
Abstract
Modified nucleosides produced by Streptomyces and related actinomycetes are widely used in agriculture and medicine as antibacterial, antifungal, anticancer and antiviral agents. These specialized small-molecule metabolites are biosynthesized by complex enzymatic machineries encoded within gene clusters in the genome. The past decade has witnessed a burst of reports defining the key metabolic processes involved in the biosynthesis of several distinct families of nucleoside antibiotics. Furthermore, genome sequencing of various Streptomyces species has dramatically increased over recent years. Potential biosynthetic gene clusters for novel nucleoside antibiotics are now apparent by analysis of these genomes. Here we revisit strategies for production improvement of nucleoside antibiotics that have defined mechanisms of action, and are in clinical or agricultural use. We summarize the progress for genetically manipulating biosynthetic pathways for structural diversification of nucleoside antibiotics. Microorganism-based biosynthetic examples are provided and organized under genetic principles and metabolic engineering guidelines. We show perspectives on the future of combinatorial biosynthesis, and present a working model for discovery of novel nucleoside natural products in Streptomyces.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nucleoside-containing small-molecule natural products are almost exclusively of microbial origin. These secondary metabolites, produced primarily by soil microbes such as Streptomyces, may have specialized functions in nature, but several have been used in human therapeutics and agriculture for some time because of their excellent antibacterial, antifungal and other bioactivities. Nucleoside antibiotics encompass diverse chemical structures with few features in common except a nucleoside-derived component. As amino acids are the primary building blocks of canonical bases, biosynthesis of nucleoside antibiotics is closely linked to amino acid metabolism in microbes. Often the structures are modified with additional amino acid-derived components that further solidifies this link. Some nucleoside metabolites are also modified with fatty acids (or polyketides) and carbohydrates. It is this structural diversity that enables nucleoside antibiotics to interact with a variety of cellular targets, interfering with biological processes including cell wall biosynthesis, protein synthesis, and nucleic acid replication. As the quest for new antibiotics continues, and the cost of microbial genome sequencing and engineering is no longer a barrier, it is expected that more nucleoside antibiotics with novel structures and unique mechanism of actions will be discovered in the coming years.
A comprehensive review on natural nucleoside antibiotics was reported over 25 years ago, which in total covered ~236 structures (Isono 1991). Two reviews since that time have focused on structure–function relationship and the enzymatic mechanism of peptidyl nucleoside antibiotics, providing the first synopsis of the making of these complex structures (Walsh and Zhang 2011; Winn et al. 2010). Two more recent reviews scrutinized the molecular and genetic basis of nucleoside antibiotic biosynthesis with a first outlook on biotechnological applications (Chen et al. 2016; Niu and Tan 2015). Here, we supplement the topic with more recent updates, as well as highlight the commercial development of nucleoside antibiotics. We have limited the topic to actinomycete-produced nucleoside antibiotics. Even so, this review is by far from comprehensive but is intended to summarize recent, key findings for model systems.
Representative nucleoside antibiotics in recent development
Inhibitors of bacterial cell wall synthesis
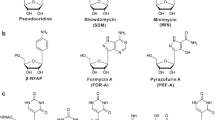
The bacterial cell wall differs from that of all other organisms by a supporting polymeric structure—peptidoglycan—that is assembled by at least 11 highly conserved enzymes. One of these essential enzymes is MraY, which transfers a UDP-N-acetylmuramic acid (UDP-MurNAc) linked pentapeptide onto a cell membrane-anchored lipid carrier, bactoprenol-phosphate, thereby initiating the lipid cycle of cell wall biosynthesis. The resulting lipid-linked MurNAc-pentapeptide (termed Lipid I), is subsequently modified with an N-acetylglucosamine in the cytoplasm to form Lipid II prior to translocation to the periplasmic side of the membrane. Although catalyzing a group transfer reaction, MraY has historically been termed translocase I. Several steps in the cell wall synthesis are known to be inhibited by natural products—perhaps best exemplified with transpeptidase inhibitors, the β-lactams—with the MraY reaction inhibited by at least five structurally distinct classes of uridyl peptides (Fig. 1). Many of the uridyl peptide antibiotics have been characterized as competitive inhibitors of MraY possibly mimicking a UDP-linked substrate or product; a crystal structure of MraY in complex with the uridyl peptide muraymycin was recently published (Chung et al. 2016). In general uridyl peptide antibiotics have very good in vivo efficacy against Gram-positive human pathogens such as methicillin-resistant Staphylococcus aureus, and vancomycin-resistant Enterococcus. Derivatives of pacidamycin and muraymycin have been pursued as drug leads against the Gram-negative pathogen, Pseudomonas aeruginosa. A U.S. company, Sequella, is currently developing a semi-synthetic capuramycin for the clinical treatment of Crohn’s disease and Clostridium difficile infections.

Structures of representative nucleoside antibiotics produced by Streptomyces species
All of the uridyl peptide antibiotics were originally discovered as secondary metabolites of Streptomyces or a related actinomycete. Like other specialized microbial products, biosynthetic genes of uridyl peptides are clustered in the genome, a phenomenon that has dramatically facilitated the identification and functional assignment of the enzymes for building complex structures from primary metabolites. Other important genes, including those involved in self-resistance and regulation of the biosynthesis pathway, are typically located within the same cluster. The first two uridyl peptide gene clusters identified were for the caprazamycins from Streptomyces sp. MK730-62F2, and A-500359s (a capuramycin type) from Streptomyces griseus SANK 60196 (Funabashi et al. 2009; Kaysser et al. 2009) (Fig. 1). Caprazamycins contain a per-methylated L-rhamnose moiety that is also found in other antibiotics, hence a sugar O-methyltransferase gene was used as a probe to identify the genetic locus. A UDP-glucose-4,6-dehydratase gene was serendipitously used as a probe to identify the A-500359s cluster because an unsaturated hexuronic acid in the structure was thought to be derived from a hexose sugar, although it is now known that this gene is likely not required. Pacidamycins, which contain a peptide with a 2,3-diaminobutyric acid residue linked to a nucleoside core (Fig. 1), were expected to be biosynthesized, in part by a non-ribosomal peptide synthetase (NRPS). Genes encoding a cysteine synthase and an argininosuccinate lyase, both demonstrated for the biosynthesis of a 2,3-diaminobutyric acid residue in other metabolites, were used as probes to search within the genome of a producing strain, Streptomyces coeruleorubidus NRRL 18370 (Zhang et al. 2010). Several NPRS genes were indeed identified in the pacidamycin gene cluster. The gene cluster for tunicamycin (Fig. 1), which contains an acylated disaccharide bonded to a nucleoside core, was identified in the genome of Streptomyces chartreusis NRRL 3882 by searching for genes for a 1 → 1-linking glycosyltransferase and a deacetylase involved in lipid processing (Wyszynski et al. 2010). The gene cluster for muraymycins (Fig. 1), which consist of a unique peptide but share a 5′-C-glycyluridine (GlyU) core structure like that in caprazamycins, was identified in the genome of Streptomyces sp. NRRL 30471 by searching for a transaldolase gene proposed to be essential for GlyU formation in the caprazamycins (Cheng et al. 2011).
The five structural classes of MraY inhibitors share a uridine-derived core but vary by the modifications of the uridine as well as the structurally diverse components—peptidyl, fatty acyl, or saccharidyl groups—connected to the core. Despite the structural differences, some similarities in the biosynthetic mechanism between two or more of the classes has been revealed from the genetic and follow-up biochemical studies (Walsh and Zhang 2011). Of note is the biosynthesis of GlyU in caprazamycins and the uridine-5′-carboxamide (CarU) found in capuramycins (Fig. 1). GlyU was initially speculated to be formed by an aldol-like reaction between uridine-5′-aldehyde and the α-carbon of glycine. Biochemical studies revealed uridine-5′-aldehyde is indeed the first intermediate yet is unexpectedly formed by a non-heme, Fe(II)-dependent α-ketoglutarate: UMP dioxygenase. Likewise, in contrast to the expectation of glycine as a co-substrate for the subsequent catalytic step, the C–C bond formation is instead catalyzed by a PLP-dependent transaldolase that uses l-threonine as a glycyl unit donor. Although the carboxamide group in the CarU of capuramycins looks different, it too was biochemically shown to be derived from l-threonine using isotopic enrichment studies, and proceed via a GlyU intermediate (Cai et al. 2016). In fact the homologous genes are found in all caprazamycin- and capuramycin-type gene clusters. The confirmation of these two novel enzymatic functions led to the employment of the genes as probes to search for new translocase I inhibitors potentially produced by rare actinomycetes. PCR-screening a library of ~2500 strains led to the identification of a novel uridyl peptide antibiotic, sphaerimicin, from Sphaerisporangium sp. SANK 60911 (Funabashi et al. 2013). A comparable genome mining strategy was most recently applied to the tunicamycin group, which resulted in the discovery of quinovosamycin that has an N-acetylquinovosamine sugar residue, produced by Streptomyces niger NRRL B-3857 (Price et al. 2016).
Inhibitors of fungal chitin synthesis
Chitin is a main component of fungal cell wall, required for the pathogenesis or survival of most fungi. Compounds targeting chitin biosynthesis often have a high therapeutic index, and can be developed as antifungals for human use. Nikkomycins and polyoxins (Fig. 1), structurally resembling UDP-N-acetylglucosamine that is a substrate of chitin synthase, are potent competitive inhibitors of the enzyme-catalyzed reaction. Nikkomycin Z has received a fast track designation from the United States Food and Drug Administration for treatment of a human fungal infection, coccidioidomycosis (valley fever), and is currently undergoing clinical trials (Nix et al. 2009). Polyoxins are non-toxic and environmentally friendly, increasingly utilized in China to control pathogenic fungi of tobacco brown spot and rice blast (Chen et al. 2016).
The nik cluster in Streptomyces tendae Tü 901 is one of the earliest studied nucleoside antibiotic gene clusters, which was discovered by a reverse genetic approach starting from a comparison of the proteomes of nikkomycin producing strains and non-producing strains generated by random mutagenesis (Bormann et al. 1996). Later, NikO was shown to catalyze the formation of 3′-enoylpyruvate-UMP from UMP and phosphoenolpyruvate (PEP), representing a branch point of nucleotide metabolism in the producing strain. Since nikkomycin and polyoxin both contain a 5′-amino-5′-carboxyl-5′-deoxyribose core structure, often referred to as an aminohexuronic acid, nikO was subsequently used as a probe to screen a genomic library of the polyoxin producer Streptomyces cacaoi. This resulted in the identification of the pol cluster and a proposed biosynthetic pathway (Chen et al. 2009). Although the framework of the two chitin synthase inhibitors is quiet similar, significant structural variations are found in the peptide portion of the two molecules (Niu and Tan 2015). While the underlying biochemical mechanism is intriguing but not well understood, robust genetic systems have been established in both nikkomycin and polyoxin producing strains, laying a foundation for the recent engineering of this class of molecules.
Other inhibitors with various modes of action
Albomycins (griseins)—Many microbial product derived antibiotics act by inhibiting protein synthesis in pathogens, including several examples of nucleoside antibiotics. Albomycin δ2, a cytosine containing peptidyl nucleoside (Fig. 1), is perhaps one of the most potent antibiotics ever discovered in nature (Reynolds and Waksman 1948). Its high antibacterial potency, exemplified by the MIC <10 nM against Escherichia coli and Streptococcus pneumoniae (Braun et al. 2009), is a result of a Janus-faced molecular structure. One face is a ferrichrome siderophore that binds iron (III) with high affinity; the other face is an unusual amino acid with several interesting features such as thioether ring, stereo-specific secondary alcohol and a methylated and carbamoylated modified cytosine base. A serine residue connects the ferrichrome, which is a nonribosomal peptide, and the modified nucleoside. The serine-linked nucleoside of albomycins can be viewed as a mimic of seryl-AMP, an intermediate of seryl-tRNA synthetase (SerRS) catalyzed reaction. This half molecule of albomycin competitively inhibits serine charging of a tRNA in the presence of ATP. When pathogens battle for iron to survive in the host, they actively recognize and take up albomycins through their iron transport system. Once inside the cell, the war face of albomycin is unveiled by proteolytic action of numerous peptidases, and the SerRS inhibitor is thereby released to block protein synthesis. The albomycin gene cluster from S. griseus ATCC 700974 was cloned by searching for biosynthetic genes of the ferrichrome-type siderophore, and a biosynthetic pathway was proposed (Zeng et al. 2012). While some steps in the biosynthetic process remain to be determined, metabolic evidence was provided to support that homocysteine is a key precursor of albomycin biosynthesis (Kulkarni et al. 2016). It was also discovered that minimally two SerRS genes are encoded in the genome of albomycin producing strain. One is located within the gene cluster and confers self-resistance to the albomycin producer. A second serRS that is albomycin-sensitive is located elsewhere in the chromosome (Zeng et al. 2009). The discovery of this genetic deployment suggests that mining for duplicated genes of aminoacyl-tRNA synthetase in Streptomyces genomes should be an innovative strategy for discovering novel antibiotic inhibitors.
Albomycin is just one example of the antibiotics containing a cytosine base. Other examples include blasticidin S (antifungal) and amicetin (antibacterial) (Fig. 1), both of which target protein synthesis by binding to the ribosome. The biosynthesis of blasticidin S and the related arginomycin, mildiomycin and gougerotin have been extensively reviewed (Chen et al. 2016). The biosynthetic gene cluster for amicetin has only recently been discovered. New members of the amicetin family have been reported, showing promising anti-mycobacterial activity (Aksoy et al. 2016; Bu et al. 2014).
A201A and ascamycin—A201A and ascamycin represent purine nucleosides that inhibit bacterial protein synthesis. A201A was recently shown to interfere with tRNA accommodation at the A site of ribosome, thereby inhibiting peptide bond formation (Polikanov et al. 2015). This mechanism is similar to puromycin, which resembles the 3′-end of aminoacylated tRNA and prematurely terminates polypeptide chain elongation on the ribosome. A201A and puromycin both consist of a 2′-amino ribose structure (Fig. 1). The puromycin gene cluster (pur) in Streptomyces alboniger was identified decades ago by reverse genetics targeting genes involved in methylation (Lacalle et al. 1993). Recent bioprospecting of marine actinomycetes led to the isolation and identification of A201A as a product of a new genus type strain, Marinoactinospora thermotolerans SCSIO 00652 (Zhu et al. 2012). Some A201A biosynthetic genes have modest sequence homology with the pur genes. Ascamycin is a narrow-spectrum antibiotic. Its gene cluster from Streptomyces sp. JCM9888 was identified by genome mining for sulfate metabolic genes associated with a gene cluster (Zhao et al. 2014). The key biosynthetic steps of A201A and ascamycin remain to be determined, including the 3′-amino substitution of ribose in A201A and chlorination of adenine in ascamycin.
Toyocamycin and sinefungin—Toyocamycin (Fig. 1) inhibits the maturation of ribosomal RNA and cleavage of stress-induced mRNA in cancer cells. It has good activity against a broad range of plant pathogenic fungi and is being pursued as a fungicide. The biosynthesis of toyacamycin starts from the nucleotide GTP, and can be converted to a fermentation congener sangivamycin by a nitrile hydratase (McCarty and Bandarian 2008). The hydratase gene was used as the probe to screen a genomic library for the identification of toy gene cluster in Streptomyces rimosus ATCC 14673. Sinefungin is structurally similar to S-adenosyl-methionine (SAM) and competitively inhibits SAM-dependent methyltransferase. Sinefungin exhibits good antifungal, antiviral and anti-parasitic activities, and more recently being explored as a chemical probe to understand epigenetic regulation (Zheng et al. 2012). To date, a sinefungin gene cluster has not been identified though Streptomyces griseolus and Streptomyces incarnatus NRRL 8089 are the known producers.
Genetic and metabolic engineering for improved production of nucleoside antibiotics
The history and engineering principles for improving antibiotic production by microbes were previously reviewed (Lauren et al. 2011; Rokem et al. 2007). Although classical methods involving random mutagenesis are effective for developing high-titer production strains, an engineering approach exploring new genomic tools and genome-wide targets, demonstrates increasing understanding of actinomycete biology and the control factors of antibiotic production at a system level. Below are the strategies that have been successfully used for nucleoside antibiotics.
Heterologous expression—An identified gene cluster can be cloned into a plasmid for heterologous expression in an actinomycete host that has a known metabolic profile, such as Streptomyces coelicolor and Streptomyces lividans. The experiment not only defines the minimal number of genes required but also facilitates gene analysis. Furthermore, the genetic chassis can be further engineered by deletion of competing pathways or increasing certain metabolic flux for improved production of the final antibiotic products. Puromycin and nikkomycin were the first two heterologously produced nucleoside antibiotics, both in S. lividans (Bormann et al. 1996; Lacalle et al. 1993). Caprazamycin, tunicamycin and gougerotin have now been heterologously produced in S. coelicolor A3(2) derived strains (Du et al. 2013; Kaysser et al. 2009; Wyszynski et al. 2010), and blasticidin S, pacidamycin, amicetin and muraymycin in S. lividans TK24 (Cone et al. 1998; Rackham et al. 2010; Xu et al. 2013; Zhang et al. 2012). Interestingly, production levels of caprazamycin and pacidamycin in the heterologous systems were reportedly higher than in the native producers. Nikkomycin and polyoxin gene clusters were separately expressed in a non-producing mutant of the other host (Zhai et al. 2012; Li et al. 2011). Both experiments resulted in successful identification and production of hybrid nucleoside-like antibiotics as well as analogs of the parent molecules.
Manipulation of regulatory genes—Antibiotic production by Streptomyces in nature is very complicated as it is governed by at least three levels of regulatory factors: global master, pleotropic and cluster-situated regulators. Molecular details on the regulatory mechanism of some nucleoside antibiotic gene clusters have been reviewed (Niu and Tan 2015). Regulations of nikkomycin biosynthesis in Streptomyces ansochromogenes and polyoxin biosynthesis in S. cacaoi subsp. asoensis, have served as excellent models for elucidating the complex regulatory cascade, and demonstrate that transcription of a gene cluster can be linked to cellular metabolic pools such as ADP/ATP. For production improvement, a substantial effort has focused on cluster-situated and pathway specific regulators. Constructing mutant strains that have an over-expressed activator gene or a deleted repressor gene has been a simple and straightforward solution to improve yields. For example, sansamycin is one of the pacidamycin-related MraY inhibitors. Its production in Streptomyces sp. strain SS increased 100% when an activator gene ssaA was expressed under the control of constitutive promoter ermE*p. The expression construct was inserted into the chromosome, generating a relatively stable strain that is a prerequisite for utility in industrial production (Li et al. 2013). In the muraymycin gene cluster, the function of activator mur33 is likely similar to ssaA, but mur33 expression in this case is negatively regulated by an additional repressor mur34. Deletion of the mur34 gene in Streptomyces sp. NRRL30471 generated a mutant that was able to produce 10-fold more muraymycins (Xu et al. 2013). At the molecular genetic level, some regulatory genes like the TetR-family regulator gouR in the gougerotin gene cluster in S. graminearus, could play a dual role in antibiotic biosynthesis. The gouR represses the transcription of 11 gou biosynthetic genes while activates an exporter gene gouM. Deletion of gouR resulted in a mutant strain producing twice as much gougerotin compared to the wild-type, but overexpression of the same gene had no effect on the production (Wei et al. 2014). For nikkomycin and polyoxin, overexpression of cluster-situated activator sanG or polR from a chromosomally inserted construct resulted in overproduction in each case. A promoter engineering strategy has also been tested in nucleoside antibiotic production systems (Du et al. 2013). Five different promoter sequences were cloned separately to direct the transcription of sanG in S. ansochromogenes. The resulting strains all showed an increase in nikkomycin production, where a strong and constitutive promoter hrdBp had the best effect. The hrdBp was then used to replace major promoters in the gou operon harbored on a single cosmid. When the cosmid was introduced into a S. coelicolor host, the construct enabled gougerotin production 10-fold higher than the control cosmid carrying an unmodified gou operon. However, not every antibiotic biosynthetic gene cluster encodes a pathway specific regulator. An old example is found with the puromycin gene cluster in Streptomyces alboniger (Tercero et al. 1998). Expression of pur genes was dependent on pleotropic regulators including bldA encoding the rare codon tRNALeu. Finally, it is noteworthy that targeting a pathway specific regulator for genetic manipulation may give a surprisingly large increase of the final product. For example, deletion of a repressor gene mtdA in M. thermotolerans SCSIO 00652 improved the yield of nucleoside antibiotic A201A 25-fold, from 12.5 mg/L in the wild-type to 312.5 mg/L in the ∆mtdA mutant strain. This range of yield improvement is critical for large scale fermentation in industrial application.
Engineering precursor supply pathway—Genetic alteration of pleotropic or global regulators would impose negative physiological consequences, possibly impeding microbial growth. A distinct strategy for improving production is to enhance the intersection of primary and secondary metabolism, and increase metabolic flux into a biosynthetic pathway. Biosynthesis of albomycin, which does not have a pathway specific regulator in the gene cluster, represents a recent example of such approach for production improvement (Kulkarni et al. 2016). Based on the analysis of albomycin production by S. griseus grown under an iron deficient condition or condition that inhibited cellular sulfur metabolism, the sulfur atom in albomycin was shown to be most likely derived from homocysteine. The results guided the ensuing genetic engineering, as genes involved in transsulfuration and direct sulfhydrylation pathways that increased intracellular homocysteine concentration were assembled and inserted into the chromosome for overexpression under the control of the hrdBp promoter. The resulting strain increased albomycin production two times, which was further improved to four times by engineering constitutive overexpression of the abmD gene into the strain. AbmD is proposed to catalyze a reaction between homocysteine and an activated serine derivative; it is likely the first step in formation of the thioether ring and should be a rate-limiting enzyme in the pathway. The preliminary but demonstrable metabolic engineering result provides an impetus for exploring strategies that aim to divert other carbon and nitrogen sources into albomycin biosynthesis, particularly those that do not negatively impact cell physiology.
Engineering growth media—Genetic engineering of primary metabolic pathways for production improvement is constricted by nutrients in the growth medium. Microbiological media are developed for simplicity, accessibility and economic reasons. When used for large scale production, the media need be modified according to manufacturing needs. Recently, only the optimization processes for nikkomycin Z (Stenland et al. 2013) and A-500359s (Muramatsu et al. 2006) have been thoroughly explored and reported in detail because of significant commercial interests. The highest yield of nikkomycin Z was ~2.3 g/L, and A-500359s >0.6 g/L, representing a maximum of 600 times improvement. Nikkomycin Z production could also be enhanced by genetic deletion of a pathway that generates side products, and further supplementing the production medium with uracil (Liao et al. 2009). Media optimization was also key to the isolation and elucidation of sphaerimicin (Funabashi et al. 2013), which was not detectable when the producer was grown under a majority of growth conditions. In this case different media were first screened for inducing the expression of biosynthetic genes that were otherwise not expressed. Finally, classical medium optimization coupled with new omics technology has just been reported for improving the fermentation process for polyoxin production (Wu et al. 2016).
Engineering whole-cell transcriptional and translational machineries—Ochi and co-workers have developed facile approaches for activating silent gene clusters in Streptomyces as well as rapidly increasing product yields (Ochi and Hosaka 2013). Drug-induced mutations in the rpoB gene (RNA polymerase β-subunit) or ribosomal protein genes were shown to dramatically increase the productivity of many Streptomyces producers, which includes the dramatic increase of the nikkomycin industrial production mentioned above. The so-called ribosome engineering approach was applied to the production of singfungin, which resulted in a seven-fold increase (Fukuda et al. 2010). Alternatively, disruption of a ribosome assembly cofactor in S. coelicolor led to enhancement of the heterologous production of polyoxin and nikkomycin (Pan et al. 2013). Production of the promising fungicide toyocamycin by S. diastatochromogenes 1628 was improved to from 152 to 684 mg/L as a result of rpoB mutations, a 450% improvement compared to the wild-type strain (Ma et al. 2016).
Discovery and generation of new nucleoside antibiotic structures
Nucleoside antibiotics are a very large family of chemical structures. New nucleoside natural products continue to be discovered, exemplified by recent isolation and structure elucidation of the strepturidin produced by Streptomyces albus DSM 40763 (Pesic et al. 2014). Streptomyces and other actinomycetes are now well appreciated for having a huge, untapped biosynthetic potential. We are just beginning to understand the ingenious mechanisms for synthesizing these structurally diverse and useful molecules. Figure 2 shows the work in progress that will lead to discovery and generation of new nucleoside antibiotic structures.

Flow chart for the discovery of microbial nucleoside natural products
Mutasynthesis is a common approach that involves isolating variants of a known microbial product from mutant strains. The first example of mutasynthesis applied to nucleoside antibiotics was the identification of nikkomycin Y/Z series (Delzer et al. 1984). More recently, mutasynthesis has evolved to a method that combines targeted gene inactivation to generate a mutant strain unable to make a building block, with feeding of an unnatural, chemically synthesized precursor as a substitute for the building block. This so-called precursor directed biosynthesis takes advantage of the relaxed substrate specificity of secondary metabolic enzymes that is often observed. The goal, therefore, is to generate a focused library of analogs for investigating the structure–activity relationship of a given microbial natural product. Some examples include the biosynthesis of nikkomycin Px and Pz, the production of halogen- or methyl- substituted pacidamycins, and the generation of sansamycins with multiple alterations (Chen et al. 2016). For the sansamycin case, the biosynthetic machinery of Streptomyces sp. SS appears particularly versatile (Shi et al. 2016; Xie et al. 2010). By in vivo analysis of the genes encoding the NRPS and inactivation of another gene required for supplying an unusual precursor, more than a dozen sansamycin analogs were prepared and produced in sufficient quantities for measuring potency against drug-resistant pathogens. One new compound was eight-fold more potent against Mycobacterium tuberculosis than the parent, and a few of the new compounds also demonstrated improved stability. Mutasynthesis of the capuramycin-group antibiotics has also been reported. Several analogs were prepared by taking advantage of an extremely substrate-flexible transacylase CapW that incorporates the aminocaprolactam component of capuramycin. Using a chemoenzymatic approach, 43 aminocaprolactam-substituted analogs were initially prepared, and some had increased antibacterial activity against M. tuberculosis. A mutasynthetic approach was subsequently adopted, first by inactivating the NRPS gene capU responsible for generating the aminocaprolactam from l-Lys, followed by feeding different building blocks to the mutant strain (Liu et al. 2016).
Mutasynthesis is only one of several approaches within combinatorial biosynthesis that can be used for generating molecular diversity originated from natural products. Combinatorial biosynthesis also incorporates a suite of synthetic biology techniques to create new small molecules using engineered strains (Kim et al. 2015). The aforementioned mix-and-match of nik and pol genes that resulted in the production of hybrid molecules by Streptomyces is the first example being applied to the biosynthesis of nucleoside antibiotics. Inspecting the structures of MraY inhibitors (Fig. 1), one can notice that nature has already been practicing combinatorial biosynthesis with different actinomycetes: relatively few UDP mimics are linked to a large variety of peptidyl, fatty acyl and saccharidyl structures that fine-tune the interactions of the peptide, lipid and sugar components of the substrates with the target MraY. Blasticidin S group antibiotics represent another example of how nature has enabled Streptomyces to diversify pharmacologically active structures. The degree of diversification is dependent on the inhibited target, either a biomolecule (e.g. MraY or ribosomal complex) or a biochemical process. Albomycins represent a third example of products that have resulted from nature’s version of combinatorial biosynthesis. The seryl-AMP mimic can be linked to not only a ferrichrome siderophore at the N-terminal in albomycin δ2, but also possibly other peptides, polysaccharides or structural motifs that will facilitate its uptake across cell membranes. Albomycin-like structures that don’t appear to contain the ferrichrome are known to be produced by a number of actinomycetes but await identification (Zeng et al. 2012).
To discover novel bioactive microbial natural products containing a nucleoside structure, we foresee two strategies that will likely have a high success rate: (1) Genome mining of microorganisms, especially Streptomyces and rare actinomycetes, for new members of the existing groups of nucleoside antibiotics. It is readily predicted that diverse structures of polyoxin-nikkomycin group and puromycin-A201A group exist, with structural novelties as those found for sphaerimicin and quinovosamycin; and (2) Screening microbial culture extracts in cell-based bioassay or biochemical reactions targeting biological pathways or steps with a nucleotide as a co-factor. Although the latter approach is relatively traditional and many nucleic acid-associated pathways or nucleotide reactions were screened in the past, new prokaryotic or eukaryotic targets and pathways involving nucleotides continue to be discovered. One such example is the universal bacterial second messenger cyclic di-GMP, as cyclic di-GMP has now been shown to regulate biofilm formation, motility, virulence, cell cycle and differentiation, etc. (Römling et al. 2013). Its biosynthesis, degradation and signaling pathways could provide biochemical screening targets; the diverse phenomena observed at an organismal level might offer convenient methods for phenotypic screening. Important for the success of both strategies is the availability of strains for sequencing and natural product extracts collection. It is noteworthy that a large and fully tractable collection of natural product-producing strains and their extracts have been made available through a non-profit organization, the Natural Products Discovery Institute (Doylestown, PA). This library, as well as the continued exploration of unique ecological niches and making these strains/extracts available to the community, give virtually endless opportunities for the discovery of new nucleoside antibiotics and development of combinatorial biosynthesis.
References
Aksoy SC, Uzel A, Bedir E (2016) Cytosine-type nucleosides from marine-derived Streptomyces rochei 06CM016. J Antibiot 69:51–56
Bormann C, Möhrle V, Bruntner C (1996) Cloning and heterologous expression of the entire set of structural genes for nikkomycin synthesis from Streptomyces tendae tü901 in Streptomyces lividans. J Bacteriol 178:1216–1218
Braun V, Pramanik A, Gwinner T, Koberle M, Bohn E (2009) Sideromycins: tools and antibiotics. Biometals 22:3–13
Bu Y-Y, Yamazaki H, Ukai K, Namikoshi M (2014) Anti-mycobacterial nucleoside antibiotics from a marine-derived Streptomyces sp. TPU1236A. Mar Drugs 12:6102
Cai W, Goswami A, Yang Z et al (2016) The biosynthesis of capuramycin-type antibiotics: identification of the A-102395 biosynthetic gene cluster, mechanism of self-resistance, and formation of uridine-5′-carboxamide. J Biol Chem 290:13710–13724
Chen W, Huang T, He X et al (2009) Characterization of the polyoxin biosynthetic gene cluster from Streptomyces cacaoi and engineered production of polyoxin H. J Biol Chem 284:10627–10638
Chen W, Qi J, Wu P, Wan D, Liu J, Feng X, Deng Z (2016) Natural and engineered biosynthesis of nucleoside antibiotics in Actinomycetes. J Ind Microbiol Biotechnol 43:401–417
Cheng L, Chen W, Zhai L et al (2011) Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol BioSyst 7:920–927
Chung BC, Mashalidis EH, Tanino T et al (2016) Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 533:557–560
Cone MC, Petrich AK, Gould SJ, Zabriskie TM (1998) Cloning and heterologous expression of blasticidin S biosynthetic genes from Streptomyces griseochromogenes. J Antibiot 51:570–578
Delzer J, Fiedler HP, Müller H, Zähner H, Rathmann R, Ernst K, König WA (1984) New nikkomycins by mutasynthesis and directed fermentation. J Antibiot 37:80–82
Du D, Zhu Y, Wei J, Tian Y, Niu G, Tan H (2013) Improvement of gougerotin and nikkomycin production by engineering their biosynthetic gene clusters. Appl Microbiol Biotechnol 97:6383–6396
Fukuda K, Tamura T, Ito H, Yamamoto S, Ochi K, Inagaki K (2010) Production improvement of antifungal, antitrypanosomal nucleoside sinefungin by rpoB mutation and optimization of resting cell system of Streptomyces incarnatus NRRL 8089. J Biosci Bioeng 109:459–465
Funabashi M, Nonaka K, Yada C, Hosobuchi M, Masuda N, Shibata T, Van Lanen SG (2009) Identification of the biosynthetic gene cluster of A-500359s in Streptomyces griseus SANK60196. J Antibiot 62:325–332
Funabashi M, Baba S, Takatsu T et al (2013) Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew Chem Int Ed 52:11607–11611
Isono K (1991) Current progress on nucleoside antibiotics. Pharmacol Ther 52:269–286
Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B (2009) Identification and manipulation of the caprazamycin gene cluster lead to new simplified liponucleoside antibiotics and give insights into the biosynthetic pathway. J Biol Chem 284:14987–14996
Kim E, Moore BS, Yoon YJ (2015) Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat Chem Biol 11:649–659
Kulkarni A, Zeng Y, Zhou W, Van Lanen S, Zhang W, Chen S (2016) A branch point of Streptomyces sulfur amino acid metabolism controls the production of albomycin. Appl Environ Microbiol 82:467–477
Lacalle RA, Tercero JA, Vara J, Jimenez A (1993) Identification of the gene encoding an N-acetylpuromycin N-acetylhydrolase in the puromycin biosynthetic gene cluster from Streptomyces alboniger. J Bacteriol 175:7474–7478
Lauren BP, Yi T, Yit-Heng C (2011) Metabolic engineering for the production of natural products. Annu Rev Chem Biomol Eng 2:211–236
Li J, Li L, Tian Y, Niu G, Tan H (2011) Hybrid antibiotics with the nikkomycin nucleoside and polyoxin peptidyl moieties. Metab Eng 13:336–344
Li Q, Wang L, Xie Y, Wang S, Chen R, Hong B (2013) SsaA, a member of a novel class of transcriptional regulators, controls sansanmycin production in Streptomyces sp. strain SS through a feedback mechanism. J Bacteriol 195:2232–2243
Liao G, Li J, Li L, Yang H, Tian Y, Tan H (2009) Selectively improving nikkomycin Z production by blocking the imidazolone biosynthetic pathway of nikkomycin X and uracil feeding in Streptomyces ansochromogenes. Microb Cell Fact 8:1–7
Liu X, Jin Y, Cai W et al (2016) A biocatalytic approach to capuramycin analogues by exploiting a substrate permissive N-transacylase CapW. Org Biomol Chem 14:3956–3962
Ma Z, Luo S, Xu X, Bechthold A, Yu X (2016) Characterization of representative rpoB gene mutations leading to a significant change in toyocamycin production of Streptomyces diastatochromogenes 1628. J Ind Microbiol Biotechnol 43:463–471
McCarty RM, Bandarian V (2008) Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem Biol 15:790–798
Muramatsu Y, Arai M, Sakaida Y, Takamatsu Y, Miyakoshi S, Inukai M (2006) Studies on novel bacterial translocase I inhibitors, A-500359s. V. Enhanced production of capuramycin and A-500359 A in Streptomyces griseus SANK 60196. J Antibiot 59:601–606
Niu G, Tan H (2015) Nucleoside antibiotics: Biosynthesis, regulation, and biotechnology. Trends Microbiol 23:110–119
Nix DE, Swezey PR, Hector R, Galgiani JN (2009) Pharmacokinetics of nikkomycin Z after single rising oral doses. Antimicrob Agents Chemother 53:2517–2521
Ochi K, Hosaka T (2013) New strategies for drug discovery: activation of silent or weakly expressed microbial gene clusters. Appl Microbiol Biotechnol 97:87–98
Pan Y, Lu C, Dong H, Yu L, Liu G, Tan H (2013) Disruption of rimP-SC, encoding a ribosome assembly cofactor, markedly enhances the production of several antibiotics in Streptomyces coelicolor. Microb Cell Fact 12:1–16
Pesic A, Steinhaus B, Kemper S, Nachtigall J, Kutzner HJ, Höfle G, Süssmuth RD (2014) Isolation and structure elucidation of the nucleoside antibiotic strepturidin from Streptomyces albus DSM 40763. J Antibiot 67:471–477
Polikanov YS, Starosta Agata L, Juette Manuel F et al (2015) Distinct tRNA accommodation intermediates observed on the ribosome with the antibiotics hygromycin A and A201A. Mol Cell 58:832–844
Price NPJ, Labeda DP, Naumann TA et al (2016) Quinovosamycins: new tunicamycin-type antibiotics in which the α, β-1″,11′-linked N-acetylglucosamine residue is replaced by N-acetylquinovosamine. J Antibiot 69:637–646
Rackham EJ, Grüschow S, Ragab AE, Dickens S, Goss RJM (2010) Pacidamycin biosynthesis: identification and heterologous expression of the first uridyl peptide antibiotic gene cluster. ChemBioChem 11:1700–1709
Reynolds DM, Waksman SA (1948) Grisein, an antibiotic produced by certain strains of Streptomyces griseus. J Bacteriol 55:739–752
Rokem JS, Lantz AE, Nielsen J (2007) Systems biology of antibiotic production by microorganisms. Nat Prod Rep 24:1262–1287
Römling U, Galperin MY, Gomelsky M (2013) Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52
Shi Y, Jiang Z, Lei X et al (2016) Improving the N-terminal diversity of sansanmycin through mutasynthesis. Microb Cell Fact 15:1–15
Stenland CJ, Lis LG, Schendel FJ et al (2013) A practical and scalable manufacturing process for an anti-fungal agent, nikkomycin Z. Org Process Res Dev 17:265–272
Tercero JA, Espinosa JC, Jiménez A (1998) Expression of the Streptomyces alboniger pur cluster in Streptomyces lividans is dependent on the bldA-encoded tRNALeu. FEBS Lett 421:221–223
Walsh CT, Zhang W (2011) Chemical logic and enzymatic machinery for biological assembly of peptidyl nucleoside antibiotics. ACS Chem Biol 6:1000–1007
Wei J, Tian Y, Niu G, Tan H (2014) GouR, a TetR family transcriptional regulator, coordinates the biosynthesis and export of gougerotin in Streptomyces graminearus. Appl Environ Microbiol 80:714–722
Winn M, Goss RJM, Kimura K-i, Bugg TDH (2010) Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat Prod Rep 27:279–304
Wu G, Zhi W, Hu Y, Liang M, Yang W (2016) Comparative proteomic analysis of Streptomyces aureochromogenes under different carbon sources and insights into polyoxin production. Appl Biochem Biotechnol 180:491–503
Wyszynski FJ, Hesketh AR, Bibb MJ, Davis BG (2010) Dissecting tunicamycin biosynthesis by genome mining: Cloning and heterologous expression of a minimal gene cluster. Chem Sci 1:581–589
Xie Y, Xu H, Sun C, Yu Y, Chen R (2010) Two novel nucleosidyl-peptide antibiotics: sansanmycin F and G produced by Streptomyces sp SS. J Antibiot 63:143–146
Xu D, Liu G, Cheng L, Lu X, Chen W, Deng Z (2013) Identification of mur34 as the novel negative regulator responsible for the biosynthesis of muraymycin in Streptomyces sp. NRRL30471. PLoS ONE 8:e76068
Zeng Y, Roy H, Patil PB, Ibba M, Chen S (2009) Characterization of two seryl-tRNA synthetases in albomycin-producing Streptomyces sp. strain ATCC 700974. Antimicrob Agents Chemother 53:4619–4627
Zeng Y, Kulkarni A, Yang Z et al (2012) Biosynthesis of albomycin δ2 provides a template for assembling siderophore and aminoacyl-tRNA synthetase inhibitor conjugates. ACS Chem Biol 7:1565–1575
Zhai L, Lin S, Qu D, Hong X, Bai L, Chen W, Deng Z (2012) Engineering of an industrial polyoxin producer for the rational production of hybrid peptidyl nucleoside antibiotics. Metab Eng 14:388–393
Zhang W, Ostash B, Walsh CT (2010) Identification of the biosynthetic gene cluster for the pacidamycin group of peptidyl nucleoside antibiotics. Proc Nat Acad Sci 107:16828–16833
Zhang G, Zhang H, Li S et al (2012) Characterization of the amicetin biosynthesis gene cluster from Streptomyces vinaceusdrappus NRRL 2363 implicates two alternative strategies for amide bond formation. Appl Environ Microbiol 78:2393–2401
Zhao C, Qi J, Tao W, He L, Xu W, Chan J, Deng Z (2014) Characterization of biosynthetic genes of ascamycin/dealanylascamycin featuring a 5′-O-sulfonamide moiety in Streptomyces sp. JCM9888. PLoS ONE 9:e114722
Zheng W, Ibanez G, Wu H et al (2012) Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J Am Chem Soc 134:18004–18014
Zhu Q, Li J, Ma J et al (2012) Discovery and engineered overproduction of antimicrobial nucleoside antibiotic A201A from the deep-sea marine actinomycete Marinactinospora thermotolerans SCSIO 00652. Antimicrob Agents Chemother 56:110–114
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, S., Kinney, W.A. & Van Lanen, S. Nature’s combinatorial biosynthesis and recently engineered production of nucleoside antibiotics in Streptomyces . World J Microbiol Biotechnol 33, 66 (2017). https://doi.org/10.1007/s11274-017-2233-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-017-2233-6