent times (T2 and T3) to give rise to the six current observed lineages.
In a second step, the historical reconstruction focused on the admixed populations. Rather than inferring past demographic parameters from a single tree composed of six branches and later events of admixtures, we ran separately three different scenarios of admixture events which best explained current population genetic composition. The details of the DIYABC analysis (prior distributions, evaluation of the simulations, selection of the most likely scenario, estimation of the demographic parameters, estimation of the bias and the precision of parameters estimation) are given in Table S2.1 and Figure S2.2 of Appendix S2.
To test whether admixed populations continued to exchange genes with their putative ancestors, we selected two admixed populations (TUR-01) and (HRV-02) and their ancestral populations (SCG-02, HRV-01, Crimea-01). We chose these admixed populations because they have in common one parental population (Crimea-01). This thus allows to test gene flow in a multi-population model while minimizing the number of parent populations. Gene flow was inferred using the coalescent-based program MIGRATE version 3.6.11 (Beerli and Palczewski 2010). The details of the analysis are given in Table S2.11. To summarize, the model of mutation was approximated to a continuous Brownian motion model where the mutation rate could vary between loci. Parameters were estimated using a Bayesian inference with uniform prior distribution for both the effective population size and the migration rate: for theta (4Neμ), min = 0, mean = 50, max = 100 and for migration (M = m/μ, with m, the fraction of the new immigrants of the population per generation) min = 0, mean = 50, max = 100. We performed two replicate Bayesian runs, which consisted of one long chain (see Table S2.11 for more details on the chains). We simulated a 5 × 5 migration matrix, M varying freely between admixed populations (TUR-01 and HRV-02) and the populations HRV-01, SCG-02 and Crimea-01, representatives of three distinct genetic lineages. Migration was fixed to zero between these three populations in order to minimize the number of parameters to estimate. The posterior distribution was generated by using the SLICE sampling which is the default method proposed in MIGRATE.
Correlations between genetic diversity and environmental variables
We computed correlations to test whether environmental characteristics could influence neutral genetic diversity patterns through demographic changes. Latitude, longitude and 19 standard bioclimatic variables were downloaded from the WorldClim database (version 1.4, Hijmans et al. 2005) in January 2016 for the present time, the Mid-Holocene, the Last Glacial Maximum and the last interglacial (Table S3.1 and S3.2 in Appendix S3). We also added three custom-made variables representative of the population isolation (Tables S3.1 and S3.2 in Appendix S3). We conducted all analyses in Rstudio (RStudio Team version 1.0.143 2016).
Results
Within-population diversity
At nSSRs, no significant linkage disequilibrium was detected among loci after the application of the Bonferroni correction at the α = 0.05 confidence level. Significant tests for the presence of null alleles were scattered among loci and populations without a clear mark indicating that a locus or an entire population should be excluded from the data set for analysis (Table S1.5 in Appendix S1). Thus, when the presence of a null allele was detected, genotypes were corrected using MICRO-CHECKER (Oosterhout et al. 2004) and this corrected data set was used in all further analyses. At candidate genes, 173 single nucleotide polymorphisms (SNPs) were detected and significant within-population linkage disequilibrium was found for each gene (Table S1.7 of Appendix S1).
All within-population diversity results are described in Table S1.8 of Appendix S1. Organelle DNA gene did not show any diversity at all in any of the preliminary samples we tested. We considered them as monomorphic in all populations throughout our data set. For candidate genes, the rarefied allelic richness averaged over 13 genes (one gene, G13484, was excluded due to missing values) was between 3.51 (ITA-01) and 4.69 (SCG-02). The rarefied allelic richness averaged over 13 nSSRs (one nSSR, SPAG_7.14, was excluded due to missing values) was between 4.85 (MAR-01) and 6.08 (ITA-02). Gene diversity corrected for sample size (HE) was high in both the nuclear genome (between 0.57 and 0.75) and the chloroplast genome (between 0.42 and 0.75). The population with the highest number of rarefied private alleles at both cpSSRs and genes was CYP-01, whereas at nSSRs it was DAZ-01. These two populations are representative of two different genetic lineages.
The multilocus analysis that computes FIS for nSSRs showed a significant excess of homozygotes in 6 out of 18 analysed populations, whereas for the candidate genes, the excess of homozygotes affected all populations. A more in depth analysis at each gene showed that three genes had significant positive FIS in more than half of the populations (G2078 (10), G10162 (17), G16810 (13)) that could be due to a selection effect. However, G2078 and G16810 are unknown proteins and only G10162 has one SNP out of 11 that is a non-synonymous mutation in a coding region (Table S1.3 in Appendix S1).
A latitudinal cline was observed at candidate genes for expected heterozygosity corrected for sample size (HE) and the number of rarefied distinct alleles (AR). Values increased from south to north (Pearson correlation between 0.42 and 0.47, P value < 0.05; Figure S1.1 in Appendix S1). We also observed significant although smaller correlations with the longitude, with the number of rarefied private alleles at cpSSRs (R2 = 0.20) and the number of pairs of SNPs within gene (averaged over a total of 14 candidate genes) displaying a significant linkage disequilibrium within population (LD), (R2 = 0.21).
Among-population structure
Genetic differentiation among populations was significant for all types of genetic markers with the strongest values at cpSSR whatever the indices used: Nei’s GST, Hedrick’s GST or Jost’s D (Table 2). Nei’s GST was 0.127, 0.064 and 0.11 for cpSSRs, nSSRs and candidate genes, respectively. Results from SPAGeDi (Hardy and Vekemans 2002) showed the presence of an isolation by distance pattern at nSSRs and five out of 14 candidate genes as shown by the positive and significant regression slope of pairwise FST on geographical distances (Figures S4.1 and S4.2 and Table S4.1 in Appendix S4). However, the linear regression slopes of pairwise NST and RST on geographical distances were never significant for any markers, indicating that allelic relatedness did not decrease with distance, thus indicating a lack of phylogeographic signal among populations (Figures S4.1 and S4.2 and Table S4.1 in Appendix S4).
Range-wide population structure
Results derived from STRUCTURE using combined nSSR, cpSSR and candidate gene data, showed that the most likely number of clusters (K) based on the delta K was K = 2 (Figures S4.3 and S4.4 in Appendix S4). However, given the probable underestimation of the number of K when using the delta K Evanno et al. (2005) method, we have used both the delta K and the distribution of the posterior log-likelihood of data for K clusters. We chose K = 6 as best number of K for the black pine because it corresponded to the second highest pic of delta K and the highest probability value with the lowest variance in the posterior distribution of L(K).
The first cluster included populations from Algeria (DZA-01), Morocco (MAR-01) and Spain (ESP-01, ESP-02). The second cluster was composed of the French (FRA-01, FRA-02) and Italian population (ITA-01). ITA-02 was admixed with the third cluster, which included the Austrian (AUT-01), the Serbian (SCG-02) and the Romanian (ROU-01) populations. The second Serbian population, SCG-01, was admixed with the fourth cluster mainly represented by the Croatian population (HRV-01). The fifth cluster was composed of the populations from Crimea (CRIMEA-01, CRIMEA-02) and Turkey (TUR-02). Finally, the sixth group was composed of the population from Cyprus (CYP-01) which constituted a highly differentiated lineage (Fig. 3).

Barplots of ancestry proportions for genetic clusters averaged over five runs for the best likely number of K (K = 6). Each individual is represented by a vertical bar divided into colour segments representing the gene pools identified by the STRUCTURE analysis. Bold types indicate the six most homogeneous populations (lineages) used in the ABC analysis. Taxonomic identification is as indicated in the “Materials and methods” section
Among the 19 populations, four (ITA-02, SCG-01, HRV-02, TUR-01) displayed a high proportion of admixture between two or more clusters (Fig. 3). The results of the analysis of STRUCTURE within the two clusters obtained at K = 2 are given in Figure S4.4, they confirm the presence of sub-structure within the main genetic clusters. The presence of two major clusters and several sub-groups is also supported by the structure of the neighbour-joining tree (Figure S4.2).
Demographic inference
Divergence patterns
The evaluations of the ABC simulations are given in the Figure S2.3 and Table S2.2 in Appendix S2, for both the PCA and a test of rank. Priors represented well the observed summary statistics and empirical data were located within the 95% of the distribution of the simulated data. The most probable scenario of divergence was scenario 1 with a probability of 0.99 (credible interval between 0.9998 and 0.9999). According to this scenario, all populations derived independently from a single common ancestor (Figure S2.4 in Appendix S2). The posterior error rate (also called “posterior predictive error”), given as a proportion of wrongly identified scenarios over the 1000 test data sets for the logistic approach, was equal to 0.105. The type I error associated with scenario S1 (the probability that scenario 1 was not selected although it was the true model) was very low (0.01 against 0.31 for S2), whereas the type II error (the probability that scenario 1 was selected, although it was not the true model) was acceptable (0.125 against 0.036 for S2) (Table S2.3 in Appendix S2).
Estimates of the original parameters (demographic size N, divergence time t, mutation rate mu) are given in Table 3. The median for the mutation rate was 0.00051 for nSSRs and 0.00023 for cpSSRs, 1.3 × 10−6 for candidate genes and 1 × 10−12 for chloroplast DNA sequences. The latter corresponds to the maximum limit of the prior and the absence of polymorphism did not allow estimation of the mutation rate. Median divergence time among the six lineages was estimated to be 628 generations ago, with a 95% credible interval of 210–2680 generations (Table 3). The divergence between the six lineages occurred between 9420 (3150–40,200) years before present for a generation time fixed at 15 years (i.e. the onset of Holocene) and 18,840 (6300–80,400) years before present for a generation time fixed at 30 years (i.e. thus at the time of Late Glacial Maximum).
Median effective population sizes for scenario 1 were 8170 for the ancestral population and between 2980 (Algeria) and 7640 (Serbia) for current populations. When comparing the mode of the ratio between current and ancestral population sizes, the ratio Ncurrent/Npast was systematically lower than 1 (between 0.11 and 0.85) (Fig. 4) and significantly so for three populations at credible interval of 90% for at least one type of marker according to the method proposed by Barthe et al. (2017) (Table S2.4 in Appendix S2). These three populations underwent a contraction at the same time as the split between populations. Populations ITA-01, HRV-01 and CYP-01 displayed demographic stability over time.

Comparison of the mode of the ratio between current and ancestral population sizes (Ncurrent/Npast) for six populations. In black, the prior distribution and in red, the posterior distribution. This ratio is systematically lower than 1, indicating population contraction
Results from the accuracy tests of parameter estimation (as measured by mean relative bias (MRB), relative root mean square (RMSE) and factor 2) and precision (as shown by coverage values) displayed, globally, reasonable values (Table S2.5 in Appendix S2). The mean relative bias was generally smaller than 1, while factor 2 values ranged from small to moderate according to the estimated parameters. The width of the 50–95% credible interval was small, showing that the parameters were reasonably well estimated.
Summary statistics computed after having simulated new data sets from the posterior distribution of parameters obtained under scenario 1 fitted well with the observed summary statistics (Table S2.6 and Figure 2.5 in Appendix S2). We found that none of the 84 summary statistics had low tail probability values when applying the model checking option to the scenario 1. The most probable scenario remained the same (S1) with a probability of 0.999 (credible interval between 0.9998 and 1.0000) when it was compared to a scenario allowing demographic resizing along the branches after divergence from the common ancestor (Figure S2.6 in Appendix S2).
Admixture patterns
The PCA and the test of rank are given in Table S2.7 and Figure S2.6 in Appendix S2. Median time of divergence between all three pairs of populations was between 304 and 872 generations (Table 4 and Table S2.8 in Appendix S2 for all parameter estimates) overlapping the estimates obtained from the six populations taken together (Table 3, median was 628 generations). Median time of admixture was between 105 and 361 generations (Table 4) with a confidence interval between 26.8 and 1150 generations. Assuming a generation time between 15 and 30 years, the admixture events between the pairs of lineages occurred between 402 and 34,500 years before present, from the Late Glacial Maximum to after the onset of Holocene. The proportion of test data sets for which the point estimate was at least half and at most twice the true value (factor 2) was rather high (mode of factor 2 > 0.70 for 8 out of 12 parameters, Table S2.9 in Appendix S2).
Summary statistics computed after having simulated new data sets from the posterior distribution of parameters fitted well with the observed summary statistics (Table S2.10 and Figure S2.8 in Appendix S2). We found that none of the 24 summary statistics had low tail probability values.
The admixed populations continued to exchange genes with the parental populations especially TUR-01 with HVR-01 and Crimea-01 and HVR-02 with SCG-02. With one exception, coalescent modelling highlighted the presence of asymmetrical gene flow more important from the parental population to the admixed population (Table S2.11 and Figure S2.11).
Correlations between genetic diversity estimates and environmental variables
Neither Pearson correlations nor partial Pearson correlations were significant at a false discovery rate (FDR) lower than 0.1 (i.e. 10% chance that the retained relationships are false positives). The rank of the explicative environmental variables obtained from the random analyses and the conditional inference tree obtained from the five most important environmental variables (Figure S3.1 of Appendix S3) highlighted mean temperature of warmest quarter (Bio10) during the Mid-Holocene (approx. 6000 years before present) as an explanatory variable for genetic diversity (He and AR) at cpSSRs (Figure S3.1). The smaller the mean temperature of the warmest Mid-Holocene quarter, the higher was the number of cpSSR alleles within populations. At nSSRs and candidate genes, the climatic variables that best explained genetic variables were for older periods: Late Glacial Maximum (approx. 22,000 years BP) and Late Interglacial (between 120,000 and 140,000 years BP) (Figure S3.1).
Discussion and conclusions
Our Bayesian clustering and demographic scenario testing study indicates convincingly that Pinus nigra is composed of six genetic lineages dating from relatively recent divergence. A simple, six-toothed rake-like demographic scenario best explains the biogeographic distribution of present-day black pine populations. The six lineages diverged from their most recent common ancestor at a very recent time, estimated between the Last Glacial Maximum and the onset of the Holocene.
This scenario agrees with the results of Rafii and Dodd (2007) who also estimated the fragmentation of European black pine lineages to be recent. Focusing on Western Europe, they were able to identify five barriers to gene flow including, similarly to ours, between the Alps and the Calabria–Corsica group and between France and Spain. Naydenov et al. (2016, 2017) identified three genetic groups from three different geographical areas that are not contradicting our own analysis: Western Mediterranean, Balkan Peninsula and Eastern Mediterranean. However, they estimated their most recent common ancestor to be older than 10 million years and dated the most recent splits between groups in the Late Pliocene. These patterns and dates were obtained using cpDNA sequence and cpSSR length polymorphism data, assuming an average mutation rate per generation of 5.6 × 10−5. This low mutation rate, identical for both length polymorphism and sequence variation in cpDNA, could be one of the reasons why our results are so different. Also, our ancestral effective population size is 30 times lower than that of Naydenov et al. (2016, 2017). Finally, as our study uses a suite of different molecular markers and because an old Pliocene divergence time would certainly have left a mutational signature in organelle DNA genes, we are confident that Late Pleistocene or Holocene demographic events are the most likely explanations for the genetic patterns we observed.
No signature of demographic events older than the last Pleistocene glacial cycle can be found in our data. Environmental correlations with climate data demonstrate not only the importance of Mid-Holocene temperature on the genetic diversity and structure of black pine but also the importance of earlier climate events during the Late Glacial Maximum and the Late Interglacial. These Late Pleistocene climate variations may have had an effect on the genetic diversity patterns of black pine.
However, proofs of the presence of black pine ancestors throughout Europe date back to the lower Cretaceous and to the Neogene, between 23 and 2.6 million years ago (Gaussen 1949). The molecular and fossil calibrated phylogeny of Eckert and Hall (2006) dates the origin of black pine at the Miocene-Pliocene boundary. Following its migration from Polar to Central and Southern European locations during the global cooling of the late Tertiary, black pine was then often found throughout the Pleistocene in charcoal and fossil records in the Mediterranean. From the end of the Pliocene (2.6 million years ago) onwards and during the Pleistocene, it is associated with the sub-Mediterranean flora of Europe (Vernet et al. 1983). P. nigra forests played an important ecological role during the Quaternary glacial and interglacial climatic fluctuations in the Western Mediterranean region (Vernet et al. 1983).
Fossil macro-remains indicate that between the Late Pleistocene and the Holocene, P. nigra forests had a large distribution (altitude and latitude) in the north-western Mediterranean Basin (e.g. García-Amorena et al. 2011 for the Iberian Peninsula). Although Pinus nigra is a highland pine, fossil and sub-fossil remains suggest that it also grew at very low elevation, in coastal areas during cold and dry periods (Postigo-Mijarra et al. 2010). During the warmer, interglacial periods of the Pleistocene, it colonized more continental, higher altitudes, as is the case for its current Holocene distribution in the supra-Mediterranean and medio-European vegetation belts (Isajev et al. 2004). If a genetic signature was left by fragmentation during the warm intervals of the Pleistocene, our results infer that it was erased by downward migration during the longer cold climate episodes. Vegetation turnover estimates using fossil records suggest that mid-elevations were the most sensitive altitudinal belts to climate change during the Late Glacial and Early Holocene (Feurdean et al. 2012).
Human impact is known to have played a role in the past dynamics of P. nigra (Vernet 1986). The decrease of the Pinus nigra subsp. salzmannii from the Iron Age onwards and the concomitant development of Quercus ilex and Pinus halepensis, for example, may be related to fire events, logging and agro-pastoral activities (Rodrigo et al. 2004). The effective size of today’s black pine populations is generally significantly smaller than that of their ancestral population. As resizing after the emergence of current lineages was not sustained by our data, the most likely explanation for this smaller effective population size is not an effect of human impact, rather that of a contraction at the time of lineage splitting.
Admixture among the six lineages of black pine was found to be frequent, suggesting recent gene flow despite habitat fragmentation. There was no signal of a phylogeographic structure, only that of a weak isolation by distance, confirming the hypothesis of recent differentiation and emergence of distinct lineages. This pattern is reminiscent of that of P. sylvestris, a European pine with broadly similar ecological requirements as black pine (Robledo-Arnuncio et al. 2005; Pyhäjärvi et al. 2007; Tóth et al. 2017).
The moderately high within-population genetic diversity of black pine is between that of low elevation and high elevation Mediterranean pines (Robledo-Arnuncio et al. 2005; Rafii and Dodd 2007; Fady and Conord 2010). The highest genetic diversity within populations was found in populations originating from the Balkans and Turkey, the geographic area where the most extensive forests of P. nigra are found today. Within-population genetic diversity shows an east-west decreasing trend, indicating that eastern Mediterranean black pine forests probably found more favourable habitats there at the time of lineage splitting (Conord et al. 2012).
All but one population of the six lineages identified are located on islands or at isolated or rear-edge locations. It is likely that these locations correspond to glacial refugia. Most of them appear as refugia in the synthesis of Médail and Diadema (2009). To this list of 52 putative refugia of importance for Mediterranean plant species, our study adds the southern Serbian mountains and Crimea.
Finally, the six lineages we identified can contribute to the debated taxonomy of the European black pine (Nyman 1879; Fukarek 1958; Debazac 1964; Vidaković 1974; Farjon 2010; Rubio-Moraga et al. 2012). The westernmost, North African and Iberian Peninsula lineage corresponds to the taxonomic entity generally recognized as Pinus nigra subsp. salzmannii. The existence of a separated North African lineage corresponding to the taxon Pinus nigra subsp. mauretanica is not supported by our analysis. The main morphological traits of the trees in this group are their orange/brown twigs without needles at their base. Their needles are slender and light green.
Moving eastward, the second lineage is located in continental France, Corsica (France), and Calabria (Italy) and corresponds to the taxon Pinus nigra subsp. laricio. The fact that the population France belongs to this group is rather unexpected as it is generally recognized as belonging to the salzmannii group based on morphology. However, earlier taxonomic work (e.g. Debazac 1964) has often recognized Cévennes populations of black pine as a form of Corsican black pine. It is also worth noting that Pinus nigra subsp. laricio from Calabria and from Corsica are often distinguished in the forestry literature (e.g. Roman-Amat and Arbez 1986), which we could not do here despite our use of uni- and bi-parentally inherited, neutral and putatively selected markers. The main morphological traits of the trees in this group are their dull brown twigs. Their needles are slender, curly and light blue/green.
The third lineage corresponds to the taxon Pinus nigra subsp. dalmatica on the Dalmatian coast in Croatia. The fourth Central European (Serbia) lineage corresponds to the widely distributed taxon recognized as Pinus nigra subsp. nigra. The main morphological traits of the trees in these two groups are their dull greyish twigs and their very rigid, spiky dark green needles.
The fifth lineage corresponds to the widespread Pinus nigra subsp. pallasiana and is best represented in the Crimean peninsula. It has sometimes been recognized as Pinus nigra subsp. caramanica. Lastly, the easternmost sixth lineage is located in Cyprus and corresponds to a taxon generally included in Pinus nigra subsp. pallasiana, which has also sometimes been named Pinus nigra subsp. caramanica. The main morphological traits of the trees in this group are their shiny ochre/brown twigs and their long, slender needles.
A list of synonyms of Pinus nigra sub-species and varieties is available from the IUCN red list of threatened species website, at https://doi.org/10.2305/IUCN.UK.2013-1.RLTS.T42386A2976817.en.
To conclude, and somewhat contrarily to our original expectations, the six European black pine lineages we identified most likely diverged from a common ancestor at a recent time, estimated between the Last Glacial Maximum and the onset of the Holocene. Lack of earlier signature of fragmentation is possibly due to downward migration along mountainsides and admixture during the long cold climate episodes of the Pleistocene. Despite morphological differentiation, the European black pine remains a rather genetically homogeneous species where gene flow is strong and isolation by distance weak.
References
Andrén H (1994) Effects of habitat fragmentation on birds and mammals in landscapes with different proportions of suitable habitat: a review. Oikos 71:355–366. https://doi.org/10.2307/3545823
Austerlitz F, Mariette S, Machon N, Gouyon PH, Godelle B (2000) Effects of colonization processes on genetic diversity: differences between annual plants and tree species. Genetics 154:1309–1321
Barbéro M, Loisel R, Quézel P, Richardson DM, Romane F (1998) Pines of the Mediterranean Basin. In: Richardson DM (ed) Ecology and biogeography of Pinus. Cambridge University Press, Cambridge, pp 153–170
Barthe S, Binelli G, Hérault B, Scotti-Saintagne C, Sabatier D, Scotti I (2017) Tropical rainforests that persisted: inferences from the Quaternary demographic history of eight tree species in the Guiana shield. Mol Ecol 26:1161–1174. https://doi.org/10.1111/mec.13949
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185(1):313–326
Conord C, Gurevitch J, Fady B (2012) Large-scale longitudinal gradients of genetic diversity: a meta-analysis across six phyla in the Mediterranean basin. Ecol Evol 2:2600–2614. https://doi.org/10.1002/ece3.350
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin J-M, Estoup A (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Dąbrowski MJ, Pilot M, Kruczyk M, Żmihorski M, Umer HM, Gliwicz J (2014) Reliability assessment of null allele detection: inconsistencies between and within different methods. Mol Ecol Resour 14:361–373. https://doi.org/10.1111/1755-0998.12177
Debazac EF (1964) Le pin laricio de Corse dans son aire naturelle. Revue Forestière Française 3:188–215
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eckert AJ, Hall BD (2006) Phylogeny, historical biogeography, and patterns of diversification for Pinus (Pinaceae): phylogenetic tests of fossil-based hypotheses. Mol Phylogenet Evol 40(1):166–182. https://doi.org/10.1016/j.ympev.2006.03.009
Eliades NGH, Gailing O, Leinemann L, Fady B, Finkeldey R (2011) High genetic diversity and significant population structure in Cedrus brevifolia Henry, a narrow endemic Mediterranean tree from Cyprus. Plant Syst Evol 294:185–198. https://doi.org/10.1007/s00606-011-0453-z
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fady B, Conord C (2010) Macroecological patterns of species and genetic diversity in vascular plants of the Mediterranean basin. Divers Distrib 16:53–64. https://doi.org/10.1111/j.1472-4642.2009.00621.x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Farjon A (2010) A handbook of the world’s conifers. Koninklijke Brill, Leiden
Feurdean A, Tămaş T, Tanţău I, Fărcaş S (2012) Elevational variation in regional vegetation responses to Late-Glacial climate changes in the Carpathians. J Biogeogr 39:258–271. https://doi.org/10.1111/j.1365-2699.2011.02605.x
Fukarek P (1958) Prilog poznavanju crnoga bora (Pinus nigra Arn.) / A contribution to the knowledge of black pine (Pinus nigra Arn.) / . Rad Poljopr – Šumarsk Fak Univ u Sarajevu 3:3–92
Futuyma DJ (2010) Evolutionary constraint and ecological consequences. Evolution 64:1865–1884. https://doi.org/10.1111/j.1558-5646.2010.00960.x
García-Amorena I, Rubiales JM, Amat EM, Iglesias González R, Gómez-Manzaneque F (2011) New macrofossil evidence of Pinus nigra Arnold on the Northern Iberian Meseta during the Holocene. Rev Palaeobot Palynol 163:281–288. https://doi.org/10.1016/j.revpalbo.2010.10.010
Gaussen H (1949) L’influence du passé dans la repartition des Gymnospermes de la Péninsule Iberique. CR Congr Intern Geogra Lisbonne 2:805–825
Gaussen H, Haywood VH, Chater AO (1964) Pinus. In: Tutin TG (ed) Flora Europaea, vol 1. Cambridge University Press, Cambridge, pp 32–35
Giovannelli G (2017) Histoire évolutive et diversité adaptative du pin noir, Pinus nigra Arn., à l'échelle de son aire de répartition. PhD thesis, Aix-Marseille Université, France
Giovannelli G, Roig A, Spanu I, Vendramin GG, Fady B (2017) A new set of nuclear microsatellites for an ecologically and economically important conifer: the European black pine (Pinus nigra Arn.). Plant Mol Biol Report 35:379–388. https://doi.org/10.1007/s11105-017-1029-z
Goldstein DB, Pollock DD (1997) Launching microsatellites: a review of mutation processes and methods of phylogenetic inference. J Hered 88(5):335–342. https://doi.org/10.1093/oxfordjournals.jhered.a023114
Guichoux E, Garnier-Géré P, Lagache L, Lang T, Boury C, Petit RJ (2013) Outlier loci highlight the direction of introgression in oaks. Mol Ecol 22:450–462. https://doi.org/10.1111/mec.12125
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8286.2002.00305.x
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638. https://doi.org/10.1554/05-076.1
Heuertz M, Teufel J, González-Martínez SC, Soto A, Fady B, Alía R, Vendramin GG (2010) Geography determines genetic relationships between species of mountain pine (Pinus mugo complex) in Western Europe. J Biogeogr 37(3):541–556. https://doi.org/10.1111/j.1365-2699.2009.02223.x
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68(1–2):87–112. https://doi.org/10.1111/j.1095-8312.1999.tb01160.x
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25(15):1965–1978
Isajev V, Fady B, Semerci H, Andonovski V (2004) EUFORGEN technical guidelines for genetic conservation and use for European black pine (Pinus nigra). International Plant Genetic Resources Institute, Rome
Jost L (2008) GST and its relatives do not measure differentiation. Ecology 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Kalinowski ST, Taper ML (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7:991–995. https://doi.org/10.1007/s10592-006-9134-9
Kopelman N, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kremer A, Ronce O, Robledo-Arnuncio JJ, Guillaume F, Bohrer G, Nathan R, Bridle JR, Gomulkiewicz R, Klein EK, Ritland K, Kuparinen A, Gerber S, Schueler S (2012) Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15:378–392. https://doi.org/10.1111/j.1461-0248.2012.01746.x
Kress WJ, Erickson DL (2007) A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnHpsbA spacer region. PLOS ONE 2(6):e508
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Mac Arthur RH, Wilson EO (1967) The theory of island biogeography. Princeton university press
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36(7):1333–1345. https://doi.org/10.1111/j.1365-2699.2008.02051.x
Meirmans P, Hedrick P (2011) Assessing population structure: FST and related measures. Mol Ecol Resour 11:5–18. https://doi.org/10.1111/j.1755-0998.2010.02927.x
Mosca E, Eckert AJ, Liechty JD, Wegrzyn JL, La Porta N, Vendramin GG, Neale DB (2012) Contrasting patterns of nucleotide diversity for four conifers of Alpine European forests. Evol Appl 5(7):762–775. https://doi.org/10.1111/j.1752-4571.2012.00256.x
Mossman CA, Waser PM (2001) Effects of habitat fragmentation on population genetic structure in the white-footed mouse (Peromyscus leucopus). Can J Zool 79(2):285–295
Naydenov KD, Naydenov MK, Alexandrov A, Vasilevski K, Gyuleva V, Matevski V, Nikolic B, Goudiaby V, Bogunic F, Paitaridou D, Christou A, Goia I, Carcaillet C, Alcantara AE, Ture C, Gulcu S, Peruzzi L, Kamary S, Bojovic S, Hinkov G, Tsarev A (2016) Ancient split of major genetic lineages of European black pine: evidence from chloroplast DNA. Tree Genet Genomes 12(68):1–18. https://doi.org/10.1007/s11295-016-1022-y
Naydenov KD, Naydenov MK, Alexandrov A, Vasilevski K, Hinkov G, Matevski V, Nikolic B, Goudiaby V, Riegert D, Paitaridou D, Christou A, Goia I, Carcaillet C, Escudero Alcantara A, Ture C, Gulcu S, Gyuleva V, Bojovic S, Peruzzi L, Kamary S, Tsarev A, Bogunic F (2017) Ancient genetic bottleneck and Plio-Pleistocene climatic changes imprinted the phylobiogeography of European black pine populations. Eur J For Res 136:767–786. https://doi.org/10.1007/s10342-017-1069-9
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nikolić D, Tucić N (1983) Isoenzyme variation within and among populations of European black pine (Pinus nigra Arn.). Silvae Genet 32:3–4
Nyman CF (1879). Conspectus florae Europaeae. Orebro Suecia typis Officinae Bohlinianae
Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Resour 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565. https://doi.org/10.1126/science.1083264
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered vs. unordered alleles. Genetics 144:1237–1245
Postigo-Mijarra JM, Morla C, Barrón E, Morales-Molino C, García S (2010) Patterns of extinction and persistence of Arctotertiary flora in Iberia during the Quaternary. Rev Palaeobot Palynol 162(3):416–426. https://doi.org/10.1016/j.revpalbo.2010.02.015
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pyhäjärvi T, García-Gil MR, Knürr T, Mikkonen M, Wachowiak W, Savolainen O (2007) Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 177:1713–1724. https://doi.org/10.1534/genetics.107.077099
Rafii ZA, Dodd RS (2007) Chloroplast DNA supports a hypothesis of glacial refugia over postglacial recolonization in disjunct populations of black pine (Pinus nigra) in Western Europe. Mol Ecol 16:723–736. https://doi.org/10.1111/j.1365-294X.2006.03183.x
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49(6):1280–1283. https://doi.org/10.1111/j.1558-5646.1995.tb04456.x
Riginos C, Liggins L (2013) Seascape genetics: populations, individuals, and genes marooned and adrift. Geogr Compass 7:197–216. https://doi.org/10.1111/gec3.12032
Robledo-Arnuncio JJ, Collada C, Alía R, Gill L (2005) Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J Biogeogr 32:595–605. https://doi.org/10.1111/j.1365-2699.2004.01196.x
Rodrigo A, Retana J, Pico X (2004) Direct regeneration is not the only response of Mediterranean forests to largest fires. Ecology 85:716–729. https://doi.org/10.1890/02-0492
Roman-Amat B, Arbez M (1986) Pins laricio de Corse et de Calabre, quelles provenances choisir? Revue Forestière Française 37:377–388
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
RStudio Team (2016) RStudio: integrated development for R. RStudio, Inc., Boston URL http://www.rstudio.com/
Rubio-Moraga A, Candel-Perez D, Lucas-Borja ME, Tíscar PA, Viñegla B, Linares JC, Gómez-Gómez L, Ahrazem O (2012) Genetic diversity of Pinus nigra Arn. Populations in Southern Spain and Northern Morocco Revealed By Inter-Simple Sequence Repeat Profiles. Int J Mol Sci 13:5645–5658. https://doi.org/10.3390/ijms13055645
Szpiech ZA, Jakobsson J, Rosenberg NA (2008) ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics. https://doi.org/10.1093/bioinformatics/btn478
Takezaki N, Nei M, Tamura K (2014) POPTREEW: web version of POPTREE for constructing population trees from allele frequency data and computing some other quantities. Mol Biol Evol 31(6):1622–1624
Tóth EG, Vendramin GG, Bagnoli F, Cseke K, Höhn M (2017) High genetic diversity and distinct origin of recently fragmented Scots pine (Pinus sylvestris L.) populations along the Carpathians and the Pannonian Basin. Tree Genet Genomes 13:47. https://doi.org/10.1007/s11295-017-1137-9
Vendramin GG, Lelli L, Rossi P, Morgante M (1996) A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol 5:595–598. https://doi.org/10.1046/j.1365-294X.1996.00111.x
Vernet JL (1986) Travertins et végétations holocènes méditerranéennes. Méditerranée 57(1):25–29
Vernet JL, Badal E, Grau E (1983) La végétation néolithique du sud-est de l'Espagne (Valence, Alicante) d'après l'analyse anthracologique. C R Acad Sci Paris 296(III):669–672
Vidaković M (1974) Genetics of European black pine (Pinus nigra Arn.). Ann For 6:57–86
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418. https://doi.org/10.1016/0169-5347(96)10045-8
Acknowledgements
We thank O. Gilg, F. Rei, N. Turion and D. Vauthier (INRA UEFM, Avignon, France), J. Rousselet (INRA Orléans, France), S.C. Gonzalez-Martinez (INRA Bordeaux, France), E. Kakouris (Cyprus Forest Department, Nicosia, Cyprus), F. Krouchi (University of Tizi Ouzou, Algeria), G. Huber (Bavarian Office for Forest Seeding and Planting, Teisendorf, Germany), Carmen García-Barriga (National Institute for Agronomy, Madrid, Spain) and H. Sbay (Forest Research Centre, Rabat, Morocco) for the sample collection. Collection of material was made before the Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization to the Convention on Biological Diversity was legally implemented by signatory countries.
Data archiving statement
Raw DNA sequence and genotyping data are to be archived in the Open Access archive Zenodo (https://zenodo.org/).
Funding
This study was made possible by the financial support of the French Forest Service (Office National des Forêts) and the project “Programme global de conservation des populations françaises de pin de Salzmann”. We also acknowledge the support for Sanger DNA sequencing from the French “Bibliothèque du Vivant” project and for SSR genotyping from the French Ministry of Agriculture—Irstea project 2015-339 “Déterminants de la vulnérabilité du pin laricio à la maladie des bandes rouges”. G. Giovannelli was financially supported by Aix-Marseille Université (Ecole Doctorale ED251), France, and the short-term scientific mission programme of the COST Action FP1202, while working on her PhD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Z. Kaya
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scotti-Saintagne, C., Giovannelli, G., Scotti, I. et al. Recent, Late Pleistocene fragmentation shaped the phylogeographic structure of the European black pine (Pinus nigra Arnold). Tree Genetics & Genomes 15, 76 (2019). https://doi.org/10.1007/s11295-019-1381-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-019-1381-2