
Abstract
High-quality environmentally-friendly bioplastics can be produced by mixing poly-L-lactate with poly-D-lactate. On an industrial scale, this process simultaneously consumes large amounts of both optically pure lactate stereoisomers. However, because optimal growth conditions of L-lactate producers often differ from those of D-lactate producers, each stereoisomer is produced in a specialised facility, which raises cost and lowers sustainability. To address this challenge, we metabolically engineered Lactobacillus gasseri JCM 1131T, a bioprocess-friendly and genetically malleable strain of homofermentative lactic acid bacterium, to efficiently produce either pure L- or pure D-lactate under the same bioprocess conditions. Transformation of L. gasseri with plasmids carrying additional genes for L- or D-lactate dehydrogenases failed to affect the ratio of produced stereoisomers, but inactivation of the endogenous genes created strains which yielded 0.96 g of either L- or D-lactate per gram of glucose. In this study, the plasmid pHBintE, routinely used for gene disruption in Bacillus megaterium, was used for the first time to inactivate genes in lactobacilli. Strains with inactivated genes for endogenous lactate dehydrogenases efficiently fermented sugars released by enzymatic hydrolysis of alkali pre-treated wheat straw, an abundant lignocellulose-containing raw material, producing 0.37–0.42 g of lactate per gram of solid part of alkali-treated wheat straw. Thus, the constructed strains are primed to serve as producers of both optically pure L-lactate and D-lactate in the next-generation biorefineries.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lactic acid is a platform chemical indispensable for the production of foodstuffs, pharmaceutics, and bioplastics (Oliveira et al. 2018). It can be made through environmentally harsh petrochemical processes which create both L- and D-lactate, but such racemic mixture is unfit for many uses. Thus, lactic acid is today mostly supplied by modern bioprocesses. Although both Bacillus and Rhizopus genera have robust lactate producers (Alexandri et al. 2019; Mazzoli 2019), in most bioprocesses optically pure lactate is made by lactic acid bacteria (LAB) (Abdel-Rahman et al. 2013; Castillo Martinez et al. 2013).
Historically, researchers strived to produce optically pure L-lactate, which human body metabolises easily and which could thus be readily used in food industry and medicine (Hofvendahl and Hahn–Hägerdal 2000). However, nowadays the bioplastic sector is also aware of the superior properties of isotactic stereocomplexes, which are made by polymerising optically pure L-lactate and optically pure D-lactate separately and then mixing the resulting poly-L-lactate with poly-D-lactate (Tsuji 2005). Thus, it became attractive to produce both lactate stereoisomers in their optically pure form, often in comparable amounts.
At the same time, environmental concerns drove the construction of biorefineries (Moncada et al. 2016). As such facilities are impaired by a location-limited availability of a substrate (Hassan et al. 2019), they would benefit from bioprocesses that could produce large quantities of both optically pure lactate stereoisomers from the same substrate, ideally by using almost isogenic producer strains of LAB.
LAB often express several L- and D-lactate dehydrogenases (Feldman-Salit et al. 2013), as well as lactate racemases (Desguin et al. 2017), thus fermenting sugars to both L- and D-lactate. Certain Lactobacillus species at the beginning of fermentation, produce firstly L-stereoisomer of lactic acid, by enzyme L-lactate dehydrogenase, which then leads to induction of racemase enzyme (e.g. Lactobacillus curvatus, L. plantarum) and, consequently, conversion of L-lactic acid to D-lactic acid (Gottschalk 2012; Okano et al. 2018). Lactobacillus gasseri JCM 1131 belongs to the group of Lactobacillus species that, instead of racemase, have both lactate dehydrogenases. LAB produce lactate to restore cell’s redox balance (Liang et al. 2018), metabolise it when nutrients are scarce (Liu 2003), and some even embed traces of D-lactate into their cell wall (Chapot-Chartier and Kulakauskas 2014). Although the precise physiological role of each stereoisomer remains unclear, the simultaneous production of both L- and D-lactate prevents many isolates from being used as the producing strains, as the manufactured racemate can be separated into pure stereoisomers only on costly chiral chromatographic columns.
In addition to producing optically pure lactate, an adequate microbial producer should possess several other traits as well. If it exhibits most but not all of them, the missing ones could be introduced by genetic modification. However, such an intervention requires an efficient transformation protocol, an established plasmid toolbox (Karlskås et al. 2014), and the available whole genome sequence (Sun et al. 2015). To be genetically malleable, the strain should also lack restriction-modification (Forde and Fitzgerald 1999), as well as CRISPR/Cas9 systems (Crawley et al. 2018), and its plasmid toolbox should include both a high-copy-number replication origin that stably maintains the circular plasmid and a thermosensitive replication origin that is needed for gene inactivation. Regrettably, only a few LAB fulfil these conditions (Bosma et al. 2017).
Another difficulty in the biotechnological production of lactate is that LAB grow only on a complex mix of nutrients. Today, most bioprocesses utilise glucose- and starch-rich crops, thus drawing on an already scarce supply of animal feed and foodstuffs (Townsend et al. 2017). A more sustainable bioprocess would instead be based on lignocellulose-containing raw materials (Passoth and Sandgren 2019; Yuan et al. 2018), e.g. on 1.04 billion tonnes of wheat straw that were produced in 2019 (Food and Agriculture Organization of the United Nations 2019; Montane et al. 1998; Saha et al. 2005), and that were mostly ploughed back into the soil or burned (Talebnia et al. 2010). Yet, with an appropriate pre-treatment, LAB could have fermented this agricultural by-product into lactate (Cubas-Cano et al. 2018; Garde et al. 2002).
In this study, Lactobacillus gasseri JCM 1131T (Lauer and Kandler 1980) was identified as a promising, genetically malleable lactate producer which yields 0.96 g of racemic lactate per 1 g of glucose. To construct strain which produces only L-lactate and a strain which produces only D-lactate under similar bioprocess conditions, two approaches were used. First, L. gasseri was transformed with plasmids carrying additional copies for each of the three lactate dehydrogenases that the wild type encodes for. Second, each of the three endogenous genes for lactate dehydrogenases was inactivated using the plasmid pHBintE (Barg et al. 2005), showing for the first time that this plasmid, initially developed for gene disruption in Bacillus megaterium, can be employed for gene inactivation in lactobacilli. The increased number of genes coding for lactate dehydrogenases did not change the final ratio of produced L- and D-lactate, and the plasmids carrying these genes were frequently integrated into the homologous locus of the L. gasseri genome. However, inactivation of either D-lactate dehydrogenase or one L-lactate dehydrogenase created strains that produced optically pure lactate from the complex medium MRS, with a yield equal to that of a wild type. Moreover, these strains efficiently fermented enzymatic hydrolysate of alkali-treated wheat straw, which further highlighted their biotechnological potential.
Materials and methods
Media composition and growth conditions
Escherichia coli was grown in 2xYT medium (16 g l−1 bacto tryptone, 10 g l−1 yeast extract, 5 g l−1 NaCl) at 37 °C and 150 rpm. Strains that contained plasmids were grown in medium supplemented with ampicillin (50 mg l−1 in broth, 100 mg l−1 on solid media).
The entire construction of L. gasseri strains, i.e. L. gasseri transformation, genetic analyses, and initial measurements of stereoisomer production, were performed in 15 ml Falcon tubes in standard MRS medium (Biolife, Italy) at 37 °C without shaking and without air exchange, unless stated otherwise. Strains that contained plasmids were grown in media supplemented with erythromycin (10 mg l−1).
All growth curves and bioprocess measurements were obtained by cultivating L. gasseri in the laboratory-scale stirred tank reactor (STR) (B. Braun Biotech International, Berlin, Germany). The bioreactor (V = 2 l) was sterilised in situ for 20 min at 121 °C, loaded with 1.5 l of the medium, and inoculated with 75 ml of culture suspension freshly grown in MRS for 18 h at 37 °C. Modified strains of L. gasseri were cultivated in media supplemented with 10 mg l−1 of erythromycin. Cultivation was performed at 37 °C with the stirrer speed of 300 min−1. The pH was maintained with 5 M NaOH at 5.5.
Wheat straw hydrolysate-based MRS contained all the ingredients as the standard MRS, except for glucose, which was replaced with enzymatic hydrolysate of alkali-treated wheat straw.
Analytical methods
Concentrations of glucose, xylose, mannose, galactose, lactate, acetic acid, formic acid, and ethanol were monitored by high-performance liquid chromatography (HPLC) using Shimadzu CLASS-VP LC-10AVP (Shimadzu, Kyoto, Japan) with Supelcogel H precolumn (5 cm × 4.6 mm, i.d. 9 mm; Sigma-Aldrich, USA), Supelcogel C-610H column (30 cm × 7.8 mm, i.d. 9 mm; Sigma-Aldrich, USA), and a refractive index detector, as described previously (Marđetko et al. 2018).
Concentrations and ratios of D- and L-lactate were measured with D-/L-Lactic Acid Assay Kit (Megazyme, Bray, Ireland), according to the manufacturer’s instructions.
Statistical analysis
Descriptive statistics were calculated in Microsoft Excel (2019). Statistical significance was computed in the R software environment (The R Core Team 2019), using two-sided Welch’s t-test with Benjamini–Hochberg correction for multiple testing, or using Fisher’s exact test.
Whole-genome annotation
The finalised whole genome sequence of L. gasseri JCM 1131T (Makarova et al. 2006) (NCBI Reference Sequence NC_008530.1) was annotated with RAST (Overbeek et al. 2014), using ClassicRAST annotation scheme and by allowing for automatic repair of errors, frameshifts, and for backfilling of gaps. The annotation scheme identified putative lactate dehydrogenases through sequence similarity and synteny.
Plasmid construction
DNA manipulations and restriction cloning were performed as in Sambrook (2001). Restriction and modification enzymes were used as per the manufacturer’s instructions (NEB, United States). E. coli was transformed as in Miller and Nickoloff (1995), and transformants were selected with ampicillin (100 mg l−1).
Shuttle vector pEBO-AMβ1 (Fig. 1), a plasmid which can be maintained both in E. coli and in lactobacilli, was constructed by inserting both the marker for erythromycin resistance ermB (Sørvig et al. 2005) and the lactobacilli-compatible replication origin AMβ1 (Kleerebezem et al. 1997) into the standard cloning vector pBlueScript II KS(+) (Alting-Mees and Short 1989).

Construction of plasmids carrying genes encoding putative lactate dehydrogenases. The diagram shows the restriction enzymes and oligonucleotides used during the construction. bla = ampicillin resistance, ori = E. coli replication origin from plasmid pBR322, lacZα = sequence containing multiple cloning site, AMβ1 = lactobacilli-competent replication origin from plasmid pAMβ1, ermB = erythromycin resistance, ldhD = putative D-lactate dehydrogenase, ldhL1 and ldhL2 = putative L-lactate dehydrogenases
Plasmids carrying genes for each of the three lactate dehydrogenases (Fig. 1) were constructed by amplifying the genes encoding putative lactate dehydrogenases (ldhD, ldhL1, and ldhL2) from L. gasseri JCM 1131 T, using Q5 polymerase (NEB) and oligonucleotides from Table 1. Oligonucleotides were designed with Perl Primer v1.21 (Marshall 2004) and synthesised by Metabion International AG (Germany). PCR products were subcloned into pMiniT Vector (NEB) and inserted into pEBO-AMβ1, thus creating plasmids pD, pL1, and pL2.
Plasmids for gene disruption (Fig. 2) were constructed by inserting the middle third of the lactate dehydrogenase open reading frame (ORF) into the plasmid pHBintE (Barg et al. 2005), which carried lactobacilli-compatible thermosensitive replication origin. pHBintE was a gift from Dieter Jahn (Addgene plasmid # 48136; https://n2t.net/addgene:48136; RRID:Addgene_48136). Fragments of the putative lactate dehydrogenases were amplified from L. gasseri JCM 1131T by Q5 polymerase (Table 1), subcloned into pMiniT Vector and inserted into pHBintE, thus creating plasmids piD, piL1 and piL2.

Construction of plasmids used to disrupt genes encoding putative lactate dehydrogenases. The diagram shows the restriction enzymes and oligonucleotides used during the construction. bla = ampicillin resistance, ori = E. coli replication origin from plasmid pBR322, ori194ts = lactobacilli-competent thermosensitive replication origin derived from plasmid pE194, ermC = erythromycin resistance, ldhD = putative D-lactate dehydrogenase, ldhL1 and ldhL2 = putative L-lactate dehydrogenases, in-ldhD, in-ldhL1, in-ldhL2 = middle thirds of the ldhD, ldhL1, and ldhL2 ORFs
Construction of L. gasseri strains
Competent cells of L. gasseri were prepared and electroporated as described previously (Pusch et al. 2006), using Gene Pulser Xcell Electroporation System (Bio-Rad, Hercules, USA). Strains carrying additional copies of lactate dehydrogenase genes were constructed by transforming L. gasseri with each of the three plasmids, pD, pL1 and pL2, i.e. by electroporating competent cells, incubating them in MRS for 3 h at 37 °C, and plating them on MRS with 10 mg l−1 of erythromycin (MRSe). Colonies of transformants appeared after a 72-h incubation at 37 °C.
The construction of strains with disrupted lactate dehydrogenases had to account for thermosensitivity of pHBintE. Following electroporation, cells were incubated in MRS for 6 h at 28 °C, plated on MRSe, and incubated at 28 °C until the colonies appeared. Several colonies were inoculated in MRSe, grown at 28 °C until early stationary phase, reinoculated into fresh MRSe, and grown at 43 °C for 30 generations. Erythromycin-resistant colonies were isolated on MRSe plates, and gene disruption was confirmed by Southern blot.
Genomic DNA was isolated as described previously (Martín-Platero et al. 2007), with modifications. In short, the culture of L. gasseri in stationary phase was diluted 105 times in 4 ml MRS, grown to OD 0.4, and centrifuged (5 min/10,000 rpm). Traces of the media were removed completely, cells resuspended in 100 µl of TES buffer (10% sucrose, 25 mM Tris–HCl (pH 8.0), 10 mM EDTA, 10 mg ml−1 lysozyme, 40 µg ml−1 RNase) and incubated for 30 min at 37 °C. Next, the suspension was mixed by inversion with 600 µl of lysis buffer (100 mM Tris–HCl (pH 8.0), 100 mM EDTA (pH 8.0), 10 mM NaCl, 1% SDS), incubated for 15 min at room temperature, mixed with 10 µl of proteinase K buffer (10 mM Tris–HCl (pH 8.0), 5 mM EDTA (pH 8.0), 50 mM NaCl, 10 mg ml−1 proteinase K), and incubated for 5 min at 60 °C. Proteins were removed by phenol–chloroform extraction. DNA was precipitated by ammonium acetate and ethanol (1/3 v AmAc (8 M), 2 v EtOH), centrifuged (30 min/11 000 rpm/4 °C), and dissolved in TE buffer (pH 7.2). Following digestion of genomic DNA with restriction enzymes, Southern blot was performed as described previously (Štafa et al. 2017).
Pre-treatment and enzymatic hydrolysis of wheat straw
Wheat straw was treated with 2% w/w NaOH for 20 min at 180 °C in a high-pressure reactor, as described previously (Marđetko et al. 2018). Solid part of alkali-treated wheat straw was neutralized by washing out with water until it reached pH 7, and its composition was determined (61.7% glucan, 33.0% xylan, 1.6% arabinan, and 1.6% lignin). The 50 g of neutralized solid part of wheat straw was resuspended in 1 l of 60 mM sodium acetate buffer (pH 4.78), mixed for 24 h at 50 °C with 0.6 g of cellulase enzyme blend “Cellic CTec2” (Sigma-Aldrich, Germany), then mixed with additional 0.3 g of the enzyme blend for 24 h at 50 °C to increase hydrolysis yield, and finally filtered through the Buchner funnel. The obtained filtrate was stored at − 20 °C until use. Average composition of the solid part of alkali-treated wheat straw was detected by HPLC and its sugar composition was as follows: glucose, 21.07 g l−1 and together xylose, manose and galactose, 3.80 g l−1. Arabinose and cellobiose were not detected.
Calculation of bioprocess efficiency parameters
Biomass yield coefficient (YX/S = ΔX/ΔS) was calculated by dividing the grown biomass (ΔX = X − Xo) with consumed substrate (ΔS = S0 − S) concentration in the bioprocess. Product yield coefficient (YP/S = ΔP/ΔS) was calculated by dividing the produced lactate (ΔP = P − P0) with the consumed substrate (ΔS) concentration. Biomass productivity (PrX = ΔX/t) and lactate productivity (PrP = ΔP/t) indicate how much biomass and lactate were produced per hour, from the beginning of the cultivation until the substrate was depleted (t). Product formation rate (rP), substrate consumption rate (rS), and specific growth rate (µ) were calculated by standard procedures (Marđetko et al. 2018):
where X and X0 are biomass concentrations at the end and the beginning of bioprocess; P and P0 are product concentrations at the end and the beginning of bioprocess and S0 and S are substrate concentrations at the beginning and the end of bioprocess.
Results
Preliminary cultivation of Lactobacillus gasseri JCM 1131T in the stirred-tank reactor (STR)
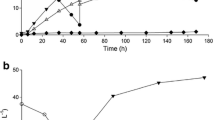
This study focused on L. gasseri JCM 1131T (Lauer and Kandler 1980), both a promising lactate producer and a genetically malleable lactic acid bacterium. In standard MRS medium, L. gasseri JCM 1131T fermented 17.2 g l−1 of glucose to 17.2 g l−1 of lactate in less than 11 h (Fig. 3, Table 2), producing lactate at a rate of 1.62 g l−1 h−1, in the D:L stereoisomer ratio of 0.51:0.49, with lactate concentration remaining unchanged throughout the stationary phase. In addition, L. gasseri JCM 1131T can grow between 28 °C and 43 °C, prefers anaerobiosis, ferments cellobiose, mannose, and galactose (Azcarate-Peril et al. 2008), lacks CRISPR/Cas9 system (Sanozky-Dawes et al. 2015), lacks endogenous plasmids (Makarova et al. 2006), can be transformed (Wegkamp et al. 2004), and is amenable to gene inactivation (Francl et al. 2010).

Cultivation of L. gasseri JCM 1131T in standard MRS in the stirred-tank reactor (STR). The diagram shows changes in concentrations of glucose (open circle), lactate (closed circle), and biomass (open square). Data points indicate mean values of duplicate fermentations
Genetic modification of L. gasseri JCM 1131T
To construct derivatives of L. gasseri JCM 1131T that produce optically pure lactate, we first searched its genome for genes involved in pyruvate metabolism. Whole-genome annotation revealed that the strain encodes two putative L- and one D-lactate dehydrogenases (ldhL1, ldhL2, and ldhD). Lactate racemase was not detected. In attempt to overexpress the identified lactate dehydrogenases genes, they were placed, together with their native promotes, on the E. coli-Lactobacillus shuttle vector pEBO-AMβ1 (Fig. 1) and transformed into L. gasseri JCM 1131T. However, both untransformed and transformed L. gasseri strains produced 17 g l−1 of lactate, in the D:L ratio of 0.55:0.45 (Fig. 4a), suggesting that the presence of additional copies of putative lactate dehydrogenases did not result in their overexpression, or that overexpression failed to affect the final concentration of D- and L-lactate.

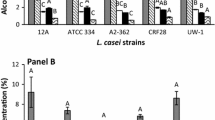
The amount and the ratio of D- and L-lactate produced a by strains of L. gasseri that carry plasmids pD, pL1, and pL2 and b by strains with disrupted ldhD, ldhL1, and ldhL2 (i-ldhD, i-ldhL1, and i-ldhL2, respectively). D-lactate is shown in white, L-lactate in grey. Error bars denote standard deviation. The ratio of stereoisomers is given under strain labels, as is the significance of its change compared to the wild type JCM 1131T (two-sided Welch’s t-test with Benjamini–Hochberg correction for multiple testing)
As the presence of additional copies of genes failed to change the stereoisomer ratio, the strains with inactivated endogenous genes for each lactate dehydrogenases were constructed. For this purpose, middle thirds of ldhD, ldhL1, and ldhL2 ORFs were placed on the E. coli-Bacillus thermosensitive shuttle vector pHBintE (Fig. 2), such constructs were introduced into L. gasseri JCM 1131T, integrands selected for, and the gene disruptions verified by Southern blot (Fig. 5). The disruptions produced two inactive copies of the targeted genes, one lacking the starting third of the ORF and one lacking the final third of the ORF. On MRS, the strain without functional ldhD (which we refer to as i-ldhD) produced only L-lactate, while the strain without functional ldhL1 (i-ldhL1) produced only D-lactate (Fig. 4b).

Genetic analysis of transformants with disrupted lactate dehydrogenases. a The diagram shows the disruption of targeted genes, enzymes used for digestion of DNA, and the size of expected bands when digoxigenin-labelled bla and phage λ DNA is used as a probe. b Southern blot of L. gasseri disruptants, with the used restriction enzymes indicated above the membranes, and the bands pointing to homologous and multiple plasmid integrations marked by black and grey arrows, respectively. M = marker (phage λ DNA digested with HindIII), piD, piL1, piL2 = plasmids piD, piL1, and piL2, respectively; C = genomic DNA of untransformed strain JCM 1131T; iD, iL1, iL2 = genomic DNA of i-ldhD, i-ldhL1, and i-ldhL2, respectively
Overexpression plasmids should have maintained several copies of lactate dehydrogenase genes in the cells, yet the transformants that carried them produced the same ratio of stereoisomers, as did the wild type. To uncover why, the transformants were analysed with Southern blot. Unexpectedly, the transformants that carried plasmids with additional copies of lactate dehidrogenase (pD, pL1, and pL2) readily integrated them into the homologous chromosomal loci (Fig. 6), and such integrands prevailed in their respective populations within fifty generations.

Genetic analysis of transformants in which the plasmids pD, pL1 and pL2 homologously integrated into the chromosomal DNA. a The diagram shows the integration of one and two copies of the plasmid into homologous chromosomal loci, enzymes used for digestion of DNA, and the size of expected bands on the Southern blot when digoxigenin-labelled ermB and phage λ DNA is used as a probe. Gene labels are identical to those in Fig. 1. b Southern blot of L. gasseri transformants, with the used restriction enzymes indicated above the membranes, the plasmids used to obtain the transformants indicated below the membranes, and the bands pointing to homologous and multiple plasmid integrations marked by black and grey arrows, respectively. M = marker (phage λ DNA digested with HindIII); pD, pL1, pL2 = plasmids pD, pL1, and pL2, respectively; C = genomic DNA of untransformed strain JCM 1131 T; 1–26 = genomic DNA of transformants
Additionally, in 8 out of 26 analysed transformants, Southern blot of digested genomic DNA uncovered another band, the size of which matched the size of the linearized plasmid. In seven cases this band marked multiple plasmid integration while in one case it originated from the circular plasmid, which we confirmed by Southern blot of the undigested DNA (data not shown). Plasmids pL1 and pL2 integrated in multiple copies significantly more often (4/8 and 3/9 transformants, respectively) than plasmid pD (0/9 transformants; p < 0.03, Fisher’s exact test). On the other hand, chromosomal integrations of plasmid pEBO-AMβ1 (plasmid which did not carry regions endogenous to L. gasseri) were not detected (data not shown). Thus, the only plasmids that integrated into the chromosome were those that carried both AMβ1 replication origin and a homologous region.
Likewise, in ldhD and ldhL1 disruptants, Southern blot uncovered additional bands, the size of which matched the size of the linearized plasmids (Fig. 5). Southern blot of the undigested DNA confirmed that these bands originated from multiple plasmid integrations (data not shown), which were likely selected for during prolonged growth in MRS with erythromycin. Such multiple integrations further stabilised gene disruptions in the constructed strains as multiple integrated plasmid copies were unlikely to excise from chromosomal DNA in one recombination event. Thus, similarly to pEBO-AMβ1 derivatives, pHBintE derivatives also integrated homologously and in tandem.
Cultivation of modified L. gasseri strains in the STR on media containing either glucose or wheat straw hydrolysate
In our further research, modified L. gasseri strains were cultivated in the STR to evaluate their fermentation capacities. In standard MRS, i-ldhD fermented 17 g l−1 of glucose to 16.5 g l−1 of optically pure L-lactate, while i-ldhL1 fermented 16.8 g l−1 of glucose to 17 g l−1 of D-lactate (Fig. 7a). Other fermentation parameters were also similar to those of the wild type (Table 2), except that i-ldhL1 suffered from lower productivity, which was caused by its prolonged lag phase.

Cultivation of strains with inactivated D-lactate dehydrogenase (i-ldhD) or L-lactate dehydrogenase (i-ldhL1) in the stirred-tank reactor (STR) a on standard MRS and b on MRS supplemented with the hydrolysate of the solid part of alkali-treated wheat straw. The diagram shows the concentrations of glucose (open circle), lactate (closed circle), biomass (open square), and the total concentration of mannose, galactose, and xylose (open triangle). Data points indicate mean values of duplicate fermentations
In a wheat straw hydrolysate-based MRS, strains performed similarly (Fig. 7b). Strain i-ldhD produced 18.7 g l−1 of lactate with a productivity of 1.92 g l−1 h−1, while i-ldhL1 produced 21.0 g l−1 of lactate with a productivity of 1.67 g l−1 h−1. Although glucose comprised over 80% of the available sugars, the strains fermented glucose, mannose, and galactose simultaneously, thus modestly improving several fermentation parameters (Table 2). As expected, xylose was not metabolised. Thus, the constructed strains fermented the hydrolysate of wheat straw efficiently, producing 0.37–0.42 g of lactate per 1 g of the solid part of alkali-treated wheat straw.
Interestingly, in the wheat straw hydrolysate-based MRS, i-ldhD continued to generate optically pure L-lactate, but i-ldhL1 no longer produced optically pure D-lactate. Rather, it produced the mixture of lactate stereoisomers in the D:L ratio 0.87:0.13 (Table 3). We investigated this anomaly by cultivating strains i-ldhL1 and i-ldhL2 in MRS in which glucose was replaced with mannose or galactose. In such media, D:L ratios suggested that ldhL1 is expressed constitutively, while ldhL2 is expressed only in the presence of mannose or galactose (data not shown). Thus, in the wheat straw hydrolysate, i-ldhL1 produced both D- and L-lactate because mannose and galactose induced the expression of ldhL2. Furthermore, the maximal lactate concentration was observed in both media (MRS supplemented with pure glucose or wheat straw hydrolysate) around 12 h of cultivation. Thus, the growth of L. gasseri is not inhibited by the wheat straw hydrolysate although it was expected. In this study, wheat straw hydrolysate was generated by the alkali-based hydrolysis which produces fewer fermentation inhibitors (e.g. phenolic compounds and furans) compared to the acid-based hydrolysis. Moreover, the produced solid part of alkali-treated wheat straw was washed with water to pH neutral what significantly decreased the content of inhibitory compounds in this substrate (Zhao et al. 2008; Ivančić Šantek et al. 2018).
Discussion
In this study, strains of L. gasseri were constructed that produce 0.96 g of either optically pure L-lactate or optically pure D-lactate per gram of glucose under identical bioprocess conditions. During their construction, it was noticed that plasmids regularly integrate into homologous chromosomal loci, and we demonstrated that thermosensitive plasmid pHBintE is functional in L. gasseri. Strains with inactivated ldhD and ldhL1 fermented glucose in MRS to optically pure lactate, but only the strain with inactivated ldhD produced optically pure lactate from the undetoxified alkali pre-treated wheat straw hydrolysate.
Lactate production by L. gasseri JCM 1131T
Although L. gasseri ferments sugars rapidly, it is currently not used in industrial lactate production. Overlooked as a fastidious organism that produces racemic lactate, it remains neglected despite its efficient and high-yielding homofermentative LAB metabolism, the advantages of which could outweigh common bioprocess difficulties (Sauer et al. 2017). Our results confirm its potential, as the strain L. gasseri JCM 1131T grew fast (0.84 h−1), as well as maintained high productivity (1.62 g l−1 h−1) and lactate yield (0.96 g g−1), irrespective of the employed medium.
The main shortcoming of L. gasseri JCM 1131T and related homofermentative LAB is their inability to ferment xylose. To remedy this problem, several studies turned to heterofermentative lactobacilli, such as L. pentosus (Garde et al. 2002; Puah et al. 2013). However, while L. pentosus ferments xylose, it grows slower (0.40 h−1) than L. gasseri (Cubas-Cano et al. 2019), and it generates unwanted by-products, such as succinate, acetate, glycerol, and dihydroxyacetone. These by-products lower lactate yield, complicate downstream processes, and build up even if glucose is the only carbon source, at rates dependent on the concentrations of oxygen and citrate (Cselovszky et al. 1992). Other xylose-fermenting bacteria, e.g. Corynebacterium glutamicum, succumb to similar drawbacks (Tsuge et al. 2019). Of course, one could metabolically engineer L. gasseri to ferment xylose (Posno et al. 1991), but the constructed strain would then produce the same by-products. Thus, none of the options available today solve adequately the problem of direct xylose-to-lactate fermentation. Furthermore, it has to be pointed out that the solid part of alkali-treated wheat straw contains lower xylose content comparing to the acid-treated wheat straw due to the higher hemicellulose dissolution during alkali treatment of lignocellulose containing raw materials (Yang et al. 2011). On the other hand, if the constructed strains of L. gasseri were used in the biorefineries, the residual pentose sugars could simply be passed to the next refining phase, onto organisms capable of metabolising them into other value-added products.
This study also offers some insight into the physiology of LAB. Compared to the wild type, transformants with integrated pEBO-AMβ1 derivatives had additional copies of lactate dehydrogenases, but they produced the same final ratio of D- and L-lactate. This result suggests that the stereoisomer ratio is important and that the cells actively regulate it. Yet, the disruptants that produced optically pure lactate suffered no apparent growth defect, although the inactivation of ldhL1 did prolong the lag phase. Thus, the lactate ratio appears to be strictly regulated, yet only mildly important both at the level of the individual cell and at the level of the laboratory-grown pure culture. Therefore, as was suggested earlier for other LAB (Desguin et al. 2017), L. gasseri seems to produce both lactate stereoisomers for reasons that are primarily ecological, to affect the makeup of nearby microbial communities.
Our future research of L. gasseri will focus on its growth in a sugar-rich medium, as lactobacilli can easily adapt to nonelectrolyte stress (Glaasker et al. 1998). For this purpose, we will use highly concentrated suspension of cells pre-cultured in the sugar-rich environment in order to increase both productivity and final concentration of lactate.
Genetic manipulation of L. gasseri JCM 1131T
This study also explores the stability and mechanisms of plasmid integration in L. gasseri. Although previous studies have focused on constructing new strains (Bruno-Bárcena et al. 2005; Pusch et al. 2006), they did not investigate why some strategies succeeded and others failed. Thus, the scientific community is forced to deduce how the plasmid construct interacts with the genome by looking at evolutionary distant model organisms. At the same time, the community recognizes that only by understanding LAB-specific genetic events can it fully and intricately exploit the LAB toolbox.
In our analysis, the plasmids integrated into the chromosome, but only when they carried a homologous region. Thus, pEBO-AMβ1 derivatives and pHBintE derivatives, but not the original pEBO-AMβ1 and pHBintE, integrated readily by homologous recombination. In this aspect, L. gasseri is similar to most bacteria. Such efficient recombination facilitates not only ongoing reductive evolution of its genome (Makarova et al. 2006) but also the construction of new strains, allowing for homologous integration of heterologous genes solely using large homologies, thus bypassing the need for temperature-sensitive replication origins. The efficient plasmid integration also nullifies the effect of plasmid copy number, in which the expression systems often differ, and implies that the plasmid integration can be avoided only by using genes that lack homology to the genome of L. gasseri.
This study is the first one to employ pHBintE in lactobacilli, a plasmid which was initially developed for gene disruption in Bacillus megaterium (Barg et al. 2005) and which carries thermosensitive replication origin derived from plasmid pE194. As such, plasmid pHBintE presents an alternative to the commonly used lactobacilli two-plasmid system pTRK669/pORI28 (Leenhouts et al. 1996; Russell and Klaenhammer 2001) which requires two selective markers and which, as it integrates, produces two repeats spaced 1.7 kb apart. In contrast, plasmid pHBintE takes up only one selective marker and probably generates more stable repeats since they are spaced 5.7 kb apart.
In the strains that integrated pHBintE derivatives, genes that encode lactate dehydrogenases were disrupted, not completely deleted. In general, it is more desirable to delete than to disrupt a gene as the disrupted gene can reassemble into its original form if the integrated plasmid excises (Biswas et al. 1993). Although rare, such an event would recreate erythromycin-sensitive, racemate-producing cells. However, we note that growth rates of constructed strains remained similar to that of the wild type, suggesting that unlikely revertants would fail to gain selective advantage, even in standard MRS. Nevertheless, if the revertants are of concern, one could eliminate them by enforcing periodic sweeps of erythromycin selection during inoculum cultivation. Although, in general more desirable than disruption strategy, the deletion strategy would entail a more elaborate, two-step genetic manipulation, as the plasmids would first need to integrate and then excise through crossing-overs (Ferain et al. 1994). The deletion strategy would be complicated further by the lack of a convenient negative selection.
Production of optically pure lactate by modified strains of L. gasseri JCM 1131T
This study is the first to engineer L. gasseri for the production of optically pure lactate. In the related attempts, others inactivated D-lactate dehydrogenase in L. helveticus (Kylä-Nikkilä et al. 2000) and L. parakasei (Kuo et al. 2015), both D-lactate dehydrogenase and lactate racemase in L. plantarum (Okano et al. 2018), or one or more L-lactate dehydrogenases in L. plantarum (Hama et al. 2015; Okano et al. 2017, 2009; Zhang et al. 2016).
The selected L. gasseri JCM 1131T proved highly resistant to the perturbations of its central carbon metabolism, as the fermentation parameters of the modified strains only mildly deviated from the wild type (Table 2). Strains i-ldhD and i-ldhL1 performed admirably, even when glucose was replaced with enzyme-hydrolysed alkali-pre-treated wheat straw, a lignocellulose feedstock which L. pentosus (Garde et al. 2002) and B. coagulans (Zhang et al. 2014) can successfully ferment to racemic lactate or L-lactate but not to both optically pure L- and optically pure D-lactate.
In this study, an L-lactate and a D-lactate producer were constructed from the same strain. This strategy is advantageous for bioprocess development as both pure stereoisomers can be made in the same facility under the identical bioprocess conditions, using the same equipment but a different producer strain. One of the first attempts to convert a microorganism into homofermentative producer of both optically pure D-lactate and optically pure L-lactate focused on the model bacterium E. coli (Chang et al. 1999), and it resulted in a strain yielding 0.9 g of D-lactate per 1 g of glucose at a productivity of 1.09 g l−1 h−1. However, to obtain such results, Chang et al. (1999) had to inactivate two genes involved in pyruvate metabolism and cultivate the producer for the first 12 h aerobically to allow for the biomass production. To obtain homofermentative L-lactate producer, Chang et al. (1999) additionally disrupted native D-lactate dehydrogenase and expressed L-lactate dehydrogenase from L. casei, but the resulting E. coli suffered severe growth defect, producing only 0.146 g l−1 h−1 of L-lactate. Thus, these experiments pointed to naturally homofermentative lactic acid bacteria as the more suitable producers of optically pure lactate which, in as shown in our study, can produce up to 1.96 g l−1 h−1 of optically pure lactate, without requiring any aerobic conditions.
More recently, Yi et al. (2016) focused on Pediococcus acidilactici and employed a strategy similar to one described in this work, inactivating species’ ldhD and ldh genes to construct producers of optically pure L- and D-lactate. However, modified strains of L. gasseri described in this study outperform modified strains of P. acidilactici when the later were cultivated in a simultaneous saccharification and fermentation bioprocess that utilised acid pre-treated but detoxified corn stover. More precisely, they surpassed P. acidilactici both in product yield coefficient (0.99 vs 0.58–0.65) and lactate yield (0.37–0.42 g g−1 of raw material vs 0.31 g g−1 of raw material).
In a more radical approach, Tsuge et al. (2019) converted C. glutamicum from L- to D-lactate producer by inactivating ldhA and expressing D-ldhA gene from L. delbrueckii. In another example, Zhang et al. (2014) refitted Bacillus coagulans from L- to D-lactate producer by deleting ldhL1 and ldhL2 genes, and by expressing LdldhD from L. delbrueckii subsp. bulgaricus. Along with them, our study underlines the merits of such sustainable approach, and it shows that as i-ldhD and i-ldhL1 fermented the hydrolysate, they retained the lactate yield (0.99 g g−1) similar to the yield that genetically modified B. coagulans (Zhang et al. 2017) and C. glutamicum (Tsuge et al. 2019) achieved in glucose-based media (0.97–0.98 g g−1). Thus, strains i-ldhD and i-ldhL1 present the next step toward sustainable production of optically pure lactate.
Conclusion
This study harnessed genetic malleability of L. gasseri in order to improve it as an industrial microorganism for lactate production. This malleability allowed us to construct strains of L. gasseri that carry inactivated lactate dehydrogenases and thus ferment glucose to both optically pure L- and D-lactate, under the same bioprocess conditions. In the process, we enriched the lactobacilli plasmid toolbox and detailed genetic events that occur during genetic manipulation of this non-model organism. Constructed strains efficiently fermented wheat straw hydrolysate and thus can be used in the sustainable bioprocesses that strive towards eco-friendly and economical production of both L- and D-lactate.
Data availability
Reported nucleotide sequence data are available in the Third Party Annotation Section of the DDBJ/ENA/GenBank databases under the accession numbers TPA: BK010903-BK010905. L. gasseri JCM 1131T is available from Japan Collection of Microorganisms (JCM).
References
Abdel-Rahman MA, Tashiro Y, Sonomoto K (2013) Recent advances in lactic acid production by microbial fermentation processes. Biotechnol Adv 31:877–902. https://doi.org/10.1016/j.biotechadv.2013.04.002
Alexandri M, Neu A-K, Schneider R, López-Gómez JP, Venus J (2019) Evaluation of various Bacillus coagulans isolates for the production of high purity L-lactic acid using defatted rice bran hydrolysates. Int J Food Sci Technol 54:1321–1329. https://doi.org/10.1111/ijfs.14086
Alting-Mees MA, Short JM (1989) pBluescript II: gene mapping vectors. Nucleic Acids Res 17:9494–9494. https://doi.org/10.1093/nar/17.22.9494
Azcarate-Peril MA et al (2008) Analysis of the genome sequence of Lactobacillus gasseri ATCC 33323 reveals the molecular basis of an autochthonous intestinal organism. Appl Environ Microbiol 74:4610. https://doi.org/10.1128/aem.00054-08
Barg H, Malten M, Jahn M, Jahn D (2005) Protein and vitamin production in Bacillus megaterium. In: Barredo J-L (ed) Microbial processes and products. Humana Press, Totowa, pp 205–223
Biswas I, Gruss A, Ehrlich SD, Maguin E (1993) High-efficiency gene inactivation and replacement system for gram-positive bacteria. J Bacteriol 175:3628. https://doi.org/10.1128/jb.175.11.3628-3635.1993
Bosma EF, Forster J, Nielsen AT (2017) Lactobacilli and pediococci as versatile cell factories: evaluation of strain properties and genetic tools. Biotechnol Adv 35:419–442. https://doi.org/10.1016/j.biotechadv.2017.04.002
Bruno-Bárcena JM, Azcárate-Peril MA, Klaenhammer TR, Hassan HM (2005) Marker-free chromosomal integration of the manganese superoxide dismutase gene (sodA) from Streptococcus thermophilus into Lactobacillus gasseri. FEMS Microbiol Lett 246:91–101. https://doi.org/10.1016/j.femsle.2005.03.044
Castillo Martinez FA, Balciunas EM, Salgado JM, Domínguez González JM, Converti A, Oliveira RPdS (2013) Lactic acid properties, applications and production: a review. Trends Food Sci Technol 30:70–83. https://doi.org/10.1016/j.tifs.2012.11.007
Chapot-Chartier M-P, Kulakauskas S (2014) Cell wall structure and function in lactic acid bacteria. Microb Cell Fact 13:S9. https://doi.org/10.1186/1475-2859-13-S1-S9
Chang DE, Jung HC, Rhee JS, Pan JG (1999) Homofermentative production of D- or L-Lactate in metabolically engineered Escherichia coli RR1. Appl Environ Microbiol 65:1384–1389. https://doi.org/10.1128/AEM.65.4.1384-1389.1999
Crawley AB, Henriksen ED, Stout E, Brandt K, Barrangou R (2018) Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci Rep 8:11544. https://doi.org/10.1038/s41598-018-29746-3
Cselovszky J, Wolf G, Hammes WP (1992) Production of formate, acetate, and succinate by anaerobic fermentation of Lactobacillus pentosus in the presence of citrate. Appl Microbiol Biotechnol 37:94–97. https://doi.org/10.1007/BF00174210
Cubas-Cano E, González-Fernández C, Ballesteros M, Tomás-Pejó E (2018) Biotechnological advances in lactic acid production by lactic acid bacteria: lignocellulose as novel substrate. Biofuels Bioprod Biorefin 12:290–303. https://doi.org/10.1002/bbb.1852
Cubas-Cano E, González-Fernández C, Ballesteros M, Tomás-Pejó E (4023T) Lactobacillus pentosus CECT 4023T co-utilizes glucose and xylose to produce lactic acid from wheat straw hydrolysate: anaerobiosis as a key factor. Biotechnol Prog 35:e2739. https://doi.org/10.1002/btpr.2739
Desguin B, Soumillion P, Hausinger RP, Hols P (2017) Unexpected complexity in the lactate racemization system of lactic acid bacteria. Fems Microbiol Rev 41:S71–S83. https://doi.org/10.1093/femsre/fux021
Feldman-Salit A et al (2013) Regulation of the activity of lactate dehydrogenases from four lactic acid bacteria. J Biol Chem 288:21295–21306. https://doi.org/10.1074/jbc.M113.458265
Ferain T, Garmyn D, Bernard N, Hols P, Delcour J (1994) Lactobacillus plantarum ldhL gene: overexpression and deletion. J Bacteriol 176:596. https://doi.org/10.1128/jb.176.3.596-601.1994
Food and Agriculture Organization of the United Nations (2019) Crop Prospects and Food Situation: Quaterly Global Report, December 2019
Forde A, Fitzgerald GF (1999) Bacteriophage defence systems in lactic acid bacteria. In: Konings WN, Kuipers OP, Veld JHJH (eds) Lactic acid bacteria: genetics, metabolism and applications: Proceedings of the Sixth Symposium on lactic acid bacteria: genetics, metabolism and applications, 19–23 September 1999, Veldhoven, The Netherlands. Springer Netherlands, Dordrecht, pp 89–113. doi: 10.1007/978-94017-2027-4_4
Francl AL, Thongaram T, Miller MJ (2010) The PTS transporters of Lactobacillus gasseri ATCC 33323. BMC Microbiol 10:77. https://doi.org/10.1186/1471-2180-10-77
Garde A, Jonsson G, Schmidt AS, Ahring BK (2002) Lactic acid production from wheat straw hemicellulose hydrolysate by Lactobacillus pentosus and Lactobacillus brevis. Bioresour Technol 81:217–223. https://doi.org/10.1016/S0960-8524(01)00135-3
Glaasker E, Tjan FSB, Steeg PFT, Konings WN, Poolman B (1998) Physiological response of Lactobacillus plantarum to salt and nonelectrolyte stress. J Bacteriol 180(17):4718
Gottschalk G (2012) Bacterial metabolism. Springer, New York, Heidlberg, Berlin
Hama S, Mizuno S, Kihara M, Tanaka T, Ogino C, Noda H, Kondo A (2015) Production of D-lactic acid from hardwood pulp by mechanical milling followed by simultaneous saccharification and fermentation using metabolically engineered Lactobacillus plantarum. Bioresour Technol 187:167–172. https://doi.org/10.1016/j.biortech.2015.03.106
Hanahan D, Jessee J, Bloom FR (1991) Plasmid transformation of Escherichia coli and other bacteria. Methods in enzymology, vol 204. Academic Press, Cambdrige, pp 63–113
Hassan SS, Williams GA, Jaiswal AK (2019) Lignocellulosic biorefineries in Europe: current state and prospects. Trends Biotechnol 37:231–234. https://doi.org/10.1016/j.tibtech.2018.07.002
Hofvendahl K, Hahn-Hägerdal B (2000) Factors affecting the fermentative lactic acid production from renewable resources. Enzyme Microb Technol 26:87–107. https://doi.org/10.1016/S0141-0229(99)00155-6
Ivančić Šantek M, Lisičar J, Mušak L, Vrana Špoljarić I, Beluhan S, Šantek B (2018) Lipid production by yeast Trichosporon oleaginosus on the enzymatic hydrolysate of alkaline pretreated corn cobs for biodiesel production. Energy Fuels 32:12501–12513. https://doi.org/10.1021/acs.energyfuels.8b02231
Karlskås IL, Maudal K, Axelsson L, Rud I, Eijsink VGH, Mathiesen G (2014) Heterologous protein secretion in lactobacilli with modified pSIP vectors. PLoS ONE 9:e91125. https://doi.org/10.1371/journal.pone.0091125
Kleerebezem M, Beerthuyzen MM, Vaughan EE, de Vos WM, Kuipers OP (1997) Controlled gene expression systems for lactic acid bacteria: transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl Environ Microbiol 63:4581
Kuo Y-C, Yuan S-F, Wang C-A, Huang Y-J, Guo G-L, Hwang W-S (2015) Production of optically pure L-lactic acid from lignocellulosic hydrolysate by using a newly isolated and D-lactate dehydrogenase gene-deficient Lactobacillus paracasei strain. Bioresour Technol 198:651–657. https://doi.org/10.1016/j.biortech.2015.09.071
Kylä-Nikkilä K, Hujanen M, Leisola M, Palva A (2000) Metabolic engineering of Lactobacillus helveticus CNRZ32 for production of pure L-(+)-lactic acid. Appl Environ Microbiol 66:3835. https://doi.org/10.1128/aem.66.9.3835-3841.2000
Lauer E, Kandler O (1980) Lactobacillus gasseri sp. nov., a new species of the subgenus Thermobacterium. Zentralblatt für Bakteriologie: I Abt Originale C: Allgemeine, angewandte und ökologische Mikrobiologie 1:75–78. https://doi.org/10.1016/S0172-5564(80)80019-4
Leenhouts K et al (1996) A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253:217–224. https://doi.org/10.1007/s004380050315
Liang S, Gao D, Liu H, Wang C, Wen J (2018) Metabolomic and proteomic analysis of D-lactate-producing Lactobacillus delbrueckii under various fermentation conditions. J Ind Microbiol Biotechnol 45:681–696. https://doi.org/10.1007/s10295-018-2048-y
Liu SQ (2003) Practical implications of lactate and pyruvate metabolism by lactic acid bacteria in food and beverage fermentations. Int J Food Microbiol 83:115–131. https://doi.org/10.1016/S0168-1605(02)00366-5
Makarova K et al (2006) Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA 103:15611. https://doi.org/10.1073/pnas.0607117103
Marđetko N, Novak M, Trontel A, Grubišić M, Galić M, Šantek B (2018) Bioethanol production from dilute-acid pre-treated wheat straw liquor hydrolysate by genetically engineered Saccharomyces cerevisiae. Chem Biochem Eng Q 32:483–499. https://doi.org/10.15255/CABEQ.2018.1409
Marshall OJ (2004) PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 20:2471–2472. https://doi.org/10.1093/bioinformatics/bth254
Martín-Platero AM, Valdivia E, Maqueda M, Martínez-Bueno M (2007) Fast, convenient, and economical method for isolating genomic DNA from lactic acid bacteria using a modification of the protein “salting-out” procedure. Anal Biochem 366:102–104. https://doi.org/10.1016/j.ab.2007.03.010
Mazzoli R (2019) Metabolic engineering strategies for consolidated production of lactic acid from lignocellulosic biomass. Biotechnol Appl Biochem. https://doi.org/10.1002/bab.1869
Miller EM, Nickoloff JA (1995) Escherichia coli electrotransformation. In: Nickoloff JA (ed) Electroporation protocols for microorganisms. Humana Press, Totowa, pp 105–113
Moncada JB, Aristizábal VM, Cardona CA (2016) Design strategies for sustainable biorefineries. Biochem Eng J 116:122–134. https://doi.org/10.1016/j.bej.2016.06.009
Montane D, Farriol X, Salvadó J, Jollez P, Chornet E (1998) Application of steam explosion to the fractionation and rapid vapor-phase alkaline pulping of wheat straw. Biomass Bioenergy 14:261–276. https://doi.org/10.1016/S0961-9534(97)10045-9
Okano K, Hama S, Kihara M, Noda H, Tanaka T, Kondo A (2017) Production of optically pure D-lactic acid from brown rice using metabolically engineered Lactobacillus plantarum. Appl Microbiol Biotechnol 101:1869–1875. https://doi.org/10.1007/s00253-016-7976-8
Okano K, Uematsu G, Hama S, Tanaka T, Noda H, Kondo A, Honda K (2018) Metabolic engineering of Lactobacillus plantarum for direct L-lactic acid production from raw corn starch. Biotechnol J 13:1700517. https://doi.org/10.1002/biot.201700517
Okano K, Zhang Q, Shinkawa S, Yoshida S, Tanaka T, Fukuda H, Kondo A (2009) Efficient production of optically pure D-lactic acid from raw corn starch by using a genetically modified L-lactate dehydrogenase gene-deficient and α-amylase-secreting Lactobacillus plantarum strain. Appl Environ Microbiol 75:462. https://doi.org/10.1128/aem.01514-08
Oliveira RAd, Komesu A, Rossell CEV, Filho RM (2018) Challenges and opportunities in lactic acid bioprocess design-From economic to production aspects. Biochem Eng J 133:219–239. https://doi.org/10.1016/j.bej.2018.03.003
Overbeek R et al (2014) The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res 42:D206–D214. https://doi.org/10.1093/nar/gkt1226
Passoth V, Sandgren M (2019) Biofuel production from straw hydrolysates: current achievements and perspectives. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-019-09863-3
Posno M, Heuvelmans PT, van Giezen MJ, Lokman BC, Leer RJ, Pouwels PH (1991) Complementation of the inability of Lactobacillus strains to utilize D-xylose with D-xylose catabolism-encoding genes of Lactobacillus pentosus. Appl Environ Microbiol 57:2764
Puah SM, Huynh HV, Wu JC (2013) Novel two-in-one bioreactor greatly improves lactic acid production from xylose by Lactobacillus pentosus. J Chem Technol Biotechnol 88:594–598. https://doi.org/10.1002/jctb.3867
Pusch O, Kalyanaraman R, Tucker LD, Wells JM, Ramratnam B, Boden D (2006) An anti-HIV microbicide engineered in commensal bacteria: secretion of HIV-1 fusion inhibitors by lactobacilli. AIDS 20:1917–1922. https://doi.org/10.1097/01.aids.0000247112.36091.f8
Russell WM, Klaenhammer TR (2001) Efficient system for directed integration into the Lactobacillus acidophilus and Lactobacillus gasseri chromosomes via homologous recombination. Appl Environ Microbiol 67:4361. https://doi.org/10.1128/aem.67.9.4361-4364.2001
Saha BC, Iten LB, Cotta MA, Wu YV (2005) Dilute acid pretreatment, enzymatic saccharification and fermentation of wheat straw to ethanol. Process Biochem 40:3693–3700. https://doi.org/10.1016/j.procbio.2005.04.006
Sambrook J (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Sanozky-Dawes R, Selle K, Flaherty S, Klaenhammer T, Barrangou R (2015) Occurrence and activity of a type II CRISPR-Cas system in Lactobacillus gasseri. Microbiology 161:1752–1761. https://doi.org/10.1099/mic.0.000129
Sauer M, Russmayer H, Grabherr R, Peterbauer CK, Marx H (2017) The efficient clade: lactic acid bacteria for industrial chemical production. Trends Biotechnol 35:756–769. https://doi.org/10.1016/j.tibtech.2017.05.002
Sørvig E, Mathiesen G, Naterstad K, Eijsink VGH, Axelsson L (2005) High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology 151:2439–2449. https://doi.org/10.1099/mic.0.28084-0
Sun Z et al (2015) Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun 6:8322. https://doi.org/10.1038/ncomms9322
Štafa A, Miklenić MS, Zandona A, Žunar B, Čadež N, Petković H, Svetec IK (2017) In Saccharomyces cerevisiae gene targeting fidelity depends on a transformation method and proportion of the overall length of the transforming and targeted DNA. FEMS Yeast Res 17:fox041. https://doi.org/10.1093/femsyr/fox041
Talebnia F, Karakashev D, Angelidaki I (2010) Production of bioethanol from wheat straw: an overview on pretreatment, hydrolysis and fermentation. Bioresour Technol 101:4744–4753. https://doi.org/10.1016/j.biortech.2009.11.080
The R Core Team (2019) R: a language and environment for statistical computing 3.5.2 edn. R Foundation for Statistical Computing, Vienna
Townsend TJ, Sparkes DL, Wilson P (2017) Food and bioenergy: reviewing the potential of dual-purpose wheat crops. GCB Bioenergy 9:525–540. https://doi.org/10.1111/gcbb.12302
Tsuge Y, Kato N, Yamamoto S, Suda M, Jojima T, Inui M (2019) Metabolic engineering of Corynebacterium glutamicum for hyperproduction of polymer-grade L- and D-lactic acid. Appl Microbiol Biotechnol 103:3381–3391. https://doi.org/10.1007/s00253-019-09737-8
Tsuji H (2005) Poly(lactide) Stereocomplexes: formation, structure, properties, degradation, and applications. Macromol Biosci 5:569–597. https://doi.org/10.1002/mabi.200500062
Wegkamp A, Starrenburg M, de Vos WM, Hugenholtz J, Sybesma W (2004) Transformation of folate-consuming Lactobacillus gasseri into a folate producer. Appl Environ Microbiol 70:3146. https://doi.org/10.1128/aem.70.5.3146-3148.2004
Yang B, Dai Z, Ding SY, Wyman CE (2011) Enzymatic hydrolysis of cellulosic biomass. Biofuels 2:421–449. https://doi.org/10.4155/BFS.11.116
Yi X, Zhang P, Sun J, Tu Y, Gao Q, Zhang J, Bao J (2016) Engineering wild-type robust Pediococcus acidilactici strain for high titer L- and D-lactic acid production from corn stover feedstock. J Biotechnol 217:112–121. https://doi.org/10.1016/j.jbiotec.2015.11.014
Yuan S-F et al (2018) Production of optically pure L(+)-lactic acid from waste plywood chips using an isolated thermotolerant Enterococcus faecalis SI at a pilot scale. J Ind Microbiol Biotechnol 45:961–970. https://doi.org/10.1007/s10295-018-2078-5
Zhang C, Zhou C, Assavasirijinda N, Yu B, Wang L, Ma Y (2017) Non-sterilized fermentation of high optically pure D-lactic acid by a genetically modified thermophilic Bacillus coagulans strain. Microb Cell Fact 16:213. https://doi.org/10.1186/s12934-017-0827-1
Zhang Y, Chen X, Luo J, Qi B, Wan Y (2014) An efficient process for lactic acid production from wheat straw by a newly isolated Bacillus coagulans strain IPE22. Bioresour Technol 158:396–399. https://doi.org/10.1016/j.biortech.2014.02.128
Zhang Y, Kumar A, Hardwidge PR, Tanaka T, Kondo A, Vadlani PV (2016) D-lactic acid production from renewable lignocellulosic biomass via genetically modified Lactobacillus plantarum. Biotechnol Prog 32:271–278. https://doi.org/10.1002/btpr.2212
Zhao Y, Wang Y, Zhu JY, Ragauskas A, Deng Y (2008) Enhanced enzymatic hydrolysis of spruce by alkaline pretreatment at low temperature. Biotechnol Bioeng 99:1320–1328. https://doi.org/10.1002/bit.21712
Funding
This work was supported by the Croatian Science Foundation (Grant Numbers IP-11-2013-9158-SPECH-LRM and IP-01-2018-9717-SPB-LCF, awarded to Prof Božidar Šantek).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study design. Material preparation, data collection and analysis were performed by BŽ, AT, JLP, MSM, AS, NM and MN. BŠ and IKS initiated and coordinated the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Žunar, B., Trontel, A., Svetec Miklenić, M. et al. Metabolically engineered Lactobacillus gasseri JCM 1131 as a novel producer of optically pure L- and D-lactate. World J Microbiol Biotechnol 36, 111 (2020). https://doi.org/10.1007/s11274-020-02887-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-020-02887-2