
Abstract
Vibrio alginolyticus (V. alginolyticus) is a common pathogen in the ocean. In addition to causing serious economic losses in aquaculture, it can also infect humans. The rapid detection of nucleic acids of V. alginolyticus with high sensitivity and specificity in the field is very important for the diagnosis and treatment of infection caused by V. alginolyticus. Here, we established a simple, fast and effective molecular method for the identification of V. alginolyticus that does not rely on expensive instruments and professionals. The method integrates recombinase polymerase amplification (RPA) technology with CRISPR system in a single PCR tube. Using this method, the results can be visualized by lateral flow dipstick (LFD) in less than 50 min, we named this method RPA-CRISPR/Cas13a-LFD. The method was confirmed to achieve high specificity for the detection of V. alginolyticus with no cross-reactivity with similar Vibrio and common clinical pathogens. This diagnostic method shows high sensitivity; the detection limit of the RPA-CRISPR/Cas13a-LFD is 10 copies/µL. We successfully identified 35 V. alginolyticus strains from a total of 55 different bacterial isolates and confirmed their identity by (Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, MALDI-TOF MS). We also applied this method on infected mice blood, and the results were both easily and rapidly obtained. In conclusion, RPA-CRISPR/Cas13a-LFD offers great potential as a useful tool for reliable and rapid diagnosis of V. alginolyticus infection, especially in limited conditions.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vibrio alginolyticus is a member of Corynebacterium that widely exists in marine, estuarine and other aquatic environments worldwide. V. alginolyticus is a zoonotic pathogen (Wang et al. 2021a). It is one of the most important pathogens causing huge economic losses in the aquaculture industry (Gong et al. 2020a, b) and is the second leading cause of human Vibrio infection (Jacobs Slifka et al. 2017; Yin et al. 2022b). With global warming, its pathogenic period has been prolonged, and the infection rate has also increased significantly (Sheahan et al. 2022). V. alginolyticus infection often occurs in summer, usually due to the consumption of food contaminated by V. alginolyticus or exposure of wounds to the seawater environment (Baker-Austin et al. 2018). Similar to most Vibrio infections, infection with V. alginolyticus can cause foodborne diarrhea. However, non-foodborne infections caused by V. alginolyticus are more common, such as conjunctivitis, otitis media (common in children), extremity infections (common in the elderly, especially in the lower extremity) (Jacobs Slifka et al. 2017; Sheahan et al. 2022; Yin et al. 2022b), often accompanied by complications (hearing impairment, skin grafting, amputation, etc.) (Zhou et al. 2021; Hoefler et al. 2022; Yin et al. 2022b; Jacobs Slifka et al. 2017). In addition, individuals with other diseases (liver disease, heart disease, diabetes, etc.) or immunocompromised patients are more susceptible to infection, and severe cases can lead to bacteremia, which can be life-threatening (Brehm et al. 2021; Sheahan et al. 2022). V. alginolyticus is not routinely detected as a clinical pathogen (Sheahan et al. 2022). Cases requiring hospitalization and even surgery due to untimely detection of V. alginolyticus have been reported (Jacobs Slifka et al. 2017). Therefore, it is necessary to establish a rapid and sensitive detection method for V. alginolyticus to preserve human health and property.
At present, the main diagnostic methods for V. alginolyticus are pathogen isolation and identification, immunological diagnosis, and molecular biology diagnosis. The Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM) recommend the use of bacterial culture to diagnose V. alginolyticus infection (Baron et al. 2013). However, bacterial culture is time-consuming and complicated and needs to be completed by professionals. Immunological diagnosis usually requires the isolation and purification of high-quality antigens or the preparation of high-specificity and high-affinity antibodies (Chen et al. 2021). In addition, during the early stages of pathogen infection, the pathogen load in the body is relatively low, and antibody production may be delayed. Therefore, false negatives may be reported during the infection window period, which is not conducive to early rapid testing (Bonney et al. 2020). Similar biochemical properties among Vibrio spp., make it difficult to achieve rapid and specific detection of Vibrio with both pathogenic diagnosis and immunological diagnosis. PCR is a nucleic acid detection technique in clinical molecular diagnostics that has high specificity (Safiabadi Tali et al. 2021). However, it requires a professional fluorescence quantitative PCR instrument and must be completed by professional personnel in a special experimental environment. It is impossible to achieve rapid detection of V. alginolyticus nucleic acids in restricted areas. In recent years, thermostatic amplification techniques have been considered an important alternative to PCR techniques to achieve in situ detection (Safiabadi Tali et al. 2021). Thermostatic amplification techniques, such as loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA), are less dependent on specialized equipment and have a relatively short amplification time. Theoretically, they are ideal for rapid nucleic acid detection. However, these techniques still face certain shortcomings. It is prone to nonspecific amplification under constant temperature conditions. Because these methods are compatible with other detection systems, we can utilize this feature to establish detection methods that are more specific and sensitive.
The clustered regularly interspaced short palindromic sequence repeats-Cas (CRISPR-Cas) system is a part of natural adaptive immune response in many species of archaea and bacteria, against foreign bacteriophage and plasmid infections by cleaving their nucleic acid (Abudayyeh et al. 2016). Currently, gene editing technologies based on CRISPR/Cas systems have been used for rapid molecular diagnosis of different pathogens (Gootenberg et al. 2017). Cas proteins play a crucial role in the CRISPR/Cas system, enabling nucleic acid detection with high sensitivity and specificity in comparatively less time. The main Cas proteins used are Cas12a and Cas13a, which target the DNA and RNA systems, respectively. Both Cas12a proteins and Cas13a proteins have bidirectional cleavage activity. Once these Cas proteins are activated by the target, its “collateral” activity can continue to cleave any single-stranded DNA or RNA after cleaving the target sequence which can amplify the detection signal (Abudayyeh et al. 2016; Wang et al. 2021b, c; Zhang et al. 2021; Yin et al. 2022a). The main difference between the two proteins is the design of the crRNA. The design of crRNA based on Cas13a protein is simpler than that of Cas12a. Furthermore, research has shown that the Cas13a protein has higher sensitivity than Cas12a proteins (Sun et al. 2021).
In this study, we established a rapid and visualized RPA-CRISPR/Cas13a-LFD based nucleic acid detection method for V. alginolyticus. We used the conserved region of the gyrB gene as a target to design specific RPA primers and crRNA and used RPA technology to amplify the gene sequence of the pathogen. Then, we combined CRISPR/Cas13a to enhance the system’s sensitivity.
Materials and methods
Reagents and instruments
RPA reagents (TwistAmp Basic kit) were purchased from TwistDx Inc. (UK). T7 polymerase was purchased from Lucigen Corporation (USA). LwaCas13a protein was purchased from Nanjing Kingsray Biotechnology Co. (China), RNAse inhibitor was purchased from Takara Bio. Inc. (Japan), rNTP was purchased from New England Biolabs Inc. (USA), MgCl2 was purchased from Sigma Aldrich LLC. (USA), SuperReal fluorescence quantification premix reagent (probe method), Magnetic Bead Method Blood Genome Extraction Kit and Centrifuge Column Bacterial Genome DNA Extraction Kit were purchased from Tiangen Biotechnology Co. (China), and lateral flow dipsticks (Milenia HybriDetect) were purchased from Milenia Biotec Co. (Germany).
Primers, crRNA and reporter RNA
The gyrB genes of the V. alginolyticus strain ATCC 17749 EU680781.1, Vibrio parahaemolyticus strain ATCC 17802 AY527390.1, Vibrio fluvialis strain ATCC 33809 KF899127.1, Vibrio harveyi strain ATCC 33842 EU672845.1, and Vibrio vulnificus strain ATCC 27562 AY705491.1 were selected for comparison to screen for regions of differences to design specific RPA primers. Sequences of gyrB genes of different strains were obtained from NCBI and aligned using DNAMAN software (Fig. S1). RPA specific primers were designed targeting the gyrB gene conserved regions of V. alginolyticus, and 28 nucleotide sequences containing PFS (protospacer flanking sequence) sites were selected as the target sites for crRNA recognition, which were located at loci 2052–2080 of the gyrB gene (Fig. 1a, Fig. S2).

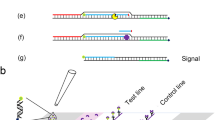
Cas13a-dependent nucleic acid detection process for Vibrio alginolyticus. a Design of crRNA. The target sites of crRNA recognized are shown in green, and the key PFS sites are shown in red. b Experimental procedure of V. alginolyticus detection. The genomic DNA of V. alginolyticus was extracted by conventional methods. RPA primers containing the T7 promoter were designed. The target fragment of the V. alginolyticus gyrB gene was amplified using RPA technology and transcribed into single-stranded RNA with the assistance of T7 polymerase. When the crRNA recognized the complementary amplification product, it was rapidly cleaved by the Cas13a protein. The “collateral” activity of the Cas13a protein was activated to cleave the reporter, enabling a signal cascade amplification effect
Oligo 7.0 software was used to design RPA primers(Li et al. 2019) for the V. alginolyticus gyrB gene conservative region. The primers, crRNA, and reporter were synthesized by Sangon Biotech (Shanghai) Co., Ltd. (China) (Table 1).
PCR system
TaqMan real-time PCR detection of the gyrB gene was carried out using an ABI 7500 (Applied Biosystems). Single-tube PCRs were prepared containing 10 μL 2× SuperReal PreMix (Probe), 1 μL primer–probe mix (primer 10 nM, probe 5 nM), 1 μL template, and 8 μL DEPC H2O. The reactions were incubated with an initial step of 95 °C for 5 min, followed by 40 cycles of 94 °C for 30 s and 58 °C for 40 s (TaqMan real-time PCR results are considered positive if the amplification peaks occur within 38 cycles).
RPA reaction system and transcription
Each RPA lyophilized reagent was added to 29.5 μL rehydration buffer, 2.1 μL forward primer (10 μM), 2.1 μL reverse primers (10 μM), 1.2 μL DEPC H2O, 3 μL T7 RNA polymerase (50 U/µL), and 4 μL RNA triphosphate mixture (25 mM), and the reagents were mixed by vortexing. Each lyophilization reagent was used for five tests in a 0.2 mL PCR tube. A reaction assembled with 1 μL DNA sample and 0.5 μL magnesium acetate (280 mM) was added as the last component to initiate the amplification reaction. The reactions were incubated at 39 °C for 20 min.
CRISPR/Cas13a detection system
Prior to the RPA reaction, the following mixture was added to the RPA reaction tube cap as the reaction system for CRISPR/Cas13a: 2.0 μL Tris (400 mM, pH 7.4), 1 μL MgCl2 (120 mM), 1 µL LwaCas13a (20 ng/μL), 1 µL recombinant RNase inhibitor (40 U/μL), 1 µL crRNA (10 ng/μL), and 1 µL lateral flow reporter (2 µM). After the RPA reaction, the sample was centrifuged briefly, mixing the reaction system on the tube cap with the RPA reaction product and enabling immediate reaction at 37 °C for 30 min. After the Cas reaction, 80 µL of HybriDetect Assay Buffer was added to each reaction tube, and then the test dipstick was inserted into the mixture, followed by reading 5 min later (Fig. 1b).
Limit of detection of the RPA-CRISPR/Cas13a-LFD
The plasmid used in this study is a plasmid fragments containing the target region of the gyrB gene of V. alginolyticus, synthesised by Sangon Biotech (Shanghai) Co., Ltd. (China). Dilute the synthesized plasmid to a concentration of 1 × 106 copies/µL to 1 copy/µL. The detection sensitivity of the RPA-CRISPR/Cas13a-LFD is based on plasmids at concentrations of 1 × 105 copies/µL to 1 copy/µL as templates.
Detection specificity of the RPA-CRISPR/Cas13a-LFD
The strains used for specificity verification in this study were V. parahaemolyticus, V. fluvialis, Vibrio melitensis, Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa, all of which were kept in the Six Medical Center of PLA General Hospital. The above strains were removed from −80 °C and inoculated on TCBS plates and blood plates for recovery. Individual colonies were picked for passaging, and then bacterial DNA was extracted according to the instructions of the Centrifuge Column Bacterial DNA Extraction Kit. The DNA was used as a template for PCR, RPA and RPA-CRISPR/Cas13a-LFD.
Verification of environmental strains
Fifty-five environmental strains (35 environmental strains of V. alginolyticus and 20 other environmental strains) were isolated from southeast China coastal waters by the Six Medical Center of PLA General Hospital, and all environmental strains were confirmed their identity by MALDI-TOF MS. The DNA extraction method for environmental strains was the same as above. All environmental strains were detected by RPA-CRISPR/Cas13a-LFD. The results were compared with the TaqMan real-time PCR method for the detection of environmental strains to confirm consistency.
Detecting V. alginolyticus in infected mice
Six to eight-week-old female BALB/c mice weighing approximately 20 g were purchased from the Bei Jing HFK bioscience Co., Ltd.. All animal procedures complied with the institutional and national guidelines prescribed by the International Council for Laboratory Animal Science (ICLAS) the ministry of health of the People’s Republic of China. The mice for were divided into two groups (sample group = 10, control group = 5). Mice of sample group were intraperitoneally injected with the V. alginolyticus above (1 × 106 CFU/mL) resuspended in normal saline, while control group was injected with normal saline. After the injection, each BALB/c mouse was fed with sterile water only. Sample group and control groups were carried out 16 h post-infection by collecting blood(Liu et al. 2014; Fu et al. 2016). Collection of blood using ep tubes containing EDTA anticoagulant, and then extract DNA from the blood for RPA-CRISPR/Cas13a-LFD detection.
Statistical analysis
The statistical analysis was performed using the SPSS software package, version 21.0 (IBM). The kappa statistic was used to compare the consistency of the two methods (Li et al. 2023). Degrees of agreement between RPA-CRISPR/Cas13a-LFD and TaqMan real-time PCR test results were measured using kappa (K) values, with K < 0.4 considered to be a poor agreement and K ≥ 0.75 considered to be a good agreement. Statistical significance was set at P < 0.05.
Results
Optimization of RPA primer concentration, RPA reaction temperature and crRNA to Cas13a protein concentration ratio
To visualize the optimization results, fluorescence signal detection was used to show the optimization results of each system. The results showed that the highest amplification efficiency was achieved when the RPA primer concentration was 10 μM (Fig. 2a) and the reaction temperature was 39 °C (Fig. 2b). The cleavage efficiency of the CRISPR system was highest when the concentration ratio of crRNA to Cas13a protein was 1:2 (Fig. 2c).

Optimization of RPA primer concentration, RPA reaction temperature and crRNA to Cas13a protein concentration ratio. a Optimization of RPA primer concentration. b Optimization of RPA reaction temperature. c Optimization of crRNA to Cas13a protein concentration ratio
Capability of the RPA-CRISPR/Cas13a-LFD for V. alginolyticus
The LOD of RPA-CRISPR/Cas13a-LFD was 10 copies/µL (Fig. 3a). The LOD of TaqMan real-time PCR was 1 × 102 copies/µL (Fig. 3c). V. alginolyticus strain was positively detected, whereas no other bacterial strain was detected by RPA-CRISPR/Cas13a-LFD (Fig. 3b).

Capability of the RPA-CRISPR/Cas13a-LFD and TaqMan real-time PCR for V. alginolyticus. a The limit of detection assessment of the RPA-CRISPR/Cas13a-LFD. b The specificity assessment of the RPA-CRISPR/Cas13a-LFD. c The limit of detection assessment of the TaqMan real-time PCR. A is 1 × 106 copies/µL, B is 1 × 105 copies/µL, C is 1 × 104 copies/µL, D is 1 × 103 copies/µL, E is 1 × 102 copies/µL, F is 1 × 101 copies/µL, and G is the negative control
Verification of environmental strains
RPA-CRISPR/Cas13a-LFD was used to detect 55 environmental strains from Chinese coastal water, of which 35 strains of environmental V. alginolyticus were all detected and 20 other strains were not detected. Fifty-five environmental strains were also tested using TaqMan real-time PCR, and their results were consistent with RPA-CRISPR/Cas13a-LFD (Fig. 4, Table 2).

Verification of the RPA-CRISPR/Cas13a-LFD for the detection of environmental V. alginolyticus
Detection of the infected blood samples
RPA-CRISPR/Cas13a-LFD was used to detect the blood of infected mice. Blood from six mice in the sample group were detected as V. alginolyticus infected, which the sensitivity of the RPA-CRISPR/Cas13a-LFD is 60%, and the specificity is 100% (Table 3). The RPA-CRISPR/Cas13a-LFD detection result was consistent with the results of TaqMan real-time PCR (the sensitivity of the TaqMan real-time PCR is 60%, and the specificity is 100%) (Table 4 and Table S1, Fig. S3), and the kappa value is 1 (Fig. 5).

The results of mice blood detection by RPA-CRISPR/Cas13a-LFD
Discussion
Vibrio alginolyticus is widely distributed in the ocean. Lack of awareness of the pathogenicity of V. alginolyticus and lack of timely diagnosis have led to huge economic losses and medical burdens (Weis et al. 2011; Sheahan et al. 2022). Rapid and accurate identification of pathogens is a prerequisite and basis for the treatment of infections. Routine detection methods are difficult to implement in nonspecialized laboratories, such as coastal and naval vessels. Therefore, it is important to establish a rapid and efficient nucleic acid on-site detection method for V. alginolyticus.
In this study, we developed a rapid and visual nucleic acid detection method for V. alginolyticus based on RPA thermostatic amplification technology and the CRISPR/Cas13a system, which can be completed in less than 50 min. The results were displayed by LFD, which showed excellent specificity with no cross-reactivity with similar species of Vibrio or other common clinical pathogens. The LOD of the RPA-CRISPR/Cas13a-LFD method was higher than that of the TaqMan real-time PCR method established, which is tenfold higher than that of the TaqMan real-time PCR method.
Selecting an appropriate detection gene as a target can improve the specificity of the entire experiment. Currently, the target genes used for molecular diagnosis of Vibrio species mainly include specific independent genes, such as the thermostable direct hemolysin (tdh) gene, TDH-related hemolysin (trh) gene, thermolabile hemolysin (tlh) gene, and toxR gene, which encodes the transmembrane transcription regulator (Vuddhakul et al. 2000; Tian et al. 2022). Housekeeping genes such as 16S ribosomal RNA (16S rRNA) and the gyrB gene (Vuddhakul et al. 2000; Liu et al. 2021) are also used. Due to the similarity of the coding sequences for virulence genes among Vibrio species, the 16S rRNA gene is commonly used as a target for nucleic acid detection. However, the highly conserved nature of the 16S rRNA gene limits the accuracy of identification between closely related species (Liu et al. 2021; Wu et al. 2022). In recent years, with the enrichment of genomic databases, the gyrB gene has been found to be significant for distinguishing closely related species (Liu et al. 2021). The gyrB gene is a commonly found single-copy gene in bacteria that encodes the DNA gyrase B subunit protein, as a housekeeping gene, which plays an important role in the DNA replication process (Tian et al. 2022). The gyrB gene is variable yet conserved, as its genetic code can undergo nucleotide substitutions without changing the translation results of the amino acid sequence. Therefore, this gene has certain value in the identification of Vibrio species (Wu et al. 2022). Ultimately, in this study, we designed and screened specific primers for the gyrB gene of V. alginolyticus.
In addition, we attempted to detect V. alginolyticus nucleic acids using the RPA technique alone (Fig. S4). The results showed that its sensitivity was comparable to that of the TaqMan real-time PCR. To improve sensitivity, we optimised the ratio of crRNA concentration to Cas13a protein concentration. The results showed that the best cleavage efficiency of the Cas13a protein was achieved when the ratio of crRNA concentration to Cas13a protein concentration was 1:2, which was consistent with the results reported by Hu et al. (2022).
Due to the late discovery of V. alginolyticus, only few studies testing V. alginolyticus in animal models and much less on mammalian models have been performed. In this study, in addition to testing pure bacterial cultures, we also tested in infected mice. Compared to the control group, the mice exhibited lethargy and decreased activity after being injected with V. alginolyticus. However, out of the ten infected mice, we only detected V. alginolyticus DNA in the blood of six mice. This could be due to the following reasons: (1) The infection model used in this study involved intraperitoneal injection, and some mice may not have developed bacteremia. (2) Blood contains phagocytes and white blood cells, which can engulf pathogens, leading to a decrease in the pathogen count in the blood.
In fact, we can further improve the above methods. In this study, we used traditional bacterial genomic kits to obtain V. alginolyticus genomic DNA, which was not conducive to field detection. Easy and rapid extraction of nucleic acids from the bacterial genome is the key to achieving rapid on-site diagnosis. Therefore, future work could attempt to use a simple and efficient crude nucleic acid extraction method combined with the RPA-CRISPR/Cas13a-LFD to complete pathogen detection. This would be better for the application of POCT-style nucleic acid rapid detection for pathogen screening in nonlaboratory conditions. Some researchers are concerned that the crude extracted genome of pathogens can introduce inhibitor-like substances that may influence the amplification results. Fortunately, the RPA technology still shows strong stability when conventional inhibitors (20 g/L haemoglobin, 0.5 U heparin, 1.25% urine) are present, which cannot be achieved by PCR (Kersting et al. 2014; Rosser et al. 2015).
In conclusion, compared to traditional molecular diagnostics, the RPA-CRISPR/Cas13a-LFD method we established offers the strengths of simplicity, rapidity, high specificity and high sensitivity. This work offers a possibility for rapid pathogen screening in nonlaboratory conditions such as within communities and aboard ships.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Abudayyeh OO, Gootenberg JS, Konermann S et al (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science (1979) 353:aaf5573. https://doi.org/10.1126/science.aaf5573
Baker-Austin C, Oliver JD, Alam M et al (2018) Vibrio spp. infections. Nat Rev Dis Primers 4:8. https://doi.org/10.1038/s41572-018-0005-8
Baron EJ, Miller JM, Weinstein MP et al (2013) A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2013 recommendations by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM)(a). Clin Infect Dis 57:e22–e121. https://doi.org/10.1093/cid/cit278
Bonney LC, Watson RJ, Slack GS et al (2020) A flexible format LAMP assay for rapid detection of Ebola virus. PLoS Negl Trop Dis 14:e0008496. https://doi.org/10.1371/journal.pntd.0008496
Brehm TT, Berneking L, Sena Martins M et al (2021) Heatwave-associated Vibrio infections in Germany, 2018 and 2019. Euro Surveill 26:2002041. https://doi.org/10.2807/1560-7917.ES.2021.26.41.2002041
Chen Z, Mao S, Zhang W et al (2021) Rapid visual detection method for barley yellow mosaic virus using reverse transcription loop-mediated isothermal amplification (RT-LAMP). Plant Dis 105:2658–2663. https://doi.org/10.1094/PDIS-06-20-1216-RE
Fu K, Li J, Wang Y et al (2016) An innovative method for rapid identification and detection of Vibrio alginolyticus in different infection models. Front Microbiol 7:651. https://doi.org/10.3389/fmicb.2016.00651
Gong Q, Yang D, Jiang M et al (2020a) l-Aspartic acid promotes fish survival against Vibrio alginolyticus infection through nitric oxide-induced phagocytosis. Fish Shellfish Immunol 97:359–366. https://doi.org/10.1016/j.fsi.2019.12.061
Gong QY, Yang MJ, Yang LF et al (2020b) Metabolic modulation of redox state confounds fish survival against Vibrio alginolyticus infection. Microb Biotechnol 13:796–812. https://doi.org/10.1111/1751-7915.13553
Gootenberg JS, Abudayyeh OO, Lee JW et al (2017) Nucleic acid detection with CRISPR-Cas13a/C2c2. Science (1979) 356:438–442. https://doi.org/10.1126/science.aam9321
Hu F, Liu Y, Zhao S et al (2022) A one-pot CRISPR/Cas13a-based contamination-free biosensor for low-cost and rapid nucleic acid diagnostics. Biosens Bioelectron 202:113994. https://doi.org/10.1016/j.bios.2022.113994
Jacobs Slifka KM, Newton AE, Mahon BE (2017) Vibrio alginolyticus infections in the USA, 1988–2012. Epidemiol Infect 145:1491–1499. https://doi.org/10.1017/S0950268817000140
Kersting S, Rausch V, Bier FF, Von Nickisch-Rosenegk M (2014) Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar J 13:99. https://doi.org/10.1186/1475-2875-13-99
Li J, Macdonald J, Von Stetten F (2019) Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 144:31–67
Li M, Gao Q, Yu T (2023) Kappa statistic considerations in evaluating inter-rater reliability between two raters: which, when and context matters. BMC Cancer 23(1):799
Liu XF, Zhang H, Liu X et al (2014) Pathogenic analysis of Vibrio alginolyticus infection in a mouse model. Folia Microbiol (praha) 59(2):167–171. https://doi.org/10.1007/s12223-013-0279-x
Liu Y, Pei T, Yi S et al (2021) Phylogenomic analysis substantiates the gyrB gene as a powerful molecular marker to efficiently differentiate the most closely related genera Myxococcus, Corallococcus, and Pyxidicoccus. Front Microbiol 12:763359. https://doi.org/10.3389/fmicb.2021.763359
Rosser A, Rollinson D, Forrest M, Webster BL (2015) Isothermal recombinase polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasit Vectors 8:446. https://doi.org/10.1186/s13071-015-1055-3
Safiabadi Tali SH, LeBlanc JJ, Sadiq Z et al (2021) Tools and techniques for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19 detection. Clin Microbiol Rev 34(3):e00228-e320. https://doi.org/10.1128/CMR.00228-20
Sheahan M, Gould CA, Neumann JE et al (2022) Examining the relationship between climate change and vibriosis in the United States: projected health and economic impacts for the 21st century. Environ Health Perspect 130:87007. https://doi.org/10.1289/EHP9999a
Sun Y, Yu L, Liu C et al (2021) One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J Transl Med 19:74. https://doi.org/10.1186/s12967-021-02741-5
Tian Z, Yang L, Qi X et al (2022) Visual LAMP method for the detection of Vibrio vulnificus in aquatic products and environmental water. BMC Microbiol 22(1):256. https://doi.org/10.1186/s12866-022-02656-1
Vuddhakul V, Nakai T, Matsumoto C et al (2000) Analysis of gyrB and toxR gene sequences of Vibrio hollisae and development of gyrB- and toxR-targeted PCR methods for isolation of V. hollisae from the environment and its identification. Appl Environ Microbiol 66(8):3506–3514. https://doi.org/10.1128/AEM.66.8.3506-3514.2000
Wang J, Ding Q, Yang Q et al (2021a) Vibrio alginolyticus triggers inflammatory response in mouse peritoneal macrophages via activation of NLRP3 inflammasome. Front Cell Infect Microbiol 11:769777. https://doi.org/10.3389/fcimb.2021.769777
Wang R, Qian C, Pang Y et al (2021b) opvCRISPR: one-pot visual RT-LAMP-CRISPR platform for SARS-cov-2 detection. Biosens Bioelectron 172:112766. https://doi.org/10.1016/j.bios.2020.112766
Wang Y, Li J, Li S et al (2021c) LAMP-CRISPR-Cas12-based diagnostic platform for detection of Mycobacterium tuberculosis complex using real-time fluorescence or lateral flow test. Mikrochim Acta 188:347. https://doi.org/10.1007/s00604-021-04985-w
Weis KE, Hammond RM, Hutchinson R, Blackmore CG (2011) Vibrio illness in Florida, 1998–2007. Epidemiol Infect 139:591–598. https://doi.org/10.1017/S0950268810001354
Wu Z, Wu Y, Gao H et al (2022) Identification and whole-genome sequencing analysis of Vibrio vulnificus strains causing pearl gentian grouper disease in China. BMC Microbiol 22(1):200. https://doi.org/10.1186/s12866-022-02610-1
Yin D, Yin L, Wang J et al (2022a) Visual detection of duck tembusu virus with CRISPR/Cas13: a sensitive and specific point-of-care detection. Front Cell Infect Microbiol 12:848365. https://doi.org/10.3389/fcimb.2022.848365
Yin Y, Yin Y, Yang H et al (2022b) Vibrio alginolyticus survives from ofloxacin stress by metabolic adjustment. Front Microbiol 13:818923. https://doi.org/10.3389/fmicb.2022.818923
Zhang W, Jiao Y, Ding C et al (2021) Rapid detection of tomato spotted wilt virus with Cas13a in tomato and Frankliniella occidentalis. Front Microbiol 12:745173. https://doi.org/10.3389/fmicb.2021.745173
Funding
This work was supported by the National Natural Science Foundation of China [No.81401311].
Author information
Authors and Affiliations
Contributions
Experimental protocol designed was performed by YW. Animal experiments was completed by YW, YH and XL. Experimental procedure was optimized by YH and XL. Environmental Vibrio alginolyticus strains identified were by NL and YD. Nucleic acid extracted from environmental Vibrio alginolyticus strains were by FL, WX and YZ. WX, JC, and CC supervised the project. YW wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Wang, Y., Hou, Y., Liu, X. et al. Rapid visual nucleic acid detection of Vibrio alginolyticus by recombinase polymerase amplification combined with CRISPR/Cas13a. World J Microbiol Biotechnol 40, 51 (2024). https://doi.org/10.1007/s11274-023-03847-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-023-03847-2