
Abstract
The compound γ-aminobutyric acid (GABA) was widely used in various fields. To enhance the production of GABA in Escherichia coli BL21(DE3), the enzymes of the regeneration pathway of the coenzyme factor pyridoxal 5’-phosphate (PLP) were engineered. The recombinant E. coli strain was screened and identified. The initial concentrations of L-monosodium glutamate (L-MSG) had an obvious influence on the production of GABA. The highest concentration of GABA in recombinant E. coli BL21/pET28a-gadA was 5.54 g/L when the initial L-MSG concentration was 10 g/L, whereas it was 8.45 g/L in recombinant E. coli BL21/pET28a-gadA-SNO1-SNZ1 at an initial L-MSG concentration of 15 g/L. The corresponding conversion yields of GABA in these two strains were 91.0% and 92.7%, respectively. When the initial concentrations of L-MSG were more than 15 g/L, the concentrations of GABA in E. coli BL21/pET28a-gadA-SNO1-SNZ1 were significantly higher as compared to those in recombinant E. coli BL21/pET28a-gadA, and it reached a maximum of 13.20 g/L at an initial L-MSG concentration of 25 g/L, demonstrating that the introduction of the enzymes of the regeneration pathway of PLP favored to enhance the production of GABA. This study provides new insight into producing GABA effectively in E. coli BL21(DE3).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The compound γ-aminobutyric acid (GABA) is a four-carbon non-protein amino acid and a natural active ingredient, which has the biological activities of improving brain blood circulation, lowering blood pressure, treating epilepsy and enhancing liver and kidney function (Li and Cao 2010; Luo et al. 2021; Shelp et al. 1999). Nowadays, GABA is used considerably in pharmaceutical, medical, cosmetics and feed industries (Barrett et al. 2012; Li et al. 2010; Luo et al. 2021; Park et al. 2005). It also was used as a major active constitute in foods, such as gammalone, cheese, gabaron tea, and shochu (Chamba and Irlinger 2004; Chemler and Koffas 2008; Chen et al. 2005).
Because GABA has numerous commercial uses, researchers have attempted to develop an efficient method for producing it (Park and Oh 2006; Shi et al. 2013; Yu et al. 2018). GABA can be not only produced by chemical synthesis and bioproduction, but also produced by plants enrichment and enzymatic method (Choi et al. 2006; Inoue et al. 2003; Luo et al. 2021). The chemical synthesis of GABA often suffers from a long process, waste of raw materials and many by-products. Thus, its biosynthesis may be more promising than the chemical synthesis since it has a simple reaction procedure, high catalytic efficiency, mild reaction condition and environmental compatibility (Huang et al. 2007).
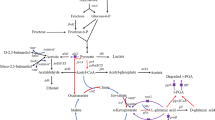
GABA can be synthesized by a one-step reaction of decarboxylating L-glutamate catalyzed via glutamate decarboxylase (Gad) (Fig. 1a) (Li and Cao 2010). However, this method has a critical problem that must be solved before becoming a competitive process for GABA production. It needs coenzyme factor pyridoxal 5′-phosphate (PLP) (Storici et al. 2004), and its supply is critical to enhance the production of GABA. PLP is the active form of vitamin B6, and is a coenzyme factor in a variety of enzymatic reactions including decarboxylation, transamination and racemization (Vacca et al. 2008). In the previous study, pyridoxal kinase was overexpressed in Lactobacillus plantarum CCTCC M209102 to enhance PLP production by the ATP- dependent phosphorylation of pyridoxal. Thus, this strain could efficiently convert L-glutamate to GABA (Zhang et al. 2014). E. coli cells overexpressing glutamate decarboxylase A (GadA) were also used as a whole-cell biocatalyst for catalyzing L-glutamate into GABA (Ke et al. 2016). However, the maintenance of the proper function of the PLP-dependent glutamate decarboxylase A (GadA) was needed during biocatalysis because of the low level of in vivo PLP, which required that the in vitro expensive PLP was supplemented. Thus, it is interesting to investigate if the introduction of the enzymes of the regeneration pathway of PLP into E. coli cells can enhance the production of GABA from L-glutamate by increasing endogenous PLP.

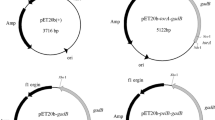
Biosynthetic pathways of γ-aminobutyric acid and pyridoxal 5’-phosphate, and conctruction of recombinant plasmids. a Bioconversion of L-glutamate to γ-aminobutyric acid; b biosynthetic pathway of pyridoxal 5’-phosphate; c construction of recombinant plasmids pET28a-gadA and pET28a-gadA-SNO1-SNZ1 The gene gadA encodes glutamate decarboxylase A (GadA). Two genes, SNZ1 and SNO1, respectively encode two subunits of pyridoxal 5’-phosphate synthase, Pdx1 and Pdx2. Three genes are in italic. PLP: pyridoxal 5’-phosphate; GABA: γ-aminobutyric acid
PLP biosynthetic pathway is widely distributed in bacteria, fungi and plants (Ehrenshaft et al. 1999; Mittenhuber 2001). However, it varies widely in different organisms. The native biosynthetic pathway of PLP in E. coli is involved in many key enzymes, and so it is difficult to engineer it for enhancing the production of PLP. In Saccharomyces cerevisiae SC288, PLP can be synthesized by only 5’-phosphate synthase that contains two subunits Pdx1 and Pdx2. PLP biosynthesis by Pdx1 requires the substrates ribose 5-phosphate (R5P) and glyceraldehyde 3-phosphate (Fitzpatrick et al. 2007). The heterocyclic nitrogen is derived from the hydrolysis of L-glutamine by Pdx2 (Tanaka et al. 2000). These two subunits form the PLP synthase complex, functionally classified as a glutamine aminotransferase (Guédez et al. 2012) (Fig. 1b). Genes SNO1 and SNZ1 respectively encode Pdx2 and Pdx1 in S. cerevisiae SC288 that participate in PLP biosynthesis (Dong et al. 2004).
In this study, the enzymes of the regeneration pathway of PLP from S. cerevisiae SC288 were attempted to introduce into E. coli BL21as a approach to synthesize endogenous PLP from its precursors in order to achieve a high production of GABA from L-glutamate. This approach was based on the PLP biosynthesis, in which Pdx1 and Pdx2, respectively encoded by SNZ1 and SNO1, were used to synthesize PLP to produce GABA, together with glutamate decarboxylase A (GadA). As a result, a system was developed using a salvage biosynthetic pathway to increase the cellular level of PLP, resulting in higher GABA production. To the best of our knowledge, this is the first report on enhancing the production of GABA in E. coli BL21 by engineering the enzymes of the regeneration pathway of PLP by introducing the pyridoxal 5’-phosphate synthase from S. cerevisiae SC288.
Materials and methods
Strains, plasmids and growth conditions
The strains and plasmids used in this study are listed in Table 1. E. coli BL21 (DE3) was used as the host strain for the expression of target genes. E. coli DH5α was used as the host strain for constructing recombinant plasmids. E. coli BL21/pET32a-gadABC was constructed before in our laboratory (Yu et al. 2018). Unless otherwise specified, all strains were cultured at 37 °C in a Luria–Bertani medium (LB: 10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl, pH 7.2). Ampicillin (100 mg/L), kanamycin (50 mg/L) and isopropyl β-D-thiogalactopyranoside (IPTG, 0.1 mM) were added for the plasmid stability maintenance and protein expression if required. IPTG, tryptone, yeast extract, NaCl, L-monosodium glutamate (MSG), ampicillin (Amp) and kanamycin (Kan) were purchased from the BBI Co. Ltd, USA. The plasmid purification kit, bacterial genomic DNA extraction kit, DNA fragment purification kit, agarose gel DNA extraction kit, restriction endonucleases and T4 DNA ligase were purchased from the TaKaRa, Co. Ltd, Japan. All other reagents were of analytical grade.
Construction and transformation of recombinant plasmids
All primer sequences are listed in Table 2, and were synthesized by Shanghai Sangon Biotech Co. Ltd, China. PCR reaction mixture includes 10 μL PCR buffer, 4 μL dNTP mixture, 2 μL template (100 ng/μL), 1 μL forward primer (20 μM), 1 μL reverse primer (20 μM), and 0.5 μL PrimeSTAR™ HS DNA polymerase. PCR amplification was performed for 30 cycles. Each cycle consisted of denaturation at 95 °C for 30 s, annealing at 55 °C for 15 s, and extension at 72 °C. The extension time varied with the length of PCR product (1 kb/min). The plasmid pET28a( +) was used to construct pET28a-gadA and pET28a-gadA-SNO1-SNZ1 (Fig. 1c).
To construct pET28a-gadA, the gene gadA was amplified using a primer set F1-gadA and R1-gadA. The plasmid pET32a-gadABC was extracted from recombinant E. coli BL21(DE3)/pET32a-gadABC (Yu et al. 2018), and was used as the template. The PCR product was digested with BamHI and SacI, and was ligated into the BamHI-SacI digested plasmid pET28a( +). The resultant plasmid was designated as pET28a-gadA (Fig. 1c).
The genomic DNA of S. cerevisiae SC288 was extracted as described by Looke et al. (2011), and was used as the template for amplifying the genes SNO1 and SNZ1. The PCR procedure was the same as that of gadA amplication except for primer sets. Primer sets, F1-SNO1/R1-SNO1 and F1-SNZ1/R1-SNZ1, were respectively used for the amplification of the genes SNO1 and SNZ1. The PCR products were sequentially ligated into pET28a-gadA to form pET28a-gadA-SNO1-SNZ1 (Fig. 1c). After being verified by DNA sequencing, the plasmid pET28a-gadA and pET28a-gadA-SNO1- SNZ1 were respectively transformed into E. coli BL21 (DE3) using CaCl2-heat shock method (Maniatis et al. 1982). The resultant strains were designated as E. coli BL21/pET28a-gadA and E. coli BL21/pET28a-gadA-SNO1-SNZ1, respectively.
Strain culture
Two hundred microliter of the glycerol-stocked cell cultures were inoculated into 10 mL LB medium, and were cultured overnight at 37 °C and 180 rpm for preparing seeds. Five percent of the cell cultures were transferred into 250 mL conical flasks pre-equipped with 50 mL fermentation medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, and different concentrations of L-MSG, pH 7.4), and were cultured for 36 h at 37 °C and 180 rpm. The pH of fermentation broth was adjusted to 7.0 by 5.0 M NaOH during the first 6 h of culture. Afterwards, IPTG (1 mM) was added when the OD600 of the cells was 0.8, and then pH was adjusted to 4.2 by 4 M HCl for producing GABA. One milliliter of the samples were taken for determining the cell optical density (OD600) and the concentrations of L-MSG and GABA. For investigating the effect of different concentrations of L-MSG on the production of GABA, L-MSG (5, 10, 15, 20 and 25 g/L) were used.
SDS-PAGE analysis of recombinant proteins
The recombinant protein SDS-PAGE analysis was performed using 15% separating gel and 5% stacking gel. Protein samples were prepared using a 10 × SDS buffer. After the sample was vortexed adequately, it was heated at 100 °C for 10 min, and was cooled in a ice water bath. The sample was centrifuged for 10 min at 15,000 × g. Twenty microliter of the samples were used for electrophoresis in a 5–15% polyacrylamide gel. After electrophoresis, the protein sample was stained with coomassie brilliant blue R250 (Laemmli 1970).
Determination of the cell optical density and concentrations of L-MSG and GABA
The cell optical density of E. coli was determined by measuring OD600 with UV-1800 spectrophotometer. The concentrations of the L-MSG and GABA were determined by HPLC after derivatization (Meeploy and Deewatthanawong 2015; Yu et al. 2019). One milliliter of the fermentation broth was taken and centrifuged for 5 min at 8000 × g. One hundred microliter of the supernatant was transferred into 1.5 mL centrifuge tube. Two hundred microliter of 1 M sodium carbonate-sodium bicarbonate buffer (pH 10.0) was added to the centrifuge tube. Afterwards, One hundred microliter of 80 g/L solution of dansyl chloride in acetone was added to the sample, followed by 600 μL of distilled water to form 1 ml of reaction system. Next, the sample derivatization was conducted by incubating it in a water bath for 40 min at 80 °C. The reaction was halted by adding 100 μL of 10% acetic acid. The sample was centrifuged for 5 min at 12,000 × g. The resultant supernatant was filtered by a 0.22 μm filter membrane.
The operation conditions for HPLC determination of L-MSG and GABA were as follows (Cho et al. 2007; Kim et al. 2009): the mobile phase A was methanol, and the mobile phase B contained 50 mM sodium acetate: methanol: tetrahydrofuran [1:15: 84, v/v] (pH 6.2). The gradient program was 80% A, 0–5 min; 80–45% A, 5–21 min; 45–0% A, 21–22 min; 0% A, 22–25 min. The flow rate of the mobile phase was 1 mL/min. The chromatographic column and UV detection wavelength were an Agilent Zorbax Eclipse Plus C18 (4.6 × 250 mm, 5 µm) and 254 nm, respectively. The injection volume was 15 µL and the column temperature was 30 °C.
The conversion yield of GABA was calculated according to the following equation:
where Y is the conversion yield of GABA from L-MSG, A is the theoretically calculated maximal mass concentration of GABA when L-MSG is depleted, and B is the mass concentration of the produced GABA.
Statistical analysis
Experiments were performed in triplicate, and the results are represented as the mean ± standard deviation. The statistical analysis of data was performed by the SPSS 17.0 software. The graphing was performed by the Origin 8.5 software.
Results
SDS-PAGE analysis of the protein expression of GadA, Pdx1 and Pdx2
The SDS-PAGE analysis of the recombinant protein expression is shown in Fig. 2. The genes gadA, SNZ1 and SNO1 encode the enzyme proteins GadA, Pdx1 and Pdx2, respectively. Their theroretically calculated molecular weights were 52.6, 38.3 and 27.8 kDa, respectively. It could be seen from Fig. 2 that the molecular weights of the expressed proteins GadA, Pdx1 and Pdx2 were in coincide with their respectively calculated ones, indicating that these three proteins were correctly expressed in recombinant strains. The enzyme GadA is responsible for the conversion of L-MSG to GABA. The other two enzymes, Pdx1 and Pdx2, are responsible for the biosynthesis of pyridoxal 5’-phosphate. The overexpression of these two enzymes could enhance the production of pyridoxal 5’-phosphate, and then improve the activity of GadA. When the activity of GadA was enhanced, it could catalyze the conversion of more L-MSG to GABA.

SDS-PAGE analysis of recombinant proteins lane M: protein standard sample; lane 1: the sample from the strain E. coli BL21; lane 2: the sample from the strain E. coli BL21/pET28a; lane 3: the sample from the strain E. coli BL21/pET28a-gadA; lane 4: the sample from the strain E. coli BL21/pET28a-gadA-SNO1-SNZ1
Production profiles of GABA in recombinant E. coli BL21/pET28a‑gadA
The cell optical density (OD600) and the concentrations of L-MSG and GABA were respectively determined during fermentation in recombinant E. coli BL21/pET28a‑gadA and E. coli BL21/pET28a‑gadA-SNO1-SNZ1. A major factor affecting GABA production by E. coli fermentation is the concentration of L-MSG. Thus, the effect of the L-MSG concentration on the production of GABA was investigated in detail.
Figure 3 shows the effect of different concentrations of L-MSG on the production of GABA in recombinant E. coli BL21/pET28a‑gadA. Overall, the concentration of L-MSG decreased as the fermentation time prolonged. When the initial concentration of L-MSG was 5.0 g/L, it was basically depleted at 9 h of fermentation. During this stage, the cell optical density and the concentration of GABA were increased, and reached 3.09 and 2.49 g/L, respectively. Afterwards, they kept relatively constant as the fermentation time prolonged (Fig. 3a). As the increase in the concentrations of L-MSG, the time when it was depleted was much prolonged. When the initial concentrations of L-MSG were 10 g/L, L-MSG was depleted at 25 h of fermentation. Meanwhile, the cell optical density and the concentration of GABA respectively reached maximum of 3.3 and 5.54 g/L (Fig. 3b), whereas they were 3.6 and 7.57 g/L at 35 h of fermentation when the initial concentration of L-MSG was 15 g/L (Fig. 3c). The highest concentrations of GABA of 9.03 and 9.76 g/L (p < 0.05) were respectively achieved at the initial L-MSG concentrations of 20 and 25 g/L (Fig. 3d-e). The conversion yield of GABA in recombinant E. coli BL21/pET28a‑gadA is shown in Fig. 3f. The conversion yield of GABA was increased when the concentrations of L-MSG varied from 0 to 10 g/L. It reached the highest value of 91% (p < 0.05) at an initial L-MSG concentration of 10 g/L. Afterwards, it was decreased as the concentrations of L-MSG were further increased.

Production profiles of GABA in E. coli BL21/pET28a-gadA at different concentrations of L-MSG. a 5 g/L of L-MSG; b 10 g/L of L-MSG; c 15 g/L of L-MSG; d 20 g/L of L-MSG; e 25 g/L of L-MSG; f the conversion yield of GABA at different concentrations of L-MSG
Production profiles of GABA in recombinant E. coli BL21/pET28a‑gadA-SNO1- SNZ1
The effect of different concentrations of L-MSG on the production of GABA was investigated in recombinant E. coli BL21/pET28a‑gadA-SNO1-SNZ1 containing the enzymes of the regeneration pathway of PLP, and the results are shown in Fig. 4. Overall, the concentrations of L-MSG had an obvious influence on the production of GABA. As the increase in the initial concentrations of L-MSG from 5 g/L to 25 g/L, the highest concentrations of GABA increased from 2.58 g/L to 13.20 g/L. When the initial concentration of L-MSG was 5 g/L, it was depleted at 12 h of fermentation. The concentration of GABA and cell optical density also reached maximum of 2.58 g/L and 2.26, respectively. Afterwards, they basically kept relatively constant (Fig. 4a). When the concentrations of L-MSG varied from 10 g/L to 20 g/L, the highest concentrations of GABA were increased from 5.41 g/L and 11.05 g/L (Fig. 4b–d). When the concentration of L-MSG was 25 g/L, it was depleted at 25 h of fermentation, and the concentration of GABA reached the highest value of 13.20 g/L (p < 0.05). The cell optical density was increased from 5 to 25 h of fermentation, and reached a maximum of 3.54 (Fig. 4e). Figure 4f shows the variation in the conversion yield of GABA at different concentrations of L-MSG. When the concentrations of L-MSG increased from 5 g/L to 15 g/L, the conversion yield of GABA increased from 84.96% to 92.7% (p < 0.05). The maximal GABA conversion yield of 92.7% was achieved at an initial L-MSG concentration of 15 g/L. Afterwards, it declined as the increase in the concentrations of L-MSG.

Production profiles of GABA in E. coli BL21/pET28a-gadA-SNO1-SNZ1 at different concentrations of L-MSG. a 5 g/L of L-MSG; b 10 g/L of L-MSG; c 15 g/L of L-MSG; d 20 g/L of L-MSG; e 25 g/L of L-MSG; f the conversion yield of GABA at different concentrations of L-MSG
Comparison of the GABA concentrations
The highest concentrations of GABA obtained at different initial concentrations of L-MSG were compared, and the result is shown in Fig. 5. When the initial concentrations of L-MSG were 5 and 10 g/L, the highest concentrations of GABA showed no obvious difference in recombinant E. coli BL21/pET28a‑gadA and E. coli BL21/pET28a‑gadA-SNO1-SNZ1. However, when the initial concentrations of L-MSG were more than 10 g/L, it was significantly observed that the concentrations of GABA were higher in recombinant E. coli BL21/pET28a‑gadA-SNO1-SNZ1 than those in recombinant E. coli BL21/pET28a‑gadA. Especially, when the initial concentration of L-MSG was 25 g/L, the concentration of GABA reached 13.20 g/L in recombinant E. coli BL21/pET28a‑gadA-SNO1-SNZ1, which was increased by 35.2% (p < 0.05) as compared to that (9.76 g/L) in recombinant E. coli BL21/pET28a‑gadA. Taken together, these results demonstrated that the introduction of the enzymes of the regeneration pathway of PLP in recombinant E. coli BL21/pET28a‑gadA-SNO1- SNZ1 was beneficial to enhance the production of GABA.

Comparison of the GABA production in E. coli BL21/pET28a-gadA and E. coli BL21/pET28a-gadA-SNO1-SNZ1
Discussion
Pyridoxal 5’-phosphate is the coenzyme factor of glutamate decarboxylase that can catalyze the conversion of the substrate L-MSG to GABA (Spink et al. 1985; Strausbauch and Fischer 1970). In S. cerevisiae SC288, pyridoxal 5’-phosphate synthase, which contained two subunits Pdx1 and Pdx2, plays a key role in the regeneration of the coenzyme factor PLP (Strohmeier et al. 2006; Zhang et al. 2010). In order to enhance the bioconversion of L-MSG to GABA, pyridoxal 5’-phosphate synthase was overexpressed in E. coli BL21 in this study by introducing its two subunits-encoding genes SNZ1 and SNO1. E. coli BL21 contains the substrates that are required for the biosynthesis of PLP, glyceraldehyde-3-phosphate and glutamine (Käck et al. 1999; Mukherjee et al. 2011). Thus, the entire regeneration pathway of PLP could be established in E. coli BL21. The regeneration of PLP could provide the adequate coenzyme factor for enhancing the activity of glutamate decarboxylase A. As a result, the recombinant strain E. coli BL21/pET28a-gadA-SNO1-SNZ1 produced 13.20 g/L of GABA, which was 35.2% (p < 0.05) higher as compared to that (9.76 g/L) in the strain E. coli BL21/pET28a-gadA. Thus, we succeed in enhancing the bioconversion of L-MSG to GABA by introducing the enzymes of the regeneration pathway of PLP in E. coli BL21. This process does not require the addition of in vitro expensive coenzyme factor PLP, and so displays an enormous potential towards reducing the production cost of GABA. Besides pyridoxal 5’-phosphate synthase used in this study, pyridoxal kinase from L. plantarum is an ideal alternative that can catalyze the conversion of pyridoxal to PLP. Corynebacterium glutamicum G01 can efficiently convert glucose to the L-glutamate, but it can not acculumate GABA directly from L-glutamate because it lacks of glutamate decarboxylase and pyridoxal kinase. In order to realize the efficient one-step production of GABA from glucose without the addition of exogenous PLP, the metabolic pathway from L-glutamate to GABA from L. plantarum was grafed into C. glutamicum by overexpressing glutamate decarboxylase and pyridoxal kinase. As a result, the recombinant C. glutamicum produced 70.6 g/L of GABA (Zhang et al. 2014). Although the production strain was different from E. coli, the idea for the GABA production was somewhat similar. Taken together, these results demonstrate that pyridoxal 5’-phosphate synthase and pyridoxal kinase are two good candidates for enhancing the biosynthesis of the endogenous PLP in microorganisms, a cofactor of glutamate decarboxylase required for the efficient conversion of L-glutamate to GABA.
Owing to considerable uses of GABA in many fields, various studies have been conducted to enhance its production. Through the expression of rice glutamate decarboxylase in Bifidobacterium longum, GABA (0.1 g/L) was obtained from 30 g/L of L-MSG (Park et al. 2005). GABA (0.4 g/L) was produced from 30 g/L of L-MSG in Bacillus subtilis by expressing glutamate decarboxylase from Lactobacillus brevis (Park and Oh 2006). By expressing glutamate decarboxylase from L. plantarum in L. sakei, the conversion yield of GABA was enhanced by 1.35-fold (Kook et al. 2010). When E. coli XL1 glutamate decarboxylase B was introduced into E. coli XB, the recombinant strain produced 5.09 g/L of GABA (Le Vo et al. 2012). In the above-mentioned studies, only glutamate decarboxylase was expressed in heterologous host cells. Moreover, only endogenous PLP from native metabolic pathways of host cells was used during the bioconversion of L-MSG to GABA. These led to a low conversion yield of GABA from L-MSG. Besides expressing glutamate decarboxylase A, the enzymes of the regeneration pathway of its coenzyme factor PLP from S. cerevisiae SC288 were introduced into E. coli BL21 in this study. This not only enhances the activity of glutamate decarboxylase A, but also eliminates the use of in vitro expensive PLP. Through these efforts, we succeeded in producing 13.20 g/L of GABA in this study.
In conclusion, the production of GABA in E. coli BL21(DE3) was enhanced by introducing the enzymes of the regeneration pathway of the coenzyme factor PLP from S. cerevisiae SC288. Thus, our study provides a new method that may be very useful for the bioconversion of L-MSG to GABA by a single recombinant E. coli strain containing the enzymes of the regeneration pathway of PLP. This study also lays good foundation for the industrial production of GABA from L-MSG by recombinant E. coli in future.
References
Barrett E, Ross R, O’toole P, Fitzgerald G, Stanton C (2012) γ-Aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol 113:411–417
Chamba J, Irlinger F (2004) Secondary and adjunct cultures. Cheese: Chem. Phys Microbiol 1:191–206
Chemler JA, Koffas MA (2008) Metabolic engineering for plant natural product biosynthesis in microbes. Curr Opin Biotechnol 19:597–605
Chen KN, Chen MJ, Liu JR, Lin CW, Chiu HY (2005) Optimization of incorporated prebiotics as coating materials for probiotic microencapsulation. J Food Sci 70:M260–M266
Cho YR, Chang JY, Chang HC (2007) Production of gamma-aminobutyric acid (GABA) by Lactobacillus buchneri isolated from kimchi and its neuroprotective effect on neuronal cells. J Microbiol Biotechnol 17:104–109
Choi SI, Lee JW, Park SM, Lee MY, Ji GE, Park MS, Heo TR (2006) Improvement of gamma-aminobutyric acid (GABA) production using cell entrapment of Lactobacillus brevis GABA 057. J Microbiol Biotechnol 16:562–568
Dong YX, Sueda S, Nikawa JI, Kondo H (2004) Characterization of the products of the genes SNO1 and SNZ1 involved in pyridoxine synthesis in Saccharomyces cerevisiae. Eur J Biochem 271:745–752
Ehrenshaft M, Bilski P, Li MY, Chignell CF, Daub ME (1999) A highly conserved sequence is a novel gene involved in de novo vitamin B6 biosynthesis. Proc Natl Acad Sci USA 96:9374–9378
Fitzpatrick TB, Amrhein N, Kappes B, Macheroux P, Tews I, Raschle T (2007) Two independent routes of de novo vitamin B6 biosynthesis: not that different after all. Biochem J 407:1–13
Guédez G, Hipp K, Windeisen V, Derrer B, Gengenbacher M, Böttcher B, Sinning I, Kappes B, Tews I (2012) Assembly of the eukaryotic PLP-synthase complex from Plasmodium and activation of the Pdx1 enzyme. Structure 20:172–184
Huang J, Mei LH, Wu H, Lin DQ (2007) Biosynthesis of γ-aminobutyric acid (GABA) using immobilized whole cells of Lactobacillus brevis. World J Microbiol Biotechnol 23:865–871
Inoue K, Shirai T, Ochiai H, Kasao M, Hayakawa K, Kimura M, Sansawa H (2003) Blood-pressure-lowering effect of a novel fermented milk containing γ-aminobutyric acid (GABA) in mild hypertensives. Eur J Clin Nutr 57:490
Käck H, Sandmark J, Gibson K, Schneider G, Lindqvist Y (1999) Crystal structure of diaminopelargonic acid synthase: evolutionary relationships between pyridoxal-5′-phosphate-dependent enzymes. J Mol Biol 291:857–876
Ke C, Yang X, Rao H, Zeng W, Hu M, Tao Y, Huang J (2016) Whole-cell conversion of L-glutamic acid into gamma-aminobutyric acid by metabolically engineered Escherichia coli. Springerplus 5:591
Kim JY, Lee MY, Ji GE, Lee YS, Hwang KT (2009) Production of γ-aminobutyric acid in black raspberry juice during fermentation by Lactobacillus brevis GABA100. Int J Food Microbiol 130:12–16
Kook MC, Seo MJ, Cheigh CI, Lee SJ, Pyun YR, Park H (2010) Enhancement of γ-amminobutyric acid production by Lactobacillus sakei B2–16 expressing glutamate decarboxylase from Lactobacillus plantarum ATCC 14917. J Kor Soc Appl Biol Chem 53:816–820
Lõoke M, Kristjuhan K, Kristjuhan A (2011) Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques 50:325–328
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Le Vo TD, Kim TW, Hong SH (2012) Effects of glutamate decarboxylase and gamma-aminobutyric acid (GABA) transporter on the bioconversion of GABA in engineered Escherichia coli. Bioproc Biosyst Eng 35:645–650
Li H, Cao Y (2010) Lactic acid bacterial cell factories for gamma-aminobutyric acid. Amino Acids 39:1107–1116
Li H, Qiu T, Huang G, Cao Y (2010) Production of gamma-aminobutyric acid by Lactobacillus brevis NCL912 using fed-batch fermentation. Microb Cell Fact 9:85
Luo H, Liu Z, Xie F, Bilal M, Liu L, Yang R, Wang Z (2021) Microbial production of gamma-aminobutyric acid: applications, state-of-the-art achievements, and future perspectives. Crit Rev Biotechnol 41:491–512
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual, vol 545. Cold spring harbor laboratory Cold Spring Harbor, NY
Meeploy M, Deewatthanawong R (2015) Determination of γ-aminobutyric acid (GABA) in rambutan fruit cv. Rongrian by HPLC-ELSD and separation of GABA from rambutan fruit using Dowex 50W–X8 column. J Chromatogr Sci 54:445–452
Mittenhuber G (2001) Phylogenetic analyses and comparative genomics of vitamin B6 (pyridoxine) and pyridoxal phosphate biosynthesis pathways. J Mol Microbiol Biotechnol 3:1–20
Mukherjee T, Hanes J, Tews I, Ealick SE, Begley TP (2011) Pyridoxal phosphate: biosynthesis and catabolism. Biochim Biophys Acta (BBA)-Proteins and Proteomics 1814:1585–1596
Park KB, Ji GE, Park MS, Oh SH (2005) Expression of rice glutamate decarboxylase in Bifidobacterium longum enhances γ-aminobutyric acid production. Biotechnol Lett 27:1681–1684
Park KB, Oh SH (2006) Enhancement of γ-aminobutyric acid production in Chungkukjang by applying a Bacillus subtilis strain expressing glutamate decarboxylase from Lactobacillus brevis. Biotechnol Lett 28:1459–1463
Shelp BJ, Bown AW, McLean MD (1999) Metabolism and functions of gamma- aminobutyric acid. Trends Plant Sci 4:446–452
Shi F, Jiang J, Li Y, Li Y, Xie Y (2013) Enhancement of γ-aminobutyric acid production in recombinant Corynebacterium glutamicum by co-expressing two glutamate decarboxylase genes from Lactobacillus brevis. J Ind Microbiol Biotechnol 40:1285–1296
Spink DC, Porter TG, Wu S, Martin DL (1985) Characterization of three kinetically distinct forms of glutamate decarboxylase from pig brain. Biochem J 231:695–703
Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, Silverman RB, Schirmer T (2004) Structures of γ-aminobutyric acid (GABA) aminotransferase, a pyridoxal 5′-phosphate, and [2Fe-2S] cluster-containing enzyme, complexed with γ-ethynyl-GABA and with the antiepilepsy drug vigabatrin. J Biol Chem 279:363–373
Strausbauch PH, Fischer EH (1970) Structure of the binding site of pyridoxal 5’-phosphate to Escherichia coli glutamate decarboxylase. Biochemistry 9:233–238
Strohmeier M, Raschle T, Mazurkiewicz J, Rippe K, Sinning I, Fitzpatrick TB, Tews I (2006) Structure of a bacterial pyridoxal 5′-phosphate synthase complex. Proc Natl Acad Sci USA 103:19284–19289
Tanaka K, Tazuya K, Yamada K, Kumaoka H (2000) Biosynthesis of pyridoxine: origin of the nitrogen atom of pyridoxine in microorganisms. J Nutr Sci Vitaminol 46:55–57
Vacca RA, Giannattasio S, Capitani G, Marra E, Christen P (2008) Molecular evolution of B 6 enzymes: Binding of pyridoxal-5’-phosphate and Lys41Arg substitution turn ribonuclease A into a model B 6 protoenzyme. BMC Biochem 9:17
Yu P, Chen K, Huang X, Wang X, Ren Q (2018) Production of γ-aminobutyric acid in Escherichia coli by engineering MSG pathway. Prep Biochem Biotechnol 48:906–913
Yu P, Ren Q, Wang XX, Huang XX (2019) Enhanced biosynthesis of gamma- aminobutyric acid (GABA) in Escherichia coli by pathway engineering. Biochem Eng J 141:252–258
Zhang R, Yang T, Rao Z, Sun H, Xu M, Zhang X, Xu Z, Yang S (2014) Efficient one-step preparation of γ-aminobutyric acid from glucose without an exogenous cofactor by the designed Corynebacterium glutamicum. Green Chem 16:4190–4197
Zhang X, Teng YB, Liu JP, He YX, Zhou K, Chen Y, Zhou CZ (2010) Structural insights into the catalytic mechanism of the yeast pyridoxal 5-phosphate synthase Snz1. Biochem J 432:445–454
Acknowledgements
This study was supported by the Natural Science Foundation of Zhejiang Province, China (No. LY21C200006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yu, P., Ma, J., Zhu, P. et al. Enhancing the production of γ-aminobutyric acid in Escherichia coli BL21 by engineering the enzymes of the regeneration pathway of the coenzyme factor pyridoxal 5’-phosphate. World J Microbiol Biotechnol 37, 130 (2021). https://doi.org/10.1007/s11274-021-03103-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-021-03103-5