
Abstract
Grape (Vitis L.) is one of the most economically valuable fruit crops in the world. The molecular basis for high genetic diversity and genetic differentiation thus conferring broad adaptability in the Vitis genus remains elusive. Genetic patterns in Vitis were comprehensively characterized by re-sequencing of 6862 nuclear SNPs in this study. Nucleotide diversity (π = 0.0073), genetic differentiation (pairwise differences, 0.24) and SNP variation in Vitis species were relatively high compared to most woody plant species. Molecular variance among accessions within species (F IS = 0.369) and among 30 species (F ST = 0.511) suggested strong population structure in Vitis (P < 0.0001). Vitis species are characterized by low frequency (4.7 %) of most minor SNPs and high proportion (>95 %) of minor SNPs to engender high differentiation among species thus high genetic diversity in the whole genus. This confers potentially broad adaptability thus a broad base for natural selection in this genus. Species from three geographical regions were discerned into independent quadrants in the principle coordinate analysis (PCoA) plot and their clustered patterns suggested that their molecular identities represented geographical origins of Vitis species. Species extinction will be an irreversible loss for breeding and genetic conservation. Species with low diversity, low heterozygosity, and high agronomical value should be given priority for conservation. Survey of species-specific SNPs will facilitate germplasm conservation and grape breeding programs. This study greatly adds to our knowledge of genetic patterns and molecular bases for ecological habits of Vitis species and provides valuable information for germplasm conservation and utilization, and grape breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grape (Vitis L.) is one of the most economically important fruit crops in the world (Reisch and Pratt 1996; Alleweldt 1997; El Oualkadi et al. 2011; Myles et al. 2011; Emanuelli et al. 2013). The Vitis genus contains approximately 60 species widely distributed across three geographical regions: (1) South Europe and Asia Minor, (2) East Asia, and (3) North and Central America (Reisch and Pratt 1996; Alleweldt 1997; Wan et al. 2008).
Both Vitis vinifera ssp. vinifera and Vitis vinifera ssp. sylvestris (refers to ‘V. vinifera’ and ‘V. sylvestris’ hereafter) originated in South Europe and Asia Minor (Reisch and Pratt 1996). V. vinifera grapes are widely cultivated in the world due to their high berry quality, productivity and multiple uses. However, high susceptibility to diseases and abiotic stresses in this species confers enormous losses to global grape production every year (Reisch and Pratt 1996; Alleweldt 1997; Emanuelli et al. 2013). Thus, extensive ongoing efforts have been put to improve resistance in V. vinifera cultivars through exploitation of wild Vitis germplasm (Reisch and Pratt 1996; Alleweldt 1997; Emanuelli et al. 2013). East Asia represents a major center of origin of Vitis species. China alone has more than 30 wild Vitis species (Wan et al. 2007). Chinese Vitis species possess good berry quality, high biotic and abiotic stress resistance, and are used for V. vinifera improvement (Wan et al. 2007). North and Central America form another major diverse Vitis gene pool with more than 30 wild Vitis species (Reisch and Pratt 1996).
Several species within the genus, e.g., Vitis rotundifolia, Vitis labrusca, Vitis amurensis, and Vitis quinquangularis, are cultivated for various grape purposes for human consumption, but most direct use of wild Vitis species has been to cross them with V. vinifera to develop cultivars with better biotic and abiotic endurance. Such interspecific crosses produce berries that are superior in quality in comparison to those in wild species that are directly cultivated (Reisch and Pratt 1996; Alleweldt 1997; El Oualkadi et al. 2011).
Wild species represent a unique, invaluable genetic resource for viticulture and grape breeding (Reisch and Pratt 1996; Alleweldt 1997; Martinez et al. 2006). However, the geographic distribution of wild grapevines has been dramatically reduced over the last 150 years (Grassi et al. 2006), particularly in China because of tremendous threats from rapid expansion of human population and industrialization therein (Wan et al. 2008). The extinction of wild species would be an irreversible loss for breeding programs and ecosystems (Grassi et al. 2006; Derero et al. 2011; Hadziabdic et al. 2012).
Characterization of molecular diversity, population structure, and molecular variation throughout the whole genus adds to knowledge of molecular basis for ecological habits of wild species and insures that we efficiently collect and conserve the germplasm in situ and choose novel genetic materials in breeding programs (Pavek et al. 2003; Emanuelli et al. 2013).
Nuclear single-nucleotide polymorphisms (SNPs) have been widely used for genetic diversity studies (Zhu et al. 2003; Lijavetzky et al. 2007; Rikkinen and Virtanen 2008; Emanuelli et al. 2013). Assessment of molecular diversity in grapevines has been reported using microsatellites (SSRs) and SNPs for V. vinfera and V. sylvestris (Grassi et al. 2006; Salmaso et al. 2004; Martinez et al. 2006; Myles et al. 2011; Emanuelli et al. 2013), cp-SSRs for V. sylvestris (Arroyo-Garcia et al. 2006; Grassi et al. 2006), and a few cp-SNPs, nuclear SSRs and amplified fragment length polymorphisms (AFLPs) for genetic relationships within the genus of Vitis (Ingrouille et al. 2002; Rossetto et al. 2002; Soejima and Wen 2006; Tröndle et al. 2010; El Oualkadi et al. 2011; Péros et al. 2011; Aradhya et al. 2012). Because of extremely low variation in cp-DNA and problematic homoplasy in SSR and AFLP markers, they have inherent limitations for study of population genetics in plants (Zhang and Hewitt 2003; DeWoody et al. 2010).
Every wild species may represent inherently unique genetic features and ecological habits, which can serve as a valuable resource for conservation and utilization (Mace and Purvis 2008; Wan et al. 2008; Pauls et al. 2013). To date, few studies have been performed concerning comprehensive assessment of genetic makeup and patterns across the Vitis genus using large numbers of nuclear SNPs (Salmaso et al. 2004; Lijavetzky et al. 2007; Myles et al. 2011; Péros et al. 2011). High morphological diversity has long been recognized in Vitis (Salmaso et al. 2004; Lijavetzky et al. 2007; Myles et al. 2011; Péros et al. 2011). High genetic diversity suggests potential broad adaptability of the population (Zhu et al. 2003). However, molecular basis for broad adaptability in Vitis is unclear. A number of strategies, e.g., based on genetic diversity, molecular heterozygosity, biodiversity-evolutionary perspective or range of the species habit, have been postulated for germplasm conservation (Mace and Purvis 2008; Pauls et al. 2013). However, these hypotheses have not yet been tested for their applicability to conservation of Vitis species. This study, based on empirical nuclear re-sequencing data, aimed to: (1) characterize patterns of genetic diversity and molecular variation in the different levels in Vitis, (2) discern genetic relationships and genetic partitioning and differentiation of Vitis species, (3) illustrate the molecular basis for high genetic diversity and genetic differentiation that confer broad adaptability in Vitis, (4) test the influence of genetic loss of one or two species on the genetic components in Vitis, and (5) comprehensively evaluate genetic features in Vitis thus to provide strategies for genetic conservation and utilization of this germplasm. This study not only greatly aids our understanding genetic patterns, molecular basis for broad adaptability of Vitis species, but also is critical to establishment of a basic molecular reference for germplasm conservation and utilization of Vitis species.
Materials and methods
Plant materials and sampling strategy
Plant materials were obtained from three germplasm repositories: (1) the Grape Germplasm Repository at the Northwest A & F University (NAFU), Yangling, Shaanxi Province, China, (2) USDA-ARS, Plant Genetic Resources Unit (PGRU), Geneva, NY, USA, and (3) USDA-ARS, National Clonal Germplasm Repository (NCGR), Davis, CA, USA (Table S1). No material collected from private/commercial vineyards was used to guarantee the trueness of accession of the materials.
Initially, 286 accessions were re-sequenced. Accessions with percentage of more than 2.5 % missing re-sequencing data and all potential hybrids between species were excluded as to minimize any experimental error. Consequently, a total of 248 accessions from 48 species or varieties (not cultivars for wild species, referred as ‘species’ hereafter) of Vitis (~80 % coverage of the extant species) were used in this study (Table S1, S2). Three large population groups were delineated in the complete set of samples according to physical geographical barriers: South Europe and Asia Minor (Eur.), East Asia, and North and Central America (NC Amer.).
DNA isolation
DNA was isolated from young leaves and apical meristems of accessions using a modified CTAB (cetyltrimethylammonium bromide) protocol (Lodhi et al. 1994) followed by NaCl and ethanol precipitation to remove polysaccharides.
Identification of polymorphic DNA regions
An in-house pipeline containing the SEAN SNP Prediction package, Phrap (Ewing and Green 1998; Ewing et al. 1998), and Primer 3 (Rozen and Skaletsky 2000) was used to sub-cluster and survey potential SNPs in V. vinifera using ESTs database in 2004 (NCBI Unigene set). This strategy for search of potential SNPs was successfully used in previous studies (Krutovsky and Neale 2005; Branca et al. 2011; Keller et al 2011; Myles et al 2011). PCR primers were designed to bind in conserved areas flanking polymorphic regions (Labate and Baldo 2005; Somers et al. 2003).
Primer design and selection
Primers were designed for gene fragments containing moderate polymorphism and 281 pairs of primers were selected to examine their PCR quality on agarose gels using DNA samples of three species representing genetic background from three major continents, e.g., V. vinifera (‘Rotberger’, DVIT2339) for Eur, V. rotundifolia (DVIT1689) for NC Amer, and V. romanetii (Jiangxi2) for East Asia. Ninety-six pairs of primers with robust, single band PCR product were selected to examine re-sequencing quality using eight species representing distinct genetic backgrounds (Vitis cinerea (PI588575), Vitis labrusca (PI588194), V. amurensis (Zuoshan1), V. quinquangularis (Weinan3), Vitis romanetii (Pingli7), Vitis davidii (Xuefeng), Vitis hancockii (Lingye_F), and Vitis yeshanensis (Yanshan_F)). Thirty gene fragments (Table S3, S4) with suitable polymorphisms and only minor sequence length variation in the eight tested accessions were re-sequenced in a total of 248 accessions. These 30 gene fragments were mapped onto 14 of 19 (or 20 for V. rotundifolia) genetic linkage groups based on alignment of the entire grapevine genome (Figure S1) and their unigene annotations were listed in Table S3.
PCR and re-sequencing protocol
PCR reactions for primer screening were performed in 25 μl volume consisting of 20 ng DNA, 3 mM MgCl2, 0.2 mM each dNTP, 8.0 pmol primer, 0.25 U GoTaq polymerase, and its commercial buffer (Promega, Madison, WI). PCR for sequencing was in 50 μl volume consisting of 20 ng DNA, 3 mM MgCl2, 0.2 mM dNTPs, 12.0 pmol primer, 0.5 U GoTaq polymerase, and its commercial buffer. The PCR conditions were: 94 °C for 4 min, followed by 35 cycles of 94 °C for 1 min, 42–56.8 °C for 1 min, 72 °C for 2 min, plus 72 °C for 7 min and 4 °C hold. Fifty microliters of PCR products were cleaned using the Edge Biosystems QuickStepTM PCR cleaning kits (www.EdgeBio.com), then concentrated to 12 μl for cycle sequencing reactions. Cycle sequencing reactions were performed in 12 μl volume consisting of 5.0 μl concentrated PCR product DNA, 3.6 μl 2.5× sequencing buffer, and 1.0 μl BigDye Ready Reaction Mix (Applied Biosystems, Foster City, CA). The PCR conditions for cycle sequencing reactions were: 95 °C for 3 min, followed by 50 cycles of 96 °C for 10 s, 58 °C for 4 min, plus 72 °C for 7 min and 4 °C hold. Cycle sequencing reaction products were cleaned using 96-well cartridge (Applied Biosystems, Foster City, CA) and dried in a speed vacuum, then resuspended in 10 μl formamide. The sequencing was performed on ABI-3100×1 sequencer (Applied Biosystems, Foster City, CA) at the USDA-ARS, Plant Genetic Resource Unit (Geneva, NY, USA).
Sequence editing and alignment
The software “Proseq v2.9” was used to visualize and analyze the sequence data (Filatov 2009). Sequencing with the forward primers produced sufficiently reliable sequences for all accessions; however, the reverse strand was also sequenced for all accessions as control. The software “Clustal W” (Thompson et al. 1994) was used to align the sequences. The software “Proseq” was used to visually identify SNPs and manually create a spreadsheet containing all scored SNPs. All sequences were released in NCBI with NCBI accession numbers [Genbank: JX952227-JX960379, EMBL: HF544510-HF544512] (Wan et al. 2013).
Assessment of genetic diversity and genetic differentiations
Nine tests with different levels were designed for estimation of genetic diversity using five statistical indices, including nucleotide diversity (π), gene diversity (θ), numbers of haplotypes, numbers of segregating sites, and mean numbers of pairwise differences between haplotypes and the statistically expected estimators for four indices above except nucleotide diversity (Nei 1987; Tajima 1983, 1989; Schneider et al. 2000). All parameters were estimated using Arlequin software version 3.1 (Nei 1987; Tajima 1983, 1989; Excoffier et al. 2005). Nucleotide diversity is the probability that two randomly chosen homologous nucleotides are different:
where d ij is an estimate of the number of mutations having occurred since the divergence of haplotypes i and j, k is the number of haplotypes, p i is the sample frequency of haplotype i, and L is the number of loci.
Gene diversity (Nei 1987) is the probability that two randomly chosen haplotypes are different:
where n, i and j, k is the number of haplotypes, p i is the sample frequency of the haplotype i.
Twelve tests with different levels were designed to manually calculate variation of SNPs based on five estimates including numbers of SNPs per gene fragment or per species (Per. SNPs), percentage of segregating sites (PSS), numbers of SNPs per segregating site (SNPs/SS), numbers of SNPs per haplotype (SNPs/Hap), numbers of haplotypes per sampled accessions (HPA).
Pairwise differences were used for assessment of the extent of genetic differentiation between pairs of 30 Vitis species. These were based on Wright’s F-statistics (F ST, number of permutations = 1,000), P value for F ST values (number of permutations = 1,000) and significance of P value (level = 0.05) as estimated using Arlequin software Version 3.1 (Excoffier et al. 2005).
Analyses of molecular variance
Ten population structures were designed to perform analyses of molecular variance (AMOVA) (based on 1,023 random permutations for P values) among the three geographic groups, among the 30 species (species containing at least three accessions were used for estimation of molecular variances), among the accessions within the species and among 248 accessions within Vitis genus using Arlequin software version 3.1 (Weir 1996; Excoffier et al. 2005; Fitzpatrick 2009). Coefficient of variation (CV = (standard variation / mean) multiplied by 100) (Hendricks and Robey 1936) was used for assessment of extent of molecular variation among accessions, among species or among gene fragments. SAS program (http://www.sas.com/) was used for calculation of correlation coefficients (r) between pairs of molecular indices and statistical significance. Permutations were performed based on the biological assumption of “expectation under random mating” and resampling the data for “pseudo-replication”.
Analysis of genetic relationships of species
Nei’s algorithms (Nei 1972) were used to estimate genetic distance between pairs of accessions and pairs of species using Arlequin software version 3.1. Principle coordinate analysis (PCoA) was used to illustrate the patterns of the genetic relationships using GenAlEx 6.0 (Peakall and Smouse 2006). Each accession (Table S1) and species was accordingly coded (Table S1 and S2), e.g., as “002N” or “S01A,” when the patterns of their relationships were discerned on the PCoA plots.
To test influence of species’ loss on genetic components of the whole dataset, one to two species were taken out from the dataset to compare molecular features between two populations: the complete population (comprising 48 species, CP) and the population without one or two species (the substracted population, SP) using software of Arlequin and GenAlEx 6.0. Mantel tests (Mantel 1967; Peakall et al. 2003) were performed to test statistical significance of relatedness between these two populations.
Results
Molecular features of nuclear sequences in Vitis
Seven DNA samples (0.09 %) did not amplify for the gene fragment ‘7393’. In total, 7,433 amplicons were successfully re-sequenced and aligned. The length of these 30 gene fragments ranged from 251 to 690 bp, averaging 413.9 ± 116.5 bp (mean ± s.d., n = 30, the statistical values in this context are all presented as mean ± s.d., except as noted). In total, ~12.4 Kb for each accession and ~3.07 million nucleotides for all 248 accessions were re-sequenced. A total of 6,862 SNPs, 2,752 segregating sites, and 4,248 haplotypes were found among 248 accessions of Vitis (Table 1).
Molecular genetic diversity in Vitis species
Nucleotide diversity and gene diversity among 248 accessions were 0.0150 and 0.927 (“T1” in Table 2), respectively. On average, these two estimates for accessions within species were 0.0073 and 0.699 (“T3” in Table 2), respectively. Numbers of segregating sites, numbers of haplotypes, and mean numbers of pairwise differences among 248 accessions were 91.7, 141.6, and 6.10 (“T1” in Table 2), respectively. These three parameters for accessions within species were 7.9, 6.1, and 2.85 (“T3” in Table 2), respectively. Genetic diversity was the highest in NC Amer among the three geographic regions (“T4–T9” in Table 2), indicating sampling size (number of total accessions or species) is an important contributor to genetic diversity.
For 248 accessions, 67 % of correlation coefficients were significant between pairs of five molecular indices above (Table S32). For 30 species, all correlation coefficients were significant (Table S32). For accessions within species, 94 % of correlation coefficients were significant (Table S33). This suggests that these five indices had similar trends of variation in Vitis.
Genetic differentiations between Vitis species
The F ST values in pairwise differences for estimation of genetic differentiation between Vitis species was mostly in the range of 0.10–0.40 (Fig. 1). On average (n 1 = 30 gene fragments, n2 = 29 pairs of comparisons), the F ST value of a species with the remaining 29 species was 0.24 (Fig. 1). In all, 86.8 % of 13,021 pairwise comparisons had significant P values (P < 0.05). Thus, the 30 Vitis species were highly genetically differentiated from each other.

Box plots of nucleotide diversity within and genetic differentiations between 30 Vitis species. a nucleotide diversity (π) of 30 Vitis species estimated from 30 gene fragments. Nucleotide diversity shows extensive variation among genes and among species in Vitis. b Average genetic differentiation of a Vitis species with the remaining 29 species (average value from 29 pairs of estimations)
Analysis of molecular variance (AMOVA)
Variance among 248 accessions was 0.684 (F IT in “Str1” of Table 3), suggesting a strong population structure across the entire genus (Excoffier et al. 2005; Peakall and Smouse 2006; Straub and Doyle 2009). On average, variance among accessions within species was 0.369 (F IS in “Str2” of Table 3), suggesting population structure within most Vitis species is also strong (Excoffier et al. 2005; Peakall and Smouse 2006; Straub and Doyle 2009). Variance among 30 species was 0.511 (F ST in ‘Str3’ of Table 3), indicating high genetic differentiation among species. For three geographic regions, estimated F ST representing variance among Euvitis species was in this order: Asia > NC Am > Eur (F ST in “Str7,”, “Str8,” and “Str10” of Table 3), suggesting the strongest structure within Asia. However, estimated F IS representing variance among accessions within Euvitis species was in this order: NC Am > Asia > Eur (F IS in “Str7,” “Str8,” and “Str10” of Table 3), indicating that NC Am species had the strongest population structure among three regions.
Variance components, i.e., percentage of variance, varied among structures in some cases, e.g., variances among accessions within species (e.g., ‘Str 2 and Str 3’ of Table 3). The reason for this is that variances for each component were the co-variances of the entire structure, and this may result in relatively different values for the same estimates among different structures (Schneider et al. 2000; Excoffier et al. 2005). However, the associated F statistics and percentage (%) of 30 gene fragments presenting significant P values for the same component were close among the different structures (e.g., in Str 2 and Str 3, Table 3), indicating that these results were consistent. Molecular variances were significant in all of the ten designed structures (Table 3). Because genotypes or species in Vitis are highly genetically differentiated from one another as analyzed above, when assessing species differentiation using a metric designed for populations, one would expect significant differentiation (Table 3).
Features of segregating sites and SNPs in Vitis
Numbers of SNPs per segregating site were similar among all different level tests (Table 4). For 248 accessions, on average, 22.0 ± 8.7 % of polymorphic sites (TL1 in Table 4, Table 1), i.e., one segregating site was found in every 3 to 8 bp among 248 accessions of Vitis. This value is relatively high compared to most plant species (Schneider et al. 2000; Tenaillon et al. 2001; Somers et al. 2003; Zhu et al. 2003; Krutovsky and Neale 2005; Heuertz et al. 2006; Pyhäjärvi et al. 2007; Chen et al. 2010; Keller et al. 2011; Wachowiak et al. 2011; Ismail et al. 2012; Emanuelli et al. 2013; Xin et al. 2013). However, after a detailed analysis, we found that 6862 SNPs were produced by 2,752 segregating sites, indicating that there was on average 2.5 ± 0.1 SNPs per segregating site. Percentage of segregating sites with which one site produced two SNPs and three SNPs, on average, accounted for 61 and 33 %, respectively (Table 1). Almost all allelic SNPs were distributed unevenly among segregating sites throughout the 248 accessions. On average, 95 % of 248 accessions shared an allelic SNP (referred as the genus major SNP). The remaining few accessions possessed other SNPs (referred as genus minor SNPs) (Table S48). Most genus minor SNPs were found within just one or a few species. Therefore, certain SNPs found as minors within the genus were identified as the majors within a species. Some of these SNPs showed no segregation within one or a few species, indicating that they may be unique to one or a few species. On average, 67.8 % of segregating sites in the genes produced SNPs with low frequency of 0.4–2.0 % (Table S45). In addition, the total frequency of the genus minor SNPs was 4.7 % (Table S44). Thus, numerous “subtle” variants were found in the Vitis gene pool.
SNPs/Hap and HPA within species were much higher than those among 248 accessions. However, percentage of segregating sites among accessions within species was greatly lower than that among 248 accessions (Table 4). Frequency of segregating sites among accessions within species, i.e., one segregating site in every 45 to 70 bp (TL2, Table 4), was higher than most reported plant species (Schneider et al. 2000; Tenaillon et al. 2001; Somers et al. 2003; Zhu et al. 2003).
Five indices, including Per. SNPs, PSS, SNPs/SS, SNPs/Hap, and HPA, were the highest in the NC Amer among three geographic groups (Table 4). With the exception of SNPs/Hap, the remaining four indices among accessions were the lowest in Eur, while five indices among accessions within species were the lowest in Asia among the three geographic groups (Table 4, Table S49 and S50).
Extent of molecular variation in Vitis
Twenty genetic structures comprising ten molecular indices (Table 5) were designed to evaluate the genetic variation (CV) in Vitis. CV in Vitis was characterized by: (1) CV among 248 accessions (SL1, Table 5) was much lower than that among 30 species (SL4, Table 5); (2) CV of nucleotide diversity (variation) was generally the highest among ten molecular indices, followed by mean numbers of pairwise differences and numbers of SNPs per gene fragment or per species; (3) CV of numbers of SNPs per segregating site and gene diversity were the lowest two indices among the ten molecular indices (Table 5, Table S51-S64), indicating that Vitis species exhibit a relatively wide extent of nucleotide variation while they maintain a relatively narrow range of gene variation (Fig. 1); (4) CV among gene fragments within species (SL2, Table 4, Table S52) was relatively the highest among the twenty tests, indicating that molecular variation ranged extensively among genes; (5) CV of numbers of haplotypes per sampled accession were within a relatively narrow range (mostly around 47 %) among the twelve tests, i.e., every two sampled accessions produced one haplotype; (6) CV among accessions within the geographic region was the highest in Eur and the lowest in Asia; however, CV among gene fragments or among species was the highest in Asia and the lowest in NC Amer in most cases among the three geographic groups (SL6-SL20 in Table 4, Table S56-S64). Thus, the geographic region comprising relatively high genetic diversity (T4–T9 in Table 2), i.e., NC Amer, may not present a wide extent of molecular variation (SL6-SL20 in Table 4).
The genetic patterns among Vitis species
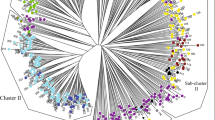
The patterns of genetic relationships of accessions and species in Vitis were characterized by: (1) two sub-genera, Muscadinia and Euvitis, were extensively differentiated from each other (Fig. 2a, c), (2) two species from Eur were discerned close to each other, but they both fell to different quadrants from American species in the PCoA plot (Fig. 2b, d), (3) Asian species were mainly discerned into two groups (Fig. 2b, d), one represented by Vitis adstricta (S01A), V. hancockii (S34A), Vitis pseudoreticulata (S08A), V. quinquangularis (S09A), Vitis bellula (S31A), Vitis ficifolia (S32A), and Vitis bashanica (S03A), the other represented by Vitis shenxiensis (S37A), V. romanetii (10A), V. davidii (S04A), Vitis piasezkii (S06A), Vitis qinlingensis (S36A), V. piasezkii var. pagnuccii (S07A), Vitis betulifolia (S47A), V. amurensis (S02A), Vitis yenshanensis (S38A), Vitis liubanensis (S35A), V. davidii var. cyanocarpa (S48A) and Vitis jacquemontii (S05A), (4) American species within Euvistis were mainly assigned into four groups (Fig. 2c), consisting of (1) Vitis arizonica (S17N), Vitis girdiana (S43N), Vitis acerifolia (S13N), Vitis riparia (S25N), Vitis blancoii (S41N), Vitis bloodworthiana (S23N), Vitis monticola (S23N), and Vitis rupestris (S27N), (2) Vitis tiliifolia (29N), Vitis nesbittiana (S44N) and Vitis shuttleworthii (S28N), (3) Vitis aestivalis var. aestivalis (39N), Vitis cinerea var. helleri (S21N), Vitis cinerea (S19N) and Vitis biformis (S40N), and (4) Vitis cinerea var. floridana (S20N), V. aestivalis (S14N), V. aestivalis var. argentifolia (S15N), V. aestivalis var. lincecumii(S16N), and Vitis labrusca (S22N).

The molecular genetic structure in Vitis based on Nei’s algorithms and PCoA methodology. a An accession-by-accession plot for the 248 accessions of the Vitis genus, the up-left five accessions were V. rotundiforlia, and the remaining 243 accessions on the right were from Euvitis; b an accession-by-accession plot for the 243 accessions of the Euvitis sub-genus, these accessions were mainly clustered into seven major groups; c a species-by-species plot for the 48 species of the Vitis genus, the left one species was V. rotundiforlia, and the remaining 47 species were from Euvitis; d a species-by-species plot for the 47 species of the Euvitis sub-genus. Comparisons between the accession-by-accession plot and the species-by-species plot support the conclusion that genetic patterns of the accessions and species were consistently and correctly discerned. Another advantage of the PCoA method is that extent of genetic differentiation among accessions or species can be easily visually identified
Effects of genetic loss on genetic component of genus
We found that haplotype frequency and genetic differentiation between CP and SP were non-significant (P = 1.00 ± 0.00) for all the tests (450 tests for one species loss (=(30 gene fragments × 30 species)/2) and 6,750 tests for two species losses (=(30 gene fragments × 30 species × 29 species)/2)). However, mean numbers of pairwise differences were extremely significant between two populations for all tests (P = 0.00 ± 0.00), suggesting each species represents unique genotype and its extinction would result in an irreversible loss of genetic variation from the gene pool of the genus.
Discussion
Comprehensive interpretation of genetic relationships of Vitis species
A precise resolution of genetic relationships is a prerequisite for genetic analyses, germplasm conservation and breeding programs (Grassi et al. 2006; Soejima and Wen 2006; Tröndle et al. 2010). In this study, all samples were obtained from governmental collections and any potential hybrids within the wild species germplasm were removed from analyses. Thus, all species are assumed to represent their natural identities. This ensured accuracy in discernment of genetic relationships among Vitis species in this study.
Although the principle of PCoA was proposed 50 years ago (Torgerson 1958), this method and its powerful attributes have not until relatively recently been recognized in the study of population genetics (Peakall and Smouse 2006). This method, unlike the tree construction methods, does not assume a hierarchical genetic structure in the population. The hierarchical assumption is rational at higher taxonomic levels. But it is not always true at the population level. Another disadvantage of tree building methods is that results may be very distinct among algorithms, among different numbers of permutation tests and among hierarchical genetic structures when the number of samples is vast for analyses (Peakall and Smouse 2006). In addition, strong evolutionary signals (historic hybridization and splitting, e.g., in Vitis (Wan et al. 2013) may lead to unwieldy trees that are difficult to interpret. PCoA uses a strategy in which the essential signals in the data were quantified and used for estimation, thus minimizing influence from conflicting signals of patterns of genetic relationships among operational taxonomic units (OTUs) (Peakall and Smouse 2006). Another advantage of PCoA is that patterns of genetic relationships and extent of differentiation among accessions or species in the PCoA plot (e.g., in Fig. 2) can be visually identified (Peakall and Smouse 2006).
The patterns between the accession-by-accession and species-by-species graphs were very similar for the whole genus (e.g., in Fig. 2a, c) and for the subgenus of Euvitis (e.g., in Fig. 2b, d), considering that coordinate orientations may vary among permutations of the different tests (Peakall and Smouse 2006). If number of accessions per species was the same for all species and molecular variation among accessions within the species was very close among species, the patterns between the accession-by-accession and species-by-species graphs would be very similar. All of these evidences suggested that the patterns in the PCoA plots in this study can reflect the intrinsic genetic relationships of the species in Vitis.
However, some differences were found between patterns between the graphs of the accession-by-accession and species-by-species (Fig. 2a and c, b and d). For example, 21 accessions from Eur overlapped with accessions from Asia in the accession-by-accession plot (Fig. 2b). However, two species from Eur were distinct from Asian species (Fig. 2d). The reason for this is that PCoA calculates the vector of each variable based on the whole matrix dataset, thus the results may vary among permutations (Peakall and Smouse 2006). The accession-by-accession analysis was a good reference with which to examine if any accession was unexpectedly assigned and for comparisons with the species-by-species results, thus to decide with confidence whether the accessions and species were consistently discerned.
The overall patterns in the PCoA plots (Fig. 2) reflected features of geographic distribution of species. The Asian species mainly fell to different quadrants from American species, indicating that genetic materials have inherited with unique molecular features to represent their identity in each of the three geographic regions since continental isolation. Our study suggested that two species from Eur were genetically closer to Asian species than American species (Fig. 2), consistently with previous studies (TrÖndle et al. 2010; Zecca et al. 2012). Fourteen accessions of V. sylvestris had relatively centralized distribution, but seven accessions of V. vinifera had relatively scattered distribution (Fig, 3a). This observation is also confirmed by the recent report (Emanuelli et al. 2013). This may indicate that high molecular variation has been maintained in the cultivated species by human selection and asexual propagation during domestication (Myles et al. 2011; Emanuelli et al. 2013).
Except for two varieties, V. aestivalis var. argentifolia and V. aestivalis var. lincecumii, that were closely related to each other, all remaining varieties were differentiated from each other and from their original species (Fig. 2d). Thus, classification for sub-species or varieties should be carefully treated, because genotypes in Vitis were genetically highly differentiated from each other. High differentiation among accessions within species can be frequently observed (Fig. 2b, SL3 in Table 5).
The overall patterns in the PCoA plots (Fig. 2) representing genetic relationship between accessions and between species in this study are very similar to those in our previous study estimated by other algorithms (Wan et al. 2013).
Comprehensive evaluation of genetic diversity in Vitis
Molecular polymorphism varied among genes and among regions within the gene in this study (Table S20). This may result from distinct conserved extent among genes and among regions within the gene (Lande 1988; Mace and Purvis 2008). Thus, choice of molecular markers is critical to assessment of genetic patterns of species. For example, nucleotide diversity in Picea abies estimated in one study (Heuertz et al. 2006) was twofold relative to a different study (Chen et al. 2010). This discrepancy may result from marker choice and sampling bias. An increase of number of accessions (≥3 accessions) per species may have little influence on assessment of molecular variance within species if there has been appropriate sampling. However, number of species (≥6 species) is very important for a precise assessment of genetic features within higher-level population structures (Fitzpatrick 2009). Linkage disequilibrium estimates suggested that the 30 gene fragments used in this study were genetically independent overall. We selected 30 gene fragments comprising moderate polymorphisms for evaluation of genetic diversity in Vitis, assuming that estimation based on these 30 gene fragments could represent the molecular make-up of a random sampling of a coding region.
There are numerous molecular indices (e.g., five basic indices in this study) for assessment of genetic diversity. Their values were significantly related to each other based on correlation analysis between pairs of five indices (P < 0.05), indicating that these molecular indices may clearly discern patterns of genetic diversity in Vitis species. Nucleotide diversity of the two sub-species V. vinifera and V. sylvestris estimated in this study was higher than a previous report (Riahia et al. 2013). However, gene diversity of these two sub-species was lower than Riahia et al. (2013), suggesting estimation of gene diversity is affected by sample number (number of accessions per species). Previous studies suggested use of nucleotide diversity for estimation of genetic diversity of species (Liu and Burke 2006; Song et al. 2009; Chen et al. 2010; Branca et al. 2011; Riahia et al. 2013). However, our estimates of gene diversity and heterozygosity were based on the same formula in Arlequin (Excoffier et al. 2005). Thus, estimates of gene diversity represent extent of heterozygosity of Vitis species in this study. Compared to reports of other species based on nuclear sequencing (Table S65), nucleotide diversity of 80 % of Vitis species was relatively high. Numbers of segregating sites, numbers of haplotypes, and mean numbers of pairwise differences were equal to or higher than their expected estimators in Vitis in most cases (Table 2, Table S30-S32). Genetic diversity in V. bashanica, V. piasezkii var. pagnuccii, V. acerifolia, V. adstricta, V. davidii, V. quinquangularis and V. labrusca was low compared to the relative high diversity in V. shuttleworthii, V. tiliifolia, V. cinerea var. floridana, V. cinerea var. helleri and V. vinifera (Table S30).
The species presenting high genetic diversity may not contribute proportionally to the genetic diversity of the genus. For example, V. rotundifolia had moderate genetic diversity and V. tiliifolia possessed relatively high genetic diversity compared to the remaining species in our study (Table S30). But contribution of V. rotundifolia to the genetic diversity of the genus was much higher than that of V. tiliifolia (Table S66 and S67). The reason for this was that a species possessing high genetic diversity may not be highly genetically differentiated from other species (Rao and Hodgkin 2002).
A molecular basis contributing to potentials broad adaptability in Vitis
Our data demonstrated that nucleotide diversity, numbers of segregating sites, and mean numbers of pairwise differences were relatively high in most Vitis species. However, most SNPs were at very low frequencies among 248 accessions (Table S48). Percentage of segregating sites in Vitis was relatively high, i.e., one segregating site was found in every 3 to 8 bp among 248 accessions within the genus and one in every 45 to 70 bp among accessions within most of species. Thus, Vitis species may be inherited with genetic characteristics of maintenance of ‘subtle’ nucleotide variation, and a high percentage of segregating sites, accumulating these subtle variations as to engender high genetic diversity in most species and high genetic differentiation among species. This molecular basis confers potential broad adaptability of Vitis species. Broad geographical distribution in this genus and extensive distinct ecological heterogeneity among species (Reisch and Pratt 1996; Alleweldt 1997) requires high genetic differentiation among species and high genetic diversity in the genus to provide potential broad ecological adaptability of this genus (Mayr 1997; Yokoyama 2002; Orr 2005; Nowak 2006).
Comprehensive interpretation of genetic diversity and genetic differentiation in Vitis species
Nucleotide diversity within a species represents genetic variability of the species (Excoffier et al. 2005). The wild Vitis species of the subgenus are out-crossing. This habit (natural hybridization) in Vitis may facilitate genetic exchange among species, thus increase their genetic diversity, which in turn may improve their fitness and confer their potential for increased adaptability (López-Pujol et al. 2012). High nucleotide diversity (Table S9) and high CV value of nucleotide diversity (Table S53) in most Vitis species suggested high genetic variability in these species. Our previous study confirmed broad hybridization among species in the evolutionary history of Vitis (Wan et al. 2013). In this case, the phylogenetic species concept may be unsuitable as suggested by López-Pujol et al. (2012). Our phylogenetic study demonstrated that the consensus phylogenetic tree in Vitis was impossible to obtain from numerous phylogenetic algorithms (Wan et al. 2013).
Extensive genetic drift may lead to a decline of genetic variability on a large scale, e.g., decrease of genetic differentiation among species (Kekkonen et al. 2011). However, both nucleotide diversity (Table S9) within and genetic differentiation (Fig. 1) among most of the Vitis species were high compared to other plant species (Table S65). Vitis species inherently showed characteristics of low frequency of most SNPs (Table S48) and a high percentage of segregating sites (Table 4), which together conferred high nucleotide diversity within species. High SNP variation derived from a high percentage of segregating sites at different loci among species can result in high genetic differentiation among Vitis species.
Strategies for germplasm conservation and improvement
Our study showed that every accession represents a unique genotype (Fig. 2b) due to highly genetic differentiation among accessions within species. This provides broad potential for improvement of the V. vinifera crop using novel germplasm. Given high morphological variation among species and high genetic diversity within Vitis, it is important to collect all species within a broad array of ecological, morphological and genetic ranges. However, an exhaustive collection is not feasible because germplasm collection and conservation are very costly for woody plants (Alleweldt 1997; Pavek et al. 2003). Previous studies demonstrated that biotic and abiotic resistance varies among species as well as among accessions within species in Vitis (Wan et al. 2007). Thus, certain accessions should be given priority for germplasm collection and conservation (Pavek et al. 2003).
High genetic diversity in taxa does not imply a low threat to the survival of species. Previous studies reported high genetic diversity in certain highly endangered species, e.g., in whorled sunflower (Helianthus verticillatus), legume species (Amorpha georgiana), giant panda (Ailuropoda melanoleuca) and Australian corroboree frogs (Pseudophryne corroboree and Pseudophryne pengilleyi) (Ellis et al. 2006; Morgan et al. 2008; Straub and Doyle 2009; Zhao et al. 2013). The reason for this phenomenon may be that most molecular studies tend to estimate genetic diversity within the status quo of the species rather than adopting a more dynamic approach to reflect changing trends within an evolutionary perspective (Mace and Purvis 2008; Pauls et al. 2013). However, ecological habit is often an important factor in influencing the survival of a species (Mace and Purvis 2008). A narrow and isolated geographical distribution may impose risk of extinction due to restricted gene flow resulting in low genetic heterozygosity thus low adaptability to changing environmental conditions (Ellis et al. 2006). Reed and Frankham (2003) reported a significant positive correlation between heterozygosity (gene diversity) and fitness in a meta-analysis of 34 plant and animal species. Lande (1988) argued that demographic factors may have more impact on population persistence than genetic factors. A number of studies proved that small-range species were more threatened by changing environments (Hering et al. 2009; Morueta-Holme et al. 2010; Pauls et al. 2013). Thus, both molecular factors (particularly potential of variability) and ecological habits of species should be comprehensively considered when planning germplasm conservation (Ellis et al. 2006; Mace and Purvis 2008; Pauls et al. 2013). Although the ecological diversity and geographical distribution of the flora of the whole genus of Vitis are broad, natural distribution of most species is limited within a mono or a few ecological regions (Reisch and Pratt 1996; Alleweldt 1997; Pavek et al. 2003; Wan et al. 2008). Considering their low genetic diversity (Table S9), low heterozygosity (Table S10), and narrow geographical distribution, these species, V. bashanica, V. piasezkii var. pagnuccii, V. acerifolia, V. adstricta, V. davidii, V. quinquangularis, should be given priority for collection and conservation. In addition to consideration of species-wide levels of diversity, extent of the partitioning of genetic variation and genetic differentiation between species is also important when designing a strategy for germplasm conservation, especially if not all species can be protected (Ellis et al. 2006). The species relatively highly distinct from other species in the PCoA plot, e.g., V. flexuosa, V. davidii var. cyanocarpa, Vitis palmate, Vitis vulpine and Vitis treleasei (Fig. 2d), may represent special genetic identities, thus these species should be highlighted for conservation. Although most species possess relatively high genetic diversity, collection and conservation of these species cannot be delayed because of increasing threats from human activity and climate changes (Pavek et al. 2003; Wan et al. 2008; Pauls et al. 2013). Collection and conservation of species from China should be emphasized because increase of human population and rapid expansion of industrialization present tremendous threats to the survival of these Chinese wild flora (Wan et al. 2008).
Grapevine is a perennial woody plant and thus it requires a relatively high cost for biodiversity conservation compared to herbaceous species (Pavek et al. 2003). Thus, a “core collection” strategy was postulated several decades ago for conservation of woody crops in order to conserve land and reduce costs (Pavek et al. 2003). However, no practical and feasible criteria and agreements have been decided on for a core collection of Vitis species due to extensive morphological diversity and high levels of interspecific hybridization in most species (Reisch and Pratt 1996; Alleweldt 1997). A number of species-specific SNPs were discovered and the patterns of relationships were clearly presented in this study. These results will provide basic tools for taxonomy, and germplasm conservation and development. Natural hybridization has been shown to commonly occur among species in Vitis (Wan et al. 2013), species unique SNPs detected in this study can be useful to classify accessions or discover whether accessions are “hybrid” or derived from several species. These SNPs can also be useful to help assemble a sample suitable for a “core collection” as a potential reference for germplasm collection, conservation, and breeding programs.
Conclusion
Although the grapevine is an economically important crop and use of wild species for improvement of the cultivated species is widely applied throughout the world (Reisch and Pratt 1996; Alleweldt 1997; Wan et al. 2008), comprehensive evaluation of genetic features in this genus using a large number of SNPs has not yet been conducted. This is first study using a large number of (6862) SNPs and a number of methods and estimates including estimation of genetic diversity, AMOVA, CV, and PCoA to comprehensively characterize genetic features among the hierarchical levels of Vitis species. Our data indicated that genetic diversity in most Vitis species is relatively high based on a comparison with that reported for other woody species. A molecular basis for high genetic diversity and genetic differentiation that together confer potential adaptability in Vitis was postulated in this study. Patterns in the PCoA plot represent genetic relationships, extent of differentiation of species and molecular identities for their origins from three geographic regions. Based on a comprehensive consideration of genetic features and ecological habits of Vitis species, the species with relatively low genetic diversity, low heterozygosity, and narrow geographic distribution should be given high priority for conservation. Given the ease of interspecific hybridization among most Vitis species and importance of novel agronomic traits for germplasm development (Reisch and Pratt 1996; Alleweldt 1997; Wan et al. 2008), the accessions with high agronomical values including resistance to multiple stresses and representing the genetic uniqueness inherent to the species should also be highlighted for conservation. Determination of species-specific SNPs in this study facilitates our identification of a “core collection” and detection of parentage in the grape breeding program. The strategy of vegetative propagation in this crop will facilitate the wide applicability of these markers in germplasm conservation and grape breeding programs. This study not only greatly adds to our knowledge of genetic features and molecular bases for ecological habits of Vitis species, but also is critical to conservation strategies and molecular tools for grape breeding programs.
References
Alleweldt G (1997) Genetics of grapevine breeding. Prog Bot 58:442–454
Aradhya M, Wang Y, Walker MA, Prins BH, Koehmstedt AM, Velasco D, Gerrath JM, Dangl GS, Preece JE (2012) Genetic diversity, structure, and patterns of differentiation in the genus Vitis. Plant Syst Evol. doi:10.1007/s00606-012-0723-4
Arroyo-Garcia R, Ruiz-Garcia L, Bolling L, Ocete R, López MA, Arnold C (2006) Multiple origin of cultivated grapevine (Vitis vinifera spp. sativa) based on chloroplast DNA polymorphism. Mol Ecol 15:3707–3714
Branca A, Paape TD, Zhou P, Briskine R, Farmer AD, Mudge J, Bhartid AK, Woodward JE, May GD, Gentzbittel L, Ben C, Denny R, Sadowsky MJ, Ronfort J, Bataillon T, Young ND, Tiffin P (2011) Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc Natl Acad Sci 108:E864–E870
Chen J, Källman T, Gyllenstrand N, Lascoux M (2010) New insights on the speciation history and nucleotide diversity of three boreal spruce species and a Tertiary relict. Heredity 104:3–14
Derero A, Gailing O, Finkeldey R (2011) Maintenance of genetic diversity in Cordia africana Lam., a declining forest tree species in Ethiopia. Tree Genet Genomes 1:1–9
DeWoody A, Bickham JW, Michler CH, Nichols KM (2010) Molecular approaches in natural resource conservation and management. Cambridge University, UK
El Oualkadi A, Ater M, Messaoudi Z, El Heit K, Laucou V, Boursiquot JM, Lacombe T, This P (2011) Genetic diversity of Moroccan grape accessions conserved ex situ compared to Maghreb and European gene pools. Tree Genet Genomes 6:1287–1298
Ellis JR, Pashley CH, Burke JM, McCauley DEM (2006) High genetic diversity in a rare and endangered sunflower as compared to a common congener. Mol Ecol 15:2345–2355
Emanuelli F, Lorenzi S, Grzeskowiak L, Catalano V, Stefanini M, Troggio M, Myles S, Martinez-Zapater JM, Zyprian E, Moreira FM, Grando MS (2013) Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol 13:39
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1:47–50
Filatov DA (2009) Processing and population genetic analysis of multigenic datasets with ProSeq3 software. Bioinformatics 25:3189–3190
Fitzpatrick EM (2009) Power and sample size for nested analysis of molecular variance. Mol Ecol 18:3961–3966
Grassi F, Labra M, Imazio S, Rubio RO, Failla O, Scienza A, Sala F (2006) Phylogeographical structure and conservation genetics of wild grapevine. Conserv Genet 6:837–845
Hadziabdic D, Wang XW, Wadl PA, Rinehart TA, Ownley BH, Trigiano RN (2012) Genetic diversity of flowering dogwood in the Great Smoky Mountains National Park. Tree Genet Genomes 4:855–871
Hendricks WA, Robey KW (1936) The sampling distribution of the coefficient of variation. Ann Math Stat 3:129–132
Hering D, Schmidt-Kloiber A, Murphy J (2009) Potential impact of climate change on aquatic insects: a sensitivity analysis for European caddisflies (Trichoptera) based on distribution patterns and ecological preferences. Aquat Sci 71:3–14
Heuertz M, Paoli ED, Källman T, Larsson H, Jurman I, Morgante M, Lascoux M, Gyllenstrand N (2006) Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174:2095–2105
Ingrouille MJ, Chase MW, Fay MF, Bowman D, van der Bank M, Bruijn ADE (2002) Systematics of Vitaceae from the viewpoint of plastid rbcl DNA sequence data. Biol J Linn Soc 138:421–432
Ismail M, Soolanayakanahally RY, Ingvarsson PK, Robert D, Guy RD, Jansson S, Silim SN, El-Kassaby YA (2012) Comparative nucleotide diversity across North American and European Populus species. J Mol Evol 74:257–272
Kekkonen J, Hanski IK, Jensen H, Väisänen RA, Brommer JE (2011) Increased genetic differentiation in house sparrows after a strong population decline: from panmixia towards structure in a common bird. Biol Conserv 144:2993–2940
Keller SR, Levsen N, Ingvarsson PK, Olson MS, Tiffin P (2011) Local selection across a latitudinal gradient shapes nucleotide diversity in balsam Poplar, Populus balsamifera L. Genetics 188:941–952
Krutovsky KV, Neale DB (2005) Nucleotide diversity and linkage disequilibrium in cold-hardiness- and wood quality-related candidate genes in Douglas fir. Genetics 171:2029–2041
Labate JA, Baldo AM (2005) Tomato SNP discovery by EST mining and resequencing. Mol Breeding 16:343–349
Lande R (1988) Genetics and demography in biological conservation. Science 241:1455–1460
Lijavetzky D, Cabezas JA, Ibanez A, Rodriguez V, Martinez-Zapater JM (2007) High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genomics 8:424
Liu A, Burke JM (2006) Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173:321–330
Lodhi MA, Ye GN, Weeden NF, Reish BI (1994) A simple and efficient method for DNA extraction from grapevine cultivars and Vitis species. Plant Mol Biol Report 12:6–13
López-Pujol J, Garcia-Jacas N, Susanna A, Vilatersana R (2012) Should we conserve pure species or hybrid species? Delimiting hybridization and introgression in the Iberian endemic Centaurea podospermifolia? Biol Conserv 152:271–279
Mace GM, Purvis A (2008) Evolutionary biology and practical conservation: bridging a widening gap. Mol Ecol 17:9–19
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Martinez LE, Cavagnaro PE, Masuelli RW, Zuniga M (2006) SSR-based assessment of genetic diversity in South American Vitis vinifera varieties. Plant Sci 6:1036–1044
Mayr E (1997) The objects of selection. Proc Natl Acad Sci U S A 6:2091–2094
Morgan MJ, Hunter D, Pietsch R, Osborne W, Keogh JS (2008) Assessment of genetic diversity in the critically endangered Australian corroboree frogs, Pseudophryne corroboree and Pseudophryne pengilleyi, identifies four evolutionarily significant units for conservation. Mol Ecol 17:3448–3463
Morueta-Holme N, Flojgaard C, Svenning JC (2010) Climate change risks and conservation implications for a threatened small-range mammal species. PLoS One 5:e10360
Myles S, Boyko AR, Owens CL, Brown PJ, Grassi F, Aradhya MK (2011) Genetic structure and domestication history of the grape. Proc Natl Acad Sci 108:3530–3535 doi:10.1073/pnas.1009363108
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nei M (1987) Molecular evolution genetics. Columbia University Press, New York
Nowak M (2006) Five rules for the evolution of cooperation. Science 5805:1560–1563
Orr H (2005) The genetic theory of adaptation, a brief history. Nat Rev Genet 2:119–127
Pauls SU, Nowak C, Bálint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol 22:925–946
Pavek DS, Lamboy WF, Garvey EJ (2003) Selecting in situ conservation sites for grape genetic resources in the USA. Genet Resour Crop Evol 50:165–173
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Ruibal L, Lindenmayer DB (2003) Spatial autocorrelation analysis offers new insights into gene flow in the Australian bush rat. Evolution 57:1182–1195
Péros JP, Berger G, Portemont A, Boursiquot JM, Lacombe T (2011) Genetic variation and biogeography of the disjunct Vitis subg. Vitis (Vitaceae). J Biogeogr 3:471–486
Pyhäjärvi T, García-Gil MR, Knürr T, Mikkonen M, Wachowiak W, Savolainen O (2007) Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 177:1713–1724
Rao VR, Hodgkin T (2002) Genetic diversity and conservation and utilization of plant genetic resources. Plant Cell Tissue Organ Cult 68:1–19
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Reisch BI, Pratt C (1996) Grapes. In: Janick J, Moore JN (eds) Fruit breeding, volume I: tree and tropical fruits. Wiley, New Jersey, pp 297–369
Riahia L, Zoghlamia N, Dereeperb A, Laucouc V, Mlikia A, This P (2013) Single nucleotide polymorphism and haplotype diversity of the gene NAC4 in grapevine. Ind Crop Prod 43:718–724
Rikkinen J, Virtanen V (2008) Genetic diversity in cyanobacterial symbionts of thalloid bryophytes. J Exp Bot:1013–1021
Rossetto M, McNally J, Henry RJ (2002) Evaluating the potential of SSR flanking regions for examining taxonomic relationships in the Vitaceae. Theor Appl Genet 104:61–66
Rozen S, Skaletsky H (2000) Primer 3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Salmaso M, Faes G, Segala C, Stefanini M, Salakhutdinov I, Zyprian E (2004) Genome diversity and gene haplotypes in the grapevine (Vitis vinifera L.), as revealed by single nucleotide polymorphisms. Mol Breeding 14:385–395
Schneider S, Roessli D, Excoffier L (2000) Arlequin ver (2.000), a software for population genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland
Soejima A, Wen J (2006) Phylogenetic analysis of the grape family (Vitaceae) based on three chloroplast markers. Am J Bot 93:278–287
Somers DJ, Kirkpatrick R, Moniwa M, Walsh A (2003) Mining single-nucleotide polymorphisms from hexaploid wheat ESTs. Genome 3:431–437
Song BH, Windsor AJ, Schmid KJ, Ramos-Onsins S, Schranz ME, Heidel AJ, Mitchell-Olds T (2009) Multilocus patterns of nucleotide diversity, population structure and linkage disequilibrium in Boechera stricta, a wild relative of Arabidopsis. Genetics 181:1021–1033
Straub SCK, Doyle JJ (2009) Conservation genetics of Amorpha georgiana (Fabaceae), an endangered legume of the Southeastern United States. Mol Ecol 18:4349–4365
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tenaillon MI, Sawkins MC, Long AD, Gaut RL, Doebley JF, Gaut BS (2001) Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays ssp. mays L.). Proc Natl Acad Sci 16:9161–9166
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W, improving the sensitivity of progressive multiple sequence alignmentthrough sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Torgerson WS (1958) Theory and methods of scaling. Wiley, New York
Tröndle D, SchrÖder S, Kassemeyer HH, Kiefer C, Koch MA, Nick P (2010) Molecular phylogeny of the genus Vitis Vitaceae. based on plastid markers. Am J Bot 97:1168–1178
Wachowiak W, Salmela MJ, Ennos RA, Iason G, Cavers S (2011) High genetic diversity at the extreme range edge: nucleotide variation at nuclear loci in Scots pine Pinus sylvestris L. in Scotland. Heredity 106:775–787
Wan Y, Li D, Schwaninger H, He P, Wang Y (2007) Comparison of resistance to powdery mildew and downy mildew in Chinese wild grapes. Vitis 3:132–136
Wan Y, Schwaninger H, Li D, Simon CJ, Wang Y, He P (2008) The eco-geographic distribution of wild grape germplasm in China. Vitis 47:77–80
Wan Y, Schwaninger H, Baldo AM, Labate JA, Zhong GY, Simon CJ (2013) A phylogenetic analysis of the grape genus (Vitis L.) reveals broad reticulation and concurrent diversification during neogene and quaternary climate change. BMC Evol Biol 13:141
Weir BS (1996) Genetic data analysis: methods for discrete population genetic DNA. Sinauer Association, Inc., Sunderland
Xin H, Zhang J, Zhu W, Wang N, Fang P, Han Y, Ming R, Li S (2013) The effects of artificial selection on sugar metabolism and transporter genes in grape. Tree Genet Genomes 9:1343–1349
Yokoyama S (2002) Evaluating adaptive evolution. Nat Genet 30:350–351
Zecca G, Abbott R, Sun WB, Spada A, Sala F, Grassi F (2012) The timing and mode of evolution of wild grapes Vitis. Mol Phylogenet Evol 62:736–747
Zhang DX, Hewitt GM (2003) Nuclear DNA analyses in genetic studies of populations, practice, problems and prospects. Mol Ecol 12:563–584
Zhao S, Zheng P, Dong S, Zhan X (2013) Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet 1:67–71
Zhu YL, Song QJ, Hyten DL, van Tassell CP, Matukumalli LK, Grimm DR (2003) Single-nucleotide polymorphisms in soybean. Genetics 3:1123–1134
Acknowledgments
We are grateful to Dr. Joanne A. Labate for making valuable comments and suggestions about this study, to Angela M. Baldo for designing primers for resequencing, to Dr. Peter Cousins (E. &. J. Gallo Winery) for providing expert advice on grape identification and classification, and to Dr. Gan-Yuan Zhong for valuable discussion. USDA provided equal contribution to this study. This work was supported by the National Yangling Bio-Tech Breeding Centre Project (2001-01), China Scholarship Council Project (22861057), the Young Scientist Foundation of NWAFU (QN2009-013), the 2012-Fund for Returnees Scientists from Foreign Study in Shaanxi Province (A289021201) and the USDA-ARS Plant Genetic Resources Unit CRIS Project 1910-21000-020-00D. USDA is an equal opportunity provider and employer.
Data archiving statement
All sequences were released in NCBI with NCBI accession numbers: [Genbank: JX952227-JX960379, EMBL: HF544510-HF544512]
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. B. Neale
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1046 kb)
Rights and permissions
About this article

Cite this article
Li, H., Wan, Yz. & Simon, C.J. Patterns of SNP distribution provide a molecular basis for high genetic diversity and genetic differentiation in Vitis species. Tree Genetics & Genomes 11, 13 (2015). https://doi.org/10.1007/s11295-015-0832-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-015-0832-7