
Abstract
Structural engineering of the recombinant thrombolytic drug, Reteplase, and its cost-effective production are important goals in the pharmaceutical industry. In this study, a single-point mutant of the protein was rationally designed and evaluated in terms of physicochemical characteristics, enzymatic activity, as well as large-scale production settings. An accurate homology model of Reteplase was used as the input to appropriate tools to identify the aggregation-prone sites, while considering the structural stability. Selected variants underwent extensive molecular dynamic simulations (total 540 ns) to assess their solvation profile and their thermal stability. The Reteplase-fibrin interaction was investigated by docking. The best variant was expressed in E. coli, and Box-Behnken design was used through response surface methodology to optimize its expression conditions. M72R mutant demonstrated appropriate stability, enhanced enzymatic activity (p < 0.05), and strengthened binding to fibrin, compared to the wild type. The optimal conditions for the variant’s production in a bioreactor was shown to be 37 ºC, induction with 0.5 mM IPTG, for 2 h of incubation. Under these conditions, the final amount of the produced enzyme was increased by about 23 mg/L compared to the wild type, with an increase in the enzymatic activity by about 2 IU/mL. This study thus offered a new Reteplase variant with nearly all favorable properties, except solubility. The impact of temperature and incubation time on its large-scale production were underlined as well.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Biopharmaceuticals production is one of the major goals considered by drug manufacturers. Protein engineering technology plays a central role to achieve this goal by improving the functional properties of protein drugs, while maintaining their effectiveness (Assenberg et al. 2013). In order to eliminate the complex and expensive stages associated with recombinant protein production, strategies such as enhancing the physicochemical properties of engineered proteins, appropriate cell line choice, and optimization of culture medium as well as the expression conditions, can help achieve quality cost-effective production with higher yields of the final product (Gupta et al. 2019; Sinha and Shukla 2019). Computational methods are a notable hallmark in the pharmaceutical biotechnology research to design new types of protein drugs with enhanced features.
Reteplase (recombinant plasminogen activator, r-PA), a 39-kDa serine protease integrating the fibrin-binding Kringle2 domain, is used to treat thromboembolic disorders such as heart attack by converting plasminogen to plasmin and subsequent blood clot lysis through breaking the crosslinks of fibrin network (Chester et al. 2019). This recombinant drug has been produced as the third generation of thrombolytic agents, with faster activity and lower bleeding tendency in comparison with the second-generation variant, Alteplase. The expression of Reteplase in Escherichia coli (E. coli) with elimination of sugar side chains is a more economical method compared to the production of glycosylated forms, such as t-PA, in mammalian cell lines (Mohammadi et al. 2021). Additionally, compared to Alteplase and Streptokinase, it has lower hepatic clearance as well as lower molecular weight, and does not create allergic reactions (Adivitiya and Khasa 2017; Nordt and Bode 2003). However, the production and storage of Reteplase is problematic due to protein aggregation, a well-known challenge in pharmaceutical biotechnology (Ghaheh et al. 2020). Therefore, production of the active enzyme requires the use of time-consuming and expensive procedures such as protein extraction from E. coli, dissolving, refolding and purification.
Designing new enzyme variants with more solubility and stability in E. coli, will help mitigate the mentioned drawbacks by minimizing the need for the complex refolding process. In this study, we apply the rational mutagenesis as an approach to improve the solubility along with increasing the stability, as well as maintaining the biological activity of Reteplase. For this purpose, aggregation-prone regions were identified bioinformatically, while monitoring the protein stability, and taking into consideration to keep the functionally important residues intact (Ghaheh et al. 2020). A novel variant was introduced and underwent experiments on its biological and physicochemical properties. To reduce the production cost of Reteplase, optimization of the expression procedure is also essential. Accordingly, we integrated investigations to find the optimal conditions for the production of Reteplase variants in large-scale by the response surface methodology (RSM).
Materials and methods
Structural modeling of Reteplase
3D structure of Reteplase was constructed by Modeler v9.18 software using the structure of t-PA catalytic and Kringle-2 domains (PDB IDs 1BDA and 1PML) as templates with 100% sequence identities with the query. The best model with the lowest value of discrete optimized protein energy (DOPE) score was selected from among 1000 contrasted models, for the next step. The quality of the model was verified by PROCHECK software (Laskowski et al. 1993), VADAR algorithm (Willard et al. 2003) and Verify3D web server (Eisenberg et al. 1997).
Exploring for mutagenesis sites
To improve the biophysical properties, amino acid substitutions with optimal effect on protein stability were first identified based on the folding free energy changes (ΔΔG), using ERIS (http://eris.dokhlab.org) (Yin et al. 2007), POPMUSIC (https://soft.dezyme.com) (Dehouck et al. 2011), DUET (http://biosig.unimelb.edu.au/duet/) (Pires et al. 2014), and Elaspic (http://elaspic.kimlab.org/) (Witvliet et al. 2016).
AggreScan3D (http://biocomp.chem.uw.edu.pl/A3D/) was subsequently used in order to detect and eliminate the effect of aggregation-prone residues on the aggregation propensity (Gil-Garcia et al. 2018). This analysis was performed in a structure-based manner in dynamic mode, setting 10 Å as the default distance in the aggregation analysis for identifying the residues involved in the formation of aggregation prone regions. Due to the importance of protecting the active site residues in the protease family and preventing alteration of the enzyme activity, the server-proposed mutations ~ 7.5 Å away from the active site of the enzyme were identified through distance analysis, and residue substitutions interacting with the catalytic site were excluded (Ghaheh et al. 2019). Point mutations approved unanimously by all bioinformatics tools, were introduced into Reteplase using Rosetta Backrub program.
Molecular dynamic simulations and analyses
Molecular dynamic simulations were performed by Gromacs-5.1 software package (Van Der Spoel et al. 2005) under GROMOS96 (G43a1) force field (Wei et al. 2019) to evaluate the structural changes of wild type and designed mutant Reteplase variants at the atomic level. The 3D structure of Reteplase wild type and mutant models were solvated using the simple point charge model of water (SPC216) as solvent in a triclinic box with the 1.0 nm distance between the edges of the box and the surface of protein, where periodic boundary conditions were applied. Cl− ions were added to neutralize the total charge of each protein system. Energy minimization was first done by steepest descent algorithm to relax the substituted residues at their position. Then, the system was equilibrated with 500 ps simulation in the canonical (NVT) ensemble followed by 1000 ps simulation in the isothermal-isobaric (NPT) ensemble. Nose–Hoover thermostat and Parrinello-Rahman barostat were adopted to keep the temperature of 300 K and pressure of 1 bar at the system, respectively. Temperature and pressure coupling constants were considered 0.1 and 1 ps, respectively. The particle mesh Ewald (PME) method was applied for controlling the electrostatic interactions at cutoff distance of 10 Å; the same cutoff was used for van der Waals (vdW) interactions. Bonds involving a hydrogen atom were constrained by LINCS algorithm. In the production step, 20 ns MD simulations were applied to evaluate the dynamic behavior of Reteplase variants, compared to wild type. Analysis of the MD outputs included root mean squared deviation (RMSD), root mean squared fluctuation (RMSF), radius of gyration (Rg), number of hydrogen bonds, and average solvent accessible surface area (SASA) (Nemaysh and Luthra 2017; Sadrjavadi et al. 2020; Yu et al. 2020). The solvation free energy of wild type and mutant structures were calculated using APBS-1.5 software from the last ten structures of the MD trajectory (Baker et al. 2001). To assess the conformational stability, lambda parameter (Λ) (Zeiske et al. 2016) was computed as follows from output of 20-ns MD simulations at three temperatures: 300, 350 and 400 K:
S2 is the squared value of the so-called order parameter, which describes the orientational fluctuations of the backbone NH vectors, as obtained from MD simulations in low, medium and high temperatures. Λ values are then calculated by linear regression, where the quality of the fit is measured by the regression coefficient, R2 (Zeiske et al. 2016).
Molecular docking of Reteplase to fibrin
Fibrin has been shown as an important cofactor for t-PA-mediated plasminogen activation (Hudson 2017). Despite the slight changes of the Reteplase variants compared to the normal protein, we investigated the affinity of the new variants for binding to fibrin, to ensure that the plasminogen activation function of the enzyme would be preserved. The interaction of Reteplase variants with fibrin was assessed using HADDOCK 2.2 web server (Van Zundert et al. 2016), where residues 37–127 of Kringle2 domain of Reteplase and residues 148–160 of fibrin αC domain (PDB ID: 3GHG) were selected as residues actively contributing to the protein–protein binding.
Gene constructs
The bio-physicochemical properties of the designed variant of Reteplase with the best profile in the computational analyses were verified through experimental tests. The wild type and mutant protein encoding sequences integrating HindIII and XbaI restriction sites were designed using Vector NTI software (Lu and Moriyama 2004) to clone into the pDEST527 vector. A 6X His-tag and the TEV protease sequence was also added at the 5’ end of the sequence and the whole construct was inserted into pDEST527 vector, ordered to be synthesized by Iranian Institute of Cell & Gene Therapy (Tehran, Iran). In order to determine the accuracy of mutant Reteplase gene, digestion with HindIII and XbaI restriction enzymes and gel electrophoresis assays were performed, in addition to DNA sequencing.
Small-scale expression of wild type and mutant Reteplase
Reteplase variants were expressed after transformation (heat shock method) of E. coli BL21 (DE3) cells with each of the pDEST527-wild type r-PA and pDEST527-mutant r-PA vectors. In order to select the positive transformants, LB-agar plate containing 100 mg/ml ampicillin was used for overnight culture at 37 °C. Selected recombinant colonies were inoculated into 300 mL fresh medium culture. After reaching an OD600 of 0.6 in each culture, the expression was induced by 1 mM IPTG for a period of 4 h at 37 °C. The protein expression was evaluated by SDS-PAGE 12% followed by western blot analysis using anti-His antibody. The solubility of protein variants was determined based on the formation of inclusion bodies, represented by the presence of the protein in sample pellets instead of supernatants, as described in the next section.
Preparation, solubilization and refolding of inclusion bodies
Inclusion body preparation, purification and refolding were carried out according to our established protocol (Esmaili et al. 2018; Ghaheh et al. 2020). In brief, about 150 g of bacterial pellets were suspended in 13 ml of solubilizing buffer and three 30-s sonication cycles were run to cell lyses. Then, lysis buffer was added to the sample and incubated at room temperature for 45 min. This was followed by centrifuging for 20 min at 4 °C (11,000×g). The pellets were washed in 10 ml Triton buffer and the sample was re-sonicated as before. The pellets were then washed with non-triton buffer. The final pellet containing inclusion bodies was obtained after sonication and centrifugation. In order to solubilize the inclusion bodies, 6 M guanidine hydrochloride, 25 mM Tris–HCl, 10 mM EDTA and 1% β-mercaptoethanol were added. Reduced glutathione 1 mM, oxidized glutathione 0.1 mM, L-arginine 0.5 M, 0.01% tween 80 and bovine serum albumin 1 mg/mL, were used to refold the solubilized inclusion bodies and incubated at 22 °C overnight. Finally, the reducing agent and buffer components were separated using dialysis buffer containing Tris 0.1 M, EDTA 1 mM and L-arginine 0.5 M (pH 8). This step was repeated every one hour for three times at 4 °C and finally overnight. Refolded samples were stored at – 20 °C for further study. In each stage, the protein expression and inclusion body extraction were analyzed by 12% (v/v) SDS-PAGE.
Refolded Reteplase bioactivity assessment
Evaluation of the plasminogen activation by mutant Reteplase in comparison to the wild type was measured by AssaySense Human t-PA Chromogenic Activity Kit (AssayPro, USA). After determining the concentration of samples using absorbance measurement at 280 nm with Take3 plate of Epoch microplate reader (BioTek Instruments Inc. Winooski VT, USA), 20 μL of tPA standard and samples in concentration of 180 µg/mL were added to 80 μL of assay mix containing assay diluents (60 μL), plasminogen (10 μL) and plasmin substrate (10 μL). Finally, the plate containing samples was incubated at 37 °C in an incubator and the absorbance was measured at 405 nm at various time intervals for each sample.
The activity test was done in three independent experiments with 2 repeats for each sample and with various standards. Analysis of variance (ANOVA) as implemented in SPSS software (version 16; Chicago, IL, USA) was performed to analyze the differences between groups of repeated measures.
Experimental design and optimization of Reteplase production
The Box-Behnken experiment design method was implemented to evaluate the effect of three independent variables (temperature, IPTG concentration, time of incubation) on the expression of Reteplase using RSM. Each of the three variables was evaluated in three levels (coded with − 1, 0, 1) to explore the optimum condition (Table 1). Expression evaluation experiments were conducted based on the data obtained from Design Expert software (version 11; Stat-Ease Inc. Miniapolis, USA), applying 3 repeatable central points to increase precision and accuracy of the experiments.
Large-scale expression of the protein in bioreactor
Large-scale production of Reteplase was performed using 2-L autoclavable glass stirred-tank fermenter (BioG-Micom, Biotron Inc. Korea) with two six-blade Rush-ton turbines. In order to prepare pre-inoculum culture, 5 ml of wild type and mutated Reteplase were added to 150 ml of fresh LB medium separately, and incubated overnight at 37 ºC while shaking at 180 rpm. Then, each of the samples was added to LB broth with ampicillin in the final volume of 150 ml of over-night culture under sterile conditions to reach the final volume of 1500 ml. After reaching an OD600 of 0.6, the protein expression was induced by adding 1 mM IPTG for 4 h. Sterile HCl or NaOH solution (0.5 N) was automatically added to control the pH of media during the process.
Results
Quality validation for the 3D model of Reteplase
Ramachandran plot of Reteplase structural model using PROCHECK web server revealed the lowest DOPE score with high percentage of residues in the most favored regions (Fig. 1a). The overall G-factor of -0.37 showed that the model quality was acceptable. Concordantly, the analysis by Verify3D indicated that 89.27% of residues had average 3D-1D score ≥ 0.2. In addition, the ERRAT web server accepted the model by reporting the overall quality factor of 80.57. The final structure is illustrated in Fig. 1b.

Validation and display of the designed Reteplase. a Ramachandran plot generated for Reteplase. The plot shows that 83.2%, 15.5%, 0.7%, and 0.7% residues were located in most favored (red color), additionally allowed (brown), generously allowed (yellow) and disallowed regions (pale yellow), respectively. b The 3D model of Reteplase structure. The Kringle2 and catalytic serine protease domains are represented in turquoise and tan color, respectively. Catalytic triad residues are shown in ball and stick mode
Reteplase variants with enhanced stability and solubility
The consensus findings from the applied tools suggested eight new variants M72R, E275V, D342M, G368R, A231I, E214I, E295M and E119I, were the most stabilizing mutations with negative ΔΔG values (Table 2). Aggregation propensity analyses identified one point mutation in the Kringle2 domain of Reteplase, as seen in M72R, which contributed significantly to the protein solubility (Table 2).
Simulations and output analyses
MD simulations were performed to allow more profound structural, dynamic, and physicochemical analyses on all the eight designed variants. The analyses included dynamic parameters for the structures from the second half of the simulations, and bioinformatics predictions for the final coordinates (Table 3). Specifically, the solvation free energy was calculated to obtain an insight of variants’ solubility (Table 3). We also carried out stability analysis in different temperatures (300, 350 and 400 K), implementing simulations for an overall duration of 540 ns (9 variants in three temperatures each for 20 ns). The obtained Λ parameter is an inverse measure of order in macromolecular structures (Zeiske et al. 2016) (Table 3). Compared to the wild type, the best profile among all variants was demonstrated by M72R, particularly in terms of solvation free energy, water interactions (hydrophilic/hydrophobic surface, hydrogen bonds, vdW and electrostatic energies), and conformational stability (Λ and associated R2).
The structures reached a stable state after nearly 10 ns of simulation time, with the M72R variant showing a more stable dynamics in comparison to the wild type (Fig. 2a, Table 3). Radius of gyration (Fig. 2b, Table 3) demonstrated the Reteplase mutant was slightly more compact than the wild type, probably due to the effect of the mutation on amplification of intermolecular interactions. Local structural fluctuations as shown by RMSF plot (Fig. 2c) indicated no difference between wild type and M72R variants at the site of mutation in Kringle2 domain. These results were consistent with SASA (Fig. 2d) and lambda (Table 3) analyses, confirming higher stability and solubility of the new Reteplase variant. Considering the promising profile shown by M72R r-PA, this variant was considered for further tests on binding properties and biological activity.

Dynamics of the best Reteplase variant, in comparison with the wild type. Plots represent the a RMSD, b Radius of gyration, c RMSF, and d SASA trends for the wild type and M72R r-PA in 300 K
Molecular docking
The best cluster among HADDOCK clusters was considered for each Reteplase/fibrin complex. The results indicated a lower docking score and more favorable binding energy for M72R r-PA/fibrin, in comparison to the wild type/fibrin complex (Table 4). This indicates that the applied mutation in Kringle2 domain enhanced the binding of Reteplase to fibrin. Electrostatic interactions were shown to be the major energy contributing to the stronger binding of new variant to fibrin, compared to the wild type. In addition, the interaction was also reinforced by van der Waals nonpolar contributions (Table 4).
Enzyme expression, and refolding of inclusion bodies
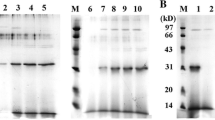
The presence of a band of about 1289 bp of the recombinant pDEST527 containing mutant Reteplase coding sequence was confirmed by restriction endonuclease digestion. The expression of mutant and wild type Reteplase with IPTG induction at 37 °C was verified by SDS-PAGE analysis with an estimated protein size about 43 kDa (Fig. 3a). As shown in Fig. 3b, there was no obvious expression of M72R Reteplase in soluble form. Furthermore, the results of the western blot analysis of the supernatant and bacterial pellets which were treated by anti-His HRP conjugated antibody, showed the related band for the pellet sample but not for the supernatant, indicating almost no soluble expression (Fig. 3c). The preparation and refolding of inclusion bodies was confirmed by SDS-PAGE analysis showing a band of about 43 kDa (Fig. 3d).

Evaluation of Reteplase expression on SDS-PAGE gel (12%). a SDS-PAGE analysis of wild type (wt) and M72R r-PA expression after induction. Lane 1: Protein marker (kDa), Lane 2: wild type r-PA, Lane 3: M72R r-PA; b SDS-PAGE analysis of wild type and M72R r-PA pellet and supernatant: Lane 1: Protein marker (kDa), Lane 2: Host cells supernatant containing pDest527-wt r-PA, Lane 3: Host cells pellet containing pDest527-wt r-PA, Lane 4: Host cells supernatant containing pDest527-M72R r-PA, Lane 5: Host cells pellet containing pDest527-M72R r-PA, Lane 6: Host cells supernatant containing BL21, Lane 7: Host cells pellet containing BL21; c Western blot analysis of wild type and M72R r-PA pellet and supernatant: Lane 1: Pre-stained protein marker (kDa), Lane 2: Host cells supernatant containing pDest527-wt r-PA, Lane 3: Host cells pellet containing pDest527-wt r-PA, Lane 4: Host cells supernatant containing pDest527-M72R r-PA, Lane 5: Host cells pellet containing pDest527-M72R r-PA, Lane 6: E. coli BL21 (DE3) cells supernatant (negative control); Lane 7: E. coli BL21 (DE3) cells Pellet (negative control); d SDS-PAGE of the inclusion bodies and refolded form from proteins extracted: Lane 1: Protein marker (kDa), Lane 2: Refolded wild type r-PA, Lane 3: Purified inclusion body of wild type r-PA, Lane 4: Refolded M72R r-PA, Lane 5: Purified inclusion body of M72R r-PA
Bioactivity assessment of wild type and mutated Reteplase
Figure 4 shows the enzymatic activity of mutant Reteplase was increased in comparison with the wild type (p < 0.05) either immediately after the dialysis or after two weeks of production. The maximum activity of wild-type and mutant Reteplase variants were 26.89 IU/mL and 28.99 IU/mL, respectively. Furthermore, the specific activity of each sample in 1.2 mg/ml concentration were obtained as 22.4 IU/mL and 24.15 IU/mL, respectively.

Comparison of the enzymatic activity up to 20 h. a Activity of Reteplase variants after dialysis. b Activity of Reteplase variants after 2 weeks of dialysis. Error bars shows mean ± SD; n = 3; p < 0.05 assumed as significant difference
RSM experimental design and modeling for expression optimization
After conducting 17 designed experiments, samples were analyzed by SDS-PAGE (Table 5, and Supplementary file 1–3). The concentration of Reteplase was considered as the chosen response to evaluate the effect of three variables i.e. temperature, IPTG concentration and induction time, on production of the enzyme. Based on the results of the experiments, the relation between production of wild type and mutated Reteplase variants and the three independent variables in fermentation (Table 1) can be explained by the following quadratic equations:
Analysis and validation of the expression models
The statistical significance of the quadratic expression models was determined by F-test and Analysis of Variance (ANOVA). The Lack of Fit test results obtained from the models showed p value of less than 0.05 for both wild type and mutant variants (Supplementary file 4 and 5, respectively). Therefore, the models were confirmed for predicting the effect of the independent variables on Reteplase expression. The R-squared values of 0.8936 and 0.8908, and F values of 15.93 and 15.51 confirmed the accuracy of the expression models, respectively for wild type r-PA and the new variant. Precision is also another index for signal to noise ratio. Precision greater than 4 shows the workability of the model for defining the design environment. This model had a proper accuracy of 14.733 and 14.791 for wild type and mutant Reteplase types, respectively, showing proper signal and suitable model.
The F-value of 15.93 (p < 0.0007) showed statistical significance of the expression model for the wild type enzyme. Based on this model, A, AB, AC, A2, B2 are significant parameters (p < 0.05), and B, C, C2 and BC are non-significant parameters (p > 0.05) in production of wild type Reteplase. Therefore, among the three independent variables, temperature had the greatest impact on the protein production (Supplementary file 4). For mutated Reteplase, the F value of 15.51 (p < 0.0007) showed the statistical significance of the relevant model. A, A2, B2, C2 and AC were significant parameters (p < 0.05), and BC, AB, B and C were non-significant (p > 0.05) in production of the new variant of Reteplase. Therefore, among the three factors investigated, the temperature of incubation and in the next rank, the time of incubation were shown to be the most contributing parameters to the expression of the new Reteplase variant (Supplementary File 5). The assumption of normality was investigated by the normal distribution plot of studentized residuals (Supplementary File 6).
Expression of Reteplase variants in bioreactor
Based on the introduced statistical models, the optimal conditions for the expression of Reteplase for the wild type and M72R variants were shown to be: induction time of 2 h, 0.55 mM IPTG, and temperature of 37 °C (Supplementary File 7). These conditions produced 375.64 mg/L of Reteplase for the wild type and 397.81 mg/L for the M72R variant in the bioreactor.
Discussion
Despite the lower probability of intra-cranial hemorrhage upon Reteplase administration and its lower hepatic clearance compared to its older ancestors (Chester et al. 2019), the production of this pharmaceutical protein is cost-intensive, and demands better strategies to produce it optimally. In addition to engineering new forms of the enzyme with improved properties, optimization of its large-scale expression is also a big step towards effective and economical production of this important bio-drug. In this study, we designed a new mutant form of Reteplase by inserting rational single-point mutations in both catalytic and Kringle2 domains, and evaluated the physicochemical profile of several variants.
Backbone deviations in the dynamics of mutant structures showed their more stable conformation than wild type r-PA. Consistent with each other, structural compaction and surface accessibility also reflected preserved structural conformation of the mutants in comparison with the normal enzyme (Kumar et al. 2014). The residue substitutions led to increased number of intra-molecular hydrogen bonds in some mutants (M72R, E214I and E295M). We also implemented extensive dynamic investigations to assess the stability of designed variants against a range of thermal conditions. These analyses revealed reduced fluctuations and increased order in the M72R and E214I mutants compared to wild type, as demonstrated by the decrease in Λ parameter value.
Protein solubility is an important characteristic to mitigate the problem of aggregation. Theoretically, the highly negative solvation free energy of M72R compared to the wild type indicated improved solubility upon replacement of the aggregation-prone residue, and this finding was consistent with other computational analyzes such as hydrophilic/hydrophobic surface and the number of inter-molecular hydrogen bonds. The new variant was thus presumably more soluble than wt r-PA. Replacing Met with the positively charged Arg residue on the protein surface could lead to strengthened hydrogen bonding and electrostatic interactions with the aqueous environment. Several studies suggested that arginine replacement is an effective strategy for enhancing the protein solubility and stability (Osire et al. 2019; Strickler et al. 2006; Turunen et al. 2002; Warwicker et al. 2014). Both mutant and wild type r-PA were expressed in E. coli BL21 (DE3) (Zhang et al. 2015) in high-level. However, contrary to the computational results and literature, our experiments indicated low solubilization of the protein (Fig. 3b, c). This may be due to the increased stabilization (Cabrita et al. 2007), specifically considering the presence of nine disulfide bonds in the r-PA structure.
The limited fluctuations in the three catalytic residues of new mutant variants of Reteplase compared with the wild type revealed that the introduced mutations will not interfere with its enzymatic activity. This was confirmed for M27R r-PA in the bioactivity assay, showing the improved bioactivity and specific activity of this variant compared to the wild type. In fact, charged hydrophilic amino acids, specifically arginine, can positively contribute to protein activity by improving its fold and strengthening the binding of clusters of water molecules to the protein surface via H-bonds and electrostatics (Mosavi and Peng, 2003; Sokalingam et al. 2012; Strickler et al. 2006; Turunen et al. 2002). In our previous study, similar results were reported regarding the important role of superficially charged amino acids in increasing protein catalytic function, which confirms the effective role of surface hydration in protein activity. In this study, fortnight storage of the enzyme led to no change in its activity trend, comparable to wild type r-PA, indicating considerable stability and conformational fidelity.
Reteplase binds to fibrin via Kringle2 domain, which is a lysine-dependent binding (Hudson 2017). The effect of the M27R mutation on Reteplase affinity to fibrin was investigated to check whether it would disturb r-PA’s binding to the αC domain fibrin. A more favorable binding of the new variant, than the wild type, to fibrin was observed, where electrostatics demonstrated the dominant contribution, probably due to the charged nature of inserted arginine residue.
Bioreactor systems allow larger-scale production of proteins, compared to shaking flasks (McNeil and Harvey 2008). Sadeghi et al. integrated the design of experiments to optimize the Reteplase expression in E. coli in bioreactors, to obtain higher protein yields (2011). Previous RSM experiments have shown that culture conditions and concentration of IPTG were the most effective factors on the protein expression (Bezerra et al. 2008; Chen et al. 2005; Gutiérrez-González et al. 2019). In the present study, the use of mathematical modeling for optimization of fermentation conditions (Shafiee et al. 2017) revealed the essential contribution of temperature and incubation time to the expression in E. coli BL21(DE3). High temperatures in microbial fermentation system can increase the reaction speed, enhance cell growth and protein synthesis, but it also can be harmful to expression rate, hence the optimum temperature for cell growth and accumulation of metabolites can be different (Zhou et al. 2018). Here, the optimal temperature was 37 °C, in accordance with similar experiments on Reteplase (Zare et al. 2019) and HSPA protein (Malik et al. 2016), and can be attributed to the effect of temperature on hydrophobic interactions and the protein aggregation (de Groot and Ventura 2006). Increasing IPTG concentrations up to 2 mM has been established to lead to increase in protein expression with no toxic effects on the producing host cells (Ramirez et al. 1994); however, the amount of IPTG is directly related to the inclusion body formation and the final production cost. In the current research, the highest yield of expressed protein was achieved with 0.5 mM of the inducer, which could reduce the final cost of the protein production.
Conclusion
The new Reteplase variant introduced by this study, M72R r-PA, features increased enzymatic activity, suitable stable structure, and more favorable interaction with fibrin than the wild type. The novel mutant, however, failed to show an efficient change in the protein solubility, which is a salient challenge in the expression of many recombinants, specifically Reteplase. On further process optimization, a high Reteplase titer of 423 mg/L was achieved. Our study also stressed the importance of the culture conditions on the r-PA expression rate at the large-scale. We suggest that the present M72R r-PA with increased activity can be considered for further modification in future experiments to design new soluble variants of this thrombolytic drug to treat acute myocardial infarction. We also hope that our research will serve as a basis for future studies on accelerating the process of optimal production of other recombinant pharmaceutical proteins.
References
Adivitiya, Khasa YP (2017) The evolution of recombinant thrombolytics: current status and future directions. Bioengineered 8(4):331–358. https://doi.org/10.1080/21655979.2016.1229718
Assenberg R, Wan PT et al (2013) Advances in recombinant protein expression for use in pharmaceutical research. Curr Opin Struct Biol 23(3):393–402. https://doi.org/10.1016/j.sbi.2013.03.008
Baker NA, Sept D et al (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci 98(18):10037–10041. https://doi.org/10.1073/pnas.181342398
Bezerra MA, Santelli RE et al (2008) Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 76(5):965–977. https://doi.org/10.1016/j.talanta.2008.05.019
Cabrita LD, Gilis D et al (2007) Enhancing the stability and solubility of TEV protease using in silico design. Protein Sci 16(11):2360–2367. https://doi.org/10.1110/ps.072822507
Chen X, Li Y et al (2005) Application of response surface methodology in medium optimization for spore production of Coniothyrium minitans in solid-state fermentation. World J Microbiol Biotechnol 21(4):593–599. https://doi.org/10.1007/s11274-004-3492-6
Chester KW, Corrigan M et al (2019) Making a case for the right ‘-ase’in acute ischemic stroke: alteplase, tenecteplase, and reteplase. Expert Opin Drug Saf 18(2):87–96. https://doi.org/10.1080/14740338.2019.1573985
de Groot NS, Ventura S (2006) Effect of temperature on protein quality in bacterial inclusion bodies. FEBS Lett 580(27):6471–6476. https://doi.org/10.1016/j.febslet.2006.10.071
Dehouck, Y., Kwasigroch, J. M., et al. (2011). PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC bioinformatics, 12(1), 1–12. https://doi.org/10.1186/1471-2105-12-151
Eisenberg D, Lüthy R et al (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 277:396–404. https://doi.org/10.1016/S0076-6879(97)77022-8
Esmaili I, Sadeghi HMM et al (2018) Effect of buffer additives on solubilization and refolding of reteplase inclusion bodies. Res Pharm Sci 13(5):413. https://doi.org/10.4103/1735-5362.236834
Ghaheh HS, Ganjalikhany MR et al (2019) Improving the solubility, activity, and stability of reteplase using in silico design of new variants. Res Pharm Sci 14(4):359. https://doi.org/10.4103/1735-5362.263560
Ghaheh HS, Ganjalikhany MR et al (2020) Investigation of supercharging as a strategy to enhance the solubility and plasminogen cleavage activity of Reteplase. Iran J Biotechnol 18(4):56. https://doi.org/10.30498/IJB.2020.2556
Gil-Garcia M, Bano-Polo M et al (2018) Combining structural aggregation propensity and stability predictions to redesign protein solubility. Mol Pharm 15(9):3846–3859. https://doi.org/10.1021/acs.molpharmaceut.8b00341
Gupta, S. K., Dangi, A. K., et al. (2019). Effectual bioprocess development for protein production. In Applied microbiology and bioengineering (pp. 203–227): Elsevier. https://doi.org/10.1016/B978-0-12-815407-6.00011-3
Gutiérrez-González M, Farías C et al (2019) Optimization of culture conditions for the expression of three different insoluble proteins in Escherichia coli. Sci Rep 9(1):1–11. https://doi.org/10.1038/s41598-019-53200-7
Hudson NE (2017) Biophysical mechanisms mediating fibrin fiber lysis. Biomed Res Int. https://doi.org/10.1155/2017/2748340
Kumar CV, Swetha RG et al (2014) Computational analysis reveals the association of threonine 118 methionine mutation in PMP22 resulting in CMT-1A. Adv Bioinform. https://doi.org/10.1155/2014/502618
Laskowski RA, MacArthur MW et al (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291. https://doi.org/10.1107/S0021889892009944
Lu G, Moriyama EN (2004) Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform 5(4):378–388. https://doi.org/10.1093/bib/5.4.378
Malik A, Alsenaidy AM et al (2016) Optimization of expression and purification of HSPA6 protein from Camelus dromedarius in E. coli. Saudi J Biol Sci 23(3):410–419. https://doi.org/10.1016/j.sjbs.2015.04.017
McNeil B, Harvey L (2008) Practical fermentation technology. Wiley, New York
Mohammadi E, Mahnam K et al (2021) Design and production of new chimeric reteplase with enhanced fibrin affinity: a theoretical and experimental study. J Biomol Struct Dyn 39(4):1321–1333. https://doi.org/10.1080/07391102.2020.1729865
Mosavi LK, Peng ZY (2003) Structure-based substitutions for increased solubility of a designed protein. Protein Eng 16(10):739–745. https://doi.org/10.1093/protein/gzg098
Nemaysh V, Luthra PM (2017) Computational analysis revealing that K634 and T681 mutations modulate the 3D-structure of PDGFR-β and lead to sunitinib resistance. RSC Adv 7(60):37612–37626. https://doi.org/10.1039/C7RA01305A
Nordt TK, Bode C (2003) Thrombolysis: newer thrombolytic agents and their role in clinical medicine. Heart 89(11):1358–1362. https://doi.org/10.1136/heart.89.11.1358
Osire T, Yang T et al (2019) Lys-Arg mutation improved the thermostability of Bacillus cereus neutral protease through increased residue interactions. World J Microbiol Biotechnol 35(11):1–11. https://doi.org/10.1007/s11274-019-2751-5
Pires DE, Ascher DB et al (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42(W1):W314–W319. https://doi.org/10.1093/nar/gku411
Ramirez O, Zamora R et al (1994) Kinetic study of penicillin acylase production by recombinant E. coli in batch cultures. Process Biochem 29(3):197–206. https://doi.org/10.1016/0032-9592(94)85004-6
Sadeghi HMM, Rabbani M et al (2011) Optimization of the expression of reteplase in Escherichia coli. Res Pharm Sci 6(2):87
Sadrjavadi K, Barzegari E et al (2020) Interactions of insulin with tragacanthic acid biopolymer: Experimental and computational study. Int J Biol Macromol 164:321–330. https://doi.org/10.1016/j.ijbiomac.2020.07.122
Shafiee F, Rabbani M et al (2017) Optimization of the expression of DT386-BR2 fusion protein in Escherichia coli using response surface methodology. Adv Biomed Res. https://doi.org/10.4103/2277-9175.201334
Sinha R, Shukla P (2019) Current trends in protein engineering: updates and progress. Curr Protein Pept Sci 20(5):398–407. https://doi.org/10.2174/1389203720666181119120120
Sokalingam S, Raghunathan G et al (2012) A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS ONE 7(7):e40410. https://doi.org/10.1371/journal.pone.0040410
Strickler SS, Gribenko AV et al (2006) Protein stability and surface electrostatics: a charged relationship. Biochemistry 45(9):2761–2766. https://doi.org/10.1021/bi0600143
Turunen O, Vuorio M et al (2002) Engineering of multiple arginines into the Ser/Thr surface of Trichoderma reesei endo-1, 4-β-xylanase II increases the thermotolerance and shifts the pH optimum towards alkaline pH. Protein Eng 15(2):141–145. https://doi.org/10.1093/protein/15.2.141
Van Der Spoel D, Lindahl E et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718. https://doi.org/10.1002/jcc.20291
Van Zundert G, Rodrigues J et al (2016) The HADDOCK2 2 web server: user-friendly integrative modeling of biomolecular complexes. Journal of molecular biology 428(4):720–725. https://doi.org/10.1016/j.jmb.2015.09.014
Warwicker J, Charonis S et al (2014) Lysine and arginine content of proteins: computational analysis suggests a new tool for solubility design. Mol Pharm 11(1):294–303. https://doi.org/10.1021/mp4004749
Wei Y, Yi Z et al (2019) Study on the binding characteristics of hydroxylated polybrominated diphenyl ethers and thyroid transporters using the multispectral technique and computational simulation. J Biomol Struct Dyn 37(6):1402–1413. https://doi.org/10.1080/07391102.2018.1461134
Willard L, Ranjan A et al (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 31(13):3316–3319. https://doi.org/10.1093/nar/gkg565
Witvliet DK, Strokach A et al (2016) ELASPIC web-server: proteome-wide structure-based prediction of mutation effects on protein stability and binding affinity. Bioinformatics 32(10):1589–1591. https://doi.org/10.1093/bioinformatics/btw031
Yin S, Ding F et al (2007) Modeling backbone flexibility improves protein stability estimation. Structure 15(12):1567–1576. https://doi.org/10.1016/j.str.2007.09.024
Yu Z, Liu Y et al (2020) Insights from molecular dynamics simulations and steered molecular dynamics simulations to exploit new trends of the interaction between HIF-1α and p300. J Biomol Struct Dyn 38(1):1–12. https://doi.org/10.1080/07391102.2019.1580616
Zare H, Sadeghi HMM et al (2019) Optimization of fermentation conditions for reteplase expression by Escherichia coli using response surface methodology. Avicenna J Med Biotechnol 11(2):162
Zeiske T, Stafford KA et al (2016) Thermostability of enzymes from molecular dynamics simulations. J Chem Theory Comput 12(6):2489–2492. https://doi.org/10.1021/acs.jctc.6b00120
Zhang Z, Kuipers G et al (2015) High-level production of membrane proteins in E. coli BL21 (DE3) by omitting the inducer IPTG. Microb Cell Factories 14(1):1–11. https://doi.org/10.1186/s12934-015-0328-z
Zhou Y, Han L-R et al (2018) Effects of agitation, aeration and temperature on production of a novel glycoprotein GP-1 by Streptomyces kanasenisi ZX01 and scale-up based on volumetric oxygen transfer coefficient. Molecules 23(1):125. https://doi.org/10.3390/molecules23010125
Acknowledgements
This study was supported by Isfahan University of Medical Sciences grant number 199477 (Ethical code: IR.MUI.RESEARCH.REC.1399.661) and the National Institute of Medical Research Development (NIMAD) grant number 958361.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Seyedhosseini Ghaheh, H., Sajjadi, S., Shafiee, F. et al. Rational design of a new variant of Reteplase with optimized physicochemical profile and large-scale production in Escherichia coli. World J Microbiol Biotechnol 38, 29 (2022). https://doi.org/10.1007/s11274-021-03204-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-021-03204-1