
Abstract
The expansion of polyhydroxyalkanoates (PHAs) into the biodegradable polymers market is mainly prevented by their production process which is still complicated with a low efficiency, resulting in relatively expensive products. In this study, we developed a method that used the lipophilic fluorescent probe Nile Red (1 mg l−1 solution in DMSO) directly into the culture broth to stain the PHA inclusions inside bacterial cells followed by detection of the emitted fluorescence by both microscopic and spectrometric techniques. Epifluorescence microscopy provides a rapid tool to distinguish producing from non-producing bacterial species and the relative fluorescence intensity (FI) determined at the maximum of emission spectra in the wavelength region of 560–710 nm (λex: 543 nm), allows a fast assessment of the cultural conditions that may enhance PHA production yield. During two-step cultivation in 500-ml flasks with glucose as the sole carbon source, the method aimed to select bacterial strains efficient for PHA synthesis among a marine collection. Subsequently, the NR assay was used to determine the C0/N0 ratio of the producing media that may improve the polymer yield as well as to follow the time course of fermentation. Characterization by GC–MS and DSC confirmed the production of the P(3-HB) homopolymer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polyhydroxyalkanoates (PHAs) are synthesized as intracellular granules by numerous bacteria, and these products are emerging as substitutes to plastics derived from fossil-fuel. Despite the great potential of PHA in the fields of packaging and biomaterials (Khanna and Srivastava 2005; Chanprateep 2010), there is still much to be done to achieve a cost-effective commercial production. One critical starting point is the isolation of suitable bacterial strains that are capable of accumulating large amounts of PHAs. This is because the polymer content can vary from 30 to >80 % of the CDW according to the strain and cultural conditions (Kim and Lenz 2001). To date, bacteria from marine environments have been little studied for PHA production even though they are recognized as promising new sources of a vast number of biopolymers and other biologically active products.
PHA yield and polymer grade (e.g. monomer composition and molecular weight distribution) are also known to be closely related to numerous nutritional and cultivation parameters including the available carbon source, the concentration of medium components and the cultivation strategy (Lee et al. 2004; Johnson et al. 2010). Hence, we require a rapid and reliable method to diagnose the efficiency of a bacterial strain as well as the optimal cultivation conditions. Likewise, an on-line tool has yet to be developed to control production during the course of fermentation since current methods do not allow for real-time diagnosis and include many time-consuming and complicated steps i.e. biomass lyophilization, chemical or enzymatic extraction, precipitation of the polymeric material, followed by characterization, which is usually performed by GCMS, FTIR or NMR, to determine the PHA monomer structures.
PHAs are stored in up to ten spherical granules within the bacteria cell cytoplasm. Each granule is 0.2 ± 0.5 µm in diameter and is surrounded by a monolayer of phospholipids (Sudesh et al. 2000). The affinity of this layer with lipophilic dyes has long been recognized, and this property has been successfully employed to stain PHA inclusions. Among these dyes, Sudan black B (Schelegel et al. 1970; Murray et al. 1994), Nile blue A (Ostle and Holt 1982; Kitamura and Doi 1994; Takagi and Yamane 1997) and Nile red (Müller et al. 1995; Degelau et al. 1995; Gorenflo et al. 1999; Spiekermann et al. 1999; Berlanga et al. 2006) have been mainly investigated for the staining of various gram-negative and, to a lesser extent, gram-positive, wild-type or recombinant PHA-producing bacteria. The most specific and sensitive visualization of PHAs granules within bacterial cells was reported using Nile red (NR) dye, the oxidized form of the basic oxazine Nile blue A. NR is a common histological fat stain that emits a bright fluorescence in lipid-rich environments visible under UV light (312/365 nm) or at excitation wavelengths of 480–490 and 510–560 nm depending on the degree of hydrophobicity of the lipids (Greenspan et al. 1985). NR-stained PHA inclusions exhibit similar fluorescence behavior, with a clear emission maximum between 570 and 605 nm at an excitation wavelength of 540–560 nm, with cells appearing coloured from golden-yellow to deep red (Greenspan et al. 1985; Gorenflo et al. 1999; Chen 2003). The mechanism of selective NR staining of PHAs is not yet completely understood, although it is assumed that the dye may diffuse through the bacterial membrane and label the cytoplasmic PHA granules by binding to the lipid molecules of the surrounding layer. Membrane and cytoplasmic lipophilic storage materials have a lower dye uptake than PHAs and induce weak background fluorescence in the same wavelength range (Ostle and Holt 1982). Although NR-staining was first considered merely as a qualitative method of PHA analysis, a good correlation was found between fluorescence intensity measured by flow cytometry and the concentration of P(3-HB) granules (Gorenflo et al. 1999). By combining Nile red staining and fluorescence spectroscopy, it was also found possible to differentiate between short-chain-length (scl-PHA) and medium-chain-length (mcl-PHAs) within the intact cells of polyhydroxyalkanoate-producing bacteria (Wu et al. 2003).
NR staining is usually carried out on bacterial colonies growing in agar medium or on cells smeared onto a glass microscope slide. However, the application of the NR-staining method to the cultivation of bacterial cells has received little attention and precise information about this procedure is scarce (Berlanga et al. 2006). When combined with appropriate fluorescence detection tools, NR-staining of bacteria cultures may even allow a qualitative and quantitative determination of the PHA production to be carried out directly in the fermenter vessel at any time.
In this study, the NR-staining method is applied by direct incorporation into the culture medium at the beginning of the PHA synthesis phase in order to stain bacterial cells throughout the cultivation experiments. The method was applied: (1) to screen for high PHA-producers obtained from marine environmental isolates, (2) to determine the associated optimal culture parameters such as nitrogen limitation, which can maximize the yield of PHAs and, finally (3) to monitor the accumulation of PHAs during the course of a batch fermentation assay.
Materials and methods
Chemicals
Nile red (9-diethylamino-5H-benzo[α]phenoxazine-5-one, dye content ~95 %) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Other chemicals were analytical reagent grade and were obtained either from Difco Laboratories (Detroit, MI, USA) or Sigma Chemical Co.
Bacterial strains
The bacteria used in this study comprised five strains of moderately halophilic gram-negative bacteria designated as SF/2003, SF/2006, SF/2066, SF/2069 and SF/2089, that were isolated from Brittany coastal sea water. The reference strains Escherichia coli (ATCC 25922) and Cupriavidus necator (DSM 428) were also used as well-described negative (PHA−) and positive (PHA+) PHA-producing bacteria, respectively. Stock cultures of each bacterial strain were prepared in marine broth at pH 7.5 containing (g l−1 of distilled water): tryptone 4.0; yeast extract 1.0; glucose 10.0, sea salts 11.0 (instant ocean salt mix); with 20 % (v/v) glycerol and stored in 2 ml vials at −80 °C until further used.
Cultivation conditions for PHA production in flasks
Biomass growth and PHAs production were performed in two separate stages. In the first stage, cells were grown in a nutrient-rich medium in order to obtain at least 109 c.f.u. ml−1. In the second stage, nitrogen-limiting conditions were established by transferring the cells into an unbalanced growth medium (with limited quantities of yeast extract) to promote redirection of the excess carbon source (glucose) to PHA synthesis.
Shake flask cultivations were carried out in cotton plugged 250-ml flasks filled with 100 ml of growth medium containing (% w/v in distilled water) tryptone 1.0, yeast extract 0.5, glucose 10.0 and sea salts 11.0 at pH 7.5. Flasks were inoculated with one vial of stock culture and incubated overnight at 25 °C with orbital shaking at 200 rev min−1. After about 17 h (early stationary phase), 10 ml of this culture were then transferred into 250-ml flasks filled with 90 ml PHA production medium containing (% w/v in distilled water) yeast extract 1.8, glucose 20.0 and sea salts 11.0 at pH 7.5. The flask was then incubated for at least 72 h in darkness and under identical temperature and shaking conditions as in the first step.
Each experiment and related fluorescence analysis was run in triplicate and the results are represented as the average and standard deviations calculated from the obtained values.
Quantification of biomass
Growth was followed by measuring the number of viable cells (c.f.u. ml−1) in the culture medium by using the spread plate technique. The samples were serially diluted (10-fold dilution) in salt-supplemented peptone water and 0.1 ml aliquots of each dilution were spread onto marine agar (% w/v in distilled water: tryptone 1.0, yeast extract 0.5, glucose 10.0, sea salts 11.0, agar 20.0; pH 7.5) followed by incubation for 48 h at 25 °C.
For fluorescence analysis, the optical density (O.D.) of the cell suspension was measured at 543 nm using a spectrophotometer (CARY 50, VARIAN).
Nile red staining procedure and fluorescence measurement
The Nile red staining procedure was adapted from Spiekermann et al. (1999). A stock solution of 0.25 mg NR per ml dimethylsulfoxide (DMSO) was used for all the assays. The staining was performed at the beginning of the second cultivation stage by mixing the dye solution with the medium to obtain a final concentration of 0.4 % (v/v). Before fluorescence measurements, the production medium was sampled (~2 ml) and stained cells were collected by centrifugation (7500 rev min−1, 10 min). Cells were washed twice with Phosphate Buffered Saline (PBS) to remove residual Nile red and thus lower the background fluorescence level. Afterwards they were resuspended in PBS until the optical density of the suspension was 0.7 and 0.1 (O.D.543nm) for microscopic and spectrometric fluorescence measurements, respectively.
Epifluorescence microscopy
Fluorescence of cells was viewed with an Olympus BX41 microscope (Olympus, America Inc., Melville, NY) equipped for epi-illumination with a 100 W high-pressure mercury lamp for excitation and UPlan 10× and 40×/0.65 Ph2 objective lenses. NR fluorescence was observed by excitation at 510–550 nm using a U-MWG excitation cube unit (Olympus) with a long-pass barrier filter (≥590 nm).
Images were captured with a CCD Color View III camera and analysis was performed with Olympus Cell^A software.
Fluorescence spectroscopy
Fluorescence measurements were performed with a SLM 8100 spectrofluorometer (SLM-Aminco, Urbana, IL, USA) using quartz cuvettes with 0.4 cm path length equipped with a magnetic stirrer to avoid cell sedimentation and fading (fluorescence extinction). Emission spectra were recorded at room temperature between 560 and 710 nm using a fixed excitation wavelength of 543 nm. Excitation and emission slits were set to 4 nm. Displayed spectra were corrected by substracting the contribution of the buffer (PBS) to the recorded spectra.
PHA extraction, purification and characterization
The steps for chemical extraction and purification of PHAs were conducted as previously described by Chardron et al. (2010). The polymer monomeric composition was determined by GCMS following the protocol of Braunegg et al. (1978). A Perkin Elmer Clarus 500 GC equipped with DB-5 ms column (polar, 60 m, 0.25 mm diameter, film thickness 0.25 mm) was used and mass data were acquisited and processed with a Perkin Elmer Clarus 500 mass spectrometer. Different 3-hydroxyalkanoates (Sigma Chemical Co.) were used as standards. The molar mass distribution of PHAs was determined by Sterical Exclusion Chromatography (SEC) using PLgel MIXED C columns (Polymer Labs.) and detection system composed of UV and refractometer detectors. Chloroform was used as an eluent with a flow rate of 0.8 ml min−1. PHA thermal properties were carried out by DSC analysis using a Mettler-Toledo DSC-882 instrument to determine melting peak (Tm), glass transition temperature (Tg) and crystalline ratio (Xc), as described in a previous study (Corre et al. 2012).
Results
Screening of PHA-accumulating bacteria using Nile red staining
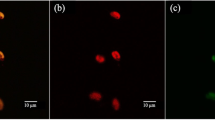
The five isolates of marine bacteria were screened for PHA accumulation after cell cultivation, following the two-stage fermentation strategy using the dye solution incorporated into the synthesis medium as described in "Cultivation conditions for PHA production in flasks" and "Quantification of biomass" sections. Living cells stained with Nile red were firstly observed by fluorescence microscopy. Among the strains investigated here, three are identified as potential PHA-producers, namely isolates SF/2006, SF/2066 and SF/2003, since these cells appear strongly fluorescent (Fig. 1). Stained cells of SF/2006 and SF/2066 exhibit a deep red fluorescence similar to that emitted by the control PHA-producing strain (PHA+). A different fluorescence behavior is observed with cells of SF/2003, which appear bright and gold yellow at the same excitation wavelength. On the contrary, no fluorescence was detected in the suspensions of the two other isolates (SF/2069 and SF/2089) or with the non-PHA producing strain (PHA−).

PHA granules within cells of a PHA− strain, b isolate SF/2003, c isolate SF/2066, d PHA+ strain, observed under fluorescent light. Bacteria were grown 72 h in synthesis medium (nitrogen-limited) including 0.4 % v/v Nile red solution. Arrows indicate gold-yellow fluorescent cells (Magnification ×100)
Qualitative assessment of PHA granule formation was carried out by measuring the fluorescence intensities of the stained cells after 72 h batch culture. Figure 2 shows the fluorescence emission spectra of the different bacterial strains. Broad spectra were recorded with varying fluorescence intensities and wavelength optima depending on the bacterial strain. The SF/2006 and SF/2066 isolates and the PHA+ strain displayed similar peaks between 615 and 618 nm. The bacterial strain SF/2006 yielded the highest FI value (1.35 a.u.). The isolate SF/2066 emitted a significantly less intense fluorescence (0.39 a.u.) which was nevertheless higher than for the PHA+ control stained cells (0.27 a.u.). Interestingly, the fluorescence displayed by the culture SF/2003 was clearly shifted towards a lower wavelength, with a peak at 595 nm and a maximum FI value (0.42 a.u.) similar to those obtained for the SF/2066 stained-cell suspension. In accordance with the microscopic observations, almost no fluorescence (0.03 a.u.<) was recorded for isolates SF/2069, SF/2089 and the PHA− control strain.

Fluorescence emission spectra of Nile-red stained bacterial suspensions of (1) SF/2006 (2) SF/2003 (3) SF/2066 (4) PHA+ (5) PHA− (6) SF/2069 (7) SF/2089 after 72 h of cultivation in PHA synthesis medium with nitrogen limitation. Fluorescence intensity (FI) was measured on cell suspension adjusted at 0.1 (OD543 nm)
Screening of nutritional conditions to optimize PHA production
PHA accumulation is well known to be triggered under nutrient-limited conditions as regards nitrogen, oxygen or phosphorus, while the carbon source is in excess. Moreover, the molar ratio of carbon to nitrogen (C0/N0) of the feed medium is a key parameter in relation to both bacterial nutritional requirements and the efficiency of PHA biosynthesis. On this basis, the NR-staining assay was applied to investigate the influence of such a parameter on PHA accumulation in the cultivation of isolate SF/2003 to define an optimized composition for the production medium that could enhance biosynthesis. For this purpose, cells were grown in modified production media with a constant glucose concentration (10 g l−1) and a yeast extract content ranging from 1.8 down to 0 g l−1. As a consequence, the C0/N0 ratio of the production media is increased. As shown in Fig. 3, PHA accumulation was detected in the bacterial cultures whatever the nitrogen levels, but becomes enhanced when there is a nitrogen deficit. Complete depletion nevertheless proves to be detrimental to the polymeric content of cells. Thus, the polymer yield may be optimized if the culture is carried out in a medium containing 0.4 g yeast extract l−1, since the fluorescence intensity signal goes through a maximum at this value.

Relative fluorescence emission intensity of Nile red-stained cells of isolate SF/2003 as a function of the yeast extract concentration in the synthesis medium formulation. Fluorescence intensity (FI) was measured after 72 h of cultivation on cell suspension adjusted at 0.1 (OD543 nm)
Application to the monitoring of PHA production
The kinetics of PHA production in SF/2003 cells was monitored over a period of 192 h by both fluorescence microscopic and fluorescence techniques. Batch cultivation was performed under optimal nitrogen-limited condition (0.4 g l−1) as mentioned above. Microscopic observations revealed that no fluorescent cells could be detected at the early stage (Fig. 4a) of the cultivation of isolate SF/2003, despite the cell density being around 108 c.f.u. ml−1 as shown in Fig. 5. After 48 h and up to 192 h of cultivation, numerous deep red and gold-yellow granules were observed that are bound to NR (Fig. 4b,c,d). The highest density of stained cells was recorded after 72 h of cultivation, which is a common finding for the maximum production of PHAs by a two-step fermentation process. A more quantitative monitoring approach can be achieved by fluorimetry. Based on the recorded IF values (Fig. 5), we can assume that the content of PHAs within the whole biomass increases steadily from the start of the second cultivation step until it reaches a maximum at 72 h. After this time, the relative fluorescence intensity remained constant or declined slightly, indicating that polymer biosynthesis had ceased.

Epifluorescence microscopy of NR-stained PHA granules within cells of isolate SF/2003 observed at a 1 h, b 48 h, c 72 h and d 192 h cultivation in synthesis medium including 0.4 % v/v Nile red and 0.4 g l−1 extract (Magnification ×100)

Variation in the relative fluorescence intensity of Nile red-stained cells (isolate SF/2003) during 192 h cultivation (square). The viable cell density in the culture medium is also plotted as a function of time with (filled circle) and without (empty circle) dye being incorporated at the beginning of the production phase
Viable cells in the culture were then determined by dilution plating onto Marine Agar. As shown in Fig. 5, the viable cell count remained fairly constant or decreased slightly (~5 × 107–108 c.f.u. ml−1) during the first 72 h of cultivation, after which it dropped sharply by more than 5 log10 c.f.u. per ml. By comparison, a similar cultivation experiment using isolate SF/2003 was carried-out without NR incorporation and the results were identical. Cell viability was found to vary over time in a similar way, regardless of whether the dye solution was added to the synthesis medium.
To confirm PHA production in isolate SF/2003, the biomass was recovered and analysed for its polymer content. The accumulation by the strain was achieved at the highest PHA content of 40 wt% of the CDW at 72 h, which is quite lower than that typically obtained 50 wt% in flask under nitrogen sufficient conditions.
A run in a 5-l fermentor under similar conditions resulted in a higher PHA content of 55 wt% of the CDW and a productivity of 0.022 g/L/h.
According to the protocol of Braunegg et al. (1978), PHA was methanolysed and the monomers were identified and quantified by GC–MS. The chromatograms indicate that the PHA produced is a homopolymer only composed of 3-hydroxybutyric units, as a unique peak with retention time of 10.05 min was detected. From SEC analyses, it was shown that the PHA exhibited an average molecular mass (M w ) of 665,000 g.mol−1, a value which lies within the usual range. Typical intended applications for PHAs would require the molecular weight to lie in the range 500,000–800,000 g mol−1, even if the molecular weight of PHAs can be also modulated by a judicious choice of experimental processing conditions. Analysis of the thermal properties of the polymer obtained in this way yielded values of T g , T m and X c are 9, 170 °C and 69 %, respectively. This polymer displays the thermal profile of poly-β-hydroxybutyrate (PHB) currently given in the literature (Noda et al. 2005). Because of the remarkable stereo-regularity of the isotactic chain configuration, the PHB homopolymer exhibited an unusually high degree of crystallinity.
Discussion
For many years, NR has been recognized for its affinity to bind to PHA granules and has been consequently accepted as a presumptive test for PHA storage in bacteria. In the present study, the NR procedure is adapted to stain bacterial cells containing PHAs during cultivation in a specific unbalanced production medium. This staining procedure is an improvement on other methods because it can detect PHAs inside living cells and uses very dilute dye solution (1 mg per litre of culture medium). Even though NR is poorly soluble in aqueous medium and its fluorescence is immediately quenched by this solvent (Greenspan et al. 1985), it has been determined that, once incorporated into the production medium, NR can subsequently enter the bacterial cells and bind to PHA inclusions giving rise to a distinctive fluorescence. Fluorescence emission of NR-stained cells was detected directly after culture sampling, thus obviating the need of an incubation step of 10–40 min to ensure complete staining according to the protocol followed. Under the microscope, PHA granules within bacterial cells appear deep red or bright yellow depending on the strain assayed. It is noteworthy that the fluorescence of NR-stained cells fades in a few seconds after exposure as previously reported by Kimura et al. (2004). Fluorescence extinction is a common problem with fluorescent dyes, with the amount of fading proportional to the intensity of the excitation light and the duration of illumination. With the fluorometric detection technique, fluorescence does not fade so rapidly. This is mainly due to a much wider spectral excitation range (40 vs 4 nm) used with the BX-60 microscope since the excitation is controlled by a dichroic filter instead of monochromator slits. Moreover, in opposite to observations performed in a standard cuvette, epifluorescence measurements imply that it is the same sample volume which is irradiated during the measurement and/or observation. The fluorescence excitation spectrum of the NR stained cells displayed a maximal excitation wavelength at 543 nm (results not shown). We find that the maximum emission occurs with excitation at around 540 nm which is different from the wavelength of 488 nm used by some authors (Müller et al. 1995; Wu et al. 2003) but similar to that reported by Degelau et al. (1995) in which 543 nm was selected for the fluorometric measurement of P(3-HB) accumulation in Alcaligenes eutrophus. This wavelength setting was consequently used in further experiments. Four out of seven stained bacterial cultures were found to emit significant fluorescence after 72 h of cultivation, including the PHA+ producer known to accumulate high levels of P(3-HB) with glucose as the sole carbon source (Ryu et al. 1997). Since little or no fluorescence could be detected by either microscopy or fluorimetry in the negative-control strain cells (E. coli) or in PHA-producing bacteria prior to biosynthesis (as shown in Fig. 4a), we can therefore assume that the staining procedure allows a specific differentiation of PHAs, i.e. non-specific binding to other cellular components is negligible.
With one of the marine bacteria identified as a PHA producer, namely isolate SF/2003, the NR emission spectrum is found to shift from ~615 to ~595 nm. The NR fluorescence maxima can vary over a range of 60 nm depending on the polarity of its environment (Greenspan and Fowler 2014). This blue shift may be related to the relative hydrophobicity of the stored PHAs, which can be affected by their monomer composition, chemical structure and length of the alkyl side-chains. Increased molecular weight may also be correlated with stronger inherent hydrophobicity. Such an approach has been used previously for the rapid differentiation between scl- and mcl-PHAs producers, even though the emission spectrum was shifted towards shorter wavelengths (Wu et al. 2003). As highly hydrophobic PHAs are more attractive as regards their potential applications, especially in the packaging field, the present investigation is mainly concerned with isolate SF/2003.
The NR-staining of these bacteria cells during cultivation is also found to be effective in defining an optimized medium composition, such as its carbon to nitrogen ratio, leading to enhanced screening speed compared to the conventional NR protocol and improved monitoring of biosynthesis. The present protocol does not affect the survival rate of stained cells, i.e. viability is not inhibited by dye uptake in cultivated cells or by exposure to DMSO used as a stain carrier. Some authors have reported a negative effect of DMSO on the efficiency of cell viability staining (Chen et al. 2009; Doan and Obbard 2011), but the concentrations used were significantly higher than in our study (15–25 % v/v vs 0.4 % v/v). It noteworthy that the purification steps (precipitations into cold methanol) allow the washing-away of the dye.
In fact, the analysis of isolate SF/2003 for polymer content confirms the production of PHAs. The accumulation of PHAs in PHA+ and SF/2006 bacterial strains is also demonstrated in other series of experiments (results not shown). The polymer yield and molecular weight of the biopolymer accumulated in isolate SF/2003 are in the upper part of the range compared with values reported for P(3-HB) synthesized by other bacteria from marine environments (González-Garciá et al. 2008; Quillaguamán et al. 2010). This finding supports the remarkable potential of halophilic archaeal and bacterial strains from diverse marine ecosystems for the biotechnological production of PHAs, as recently highlighted (Quillaguamán et al. 2010). The most promising halophilic PHA-producing bacteria have been identified in the archaeal genera, especially Haloferax, Halogeometricum, Haloarcula and Holoccus (Legat et al. 2010; Quillaguamán et al. 2010; Salgaonkar et al. 2013), as well as belonging to the eubacterial family Halomonadaceae. Among members of this family, Halomonas boliviensis attains PHA yields and volumetric productivities close to the highest levels reported so far (Simon-Coli et al. 2008; Van-Thuoc et al. 2008; Quillaguamán et al. 2010). Phenotypic and biochemical characterization of isolate SF/2003 reveals that it is a motile rod-shaped and non-spore-forming Gram-negative bacterium. On the basis of 16S rRNA gene sequence analysis, the strain is affiliated to the family Halomonadaceae, genus Cobetia, and has been deposited under accession number CNCM I-4786 at the Pasteur Institute. A major feature of most species in this genus is their metabolic versatility, which allows us to consider the production of PHAs from inexpensive renewable carbon sources. In turn, this may provide significant cost savings in production because raw materials account for up to 50 % of the expenses of the entire PHA production process. Ongoing research is being carried out in our laboratory using agro-industrial wastewaters as carbon feedstock.
Abbreviations
- NR:
-
Nile red
- PHAs:
-
Polyhydroxyalkanoates
- CDW:
-
Cell dry weight
- FI:
-
Fluorescence intensity
References
Berlanga M, Montero MT, Hernandez-Borell J, Guerrero R (2006) Rapid spectrofluorometric screening of poly-hydroxyalkanoate-producing bacteria from microbial mats. Int Microbiol 9:95–102
Braunegg G, Sonnleitner B, Lafferty RM (1978) A rapid gas chromatographic method for the determination of poly-3-hydroxybutyric acid in microbial biomass. Eur J Microbiol Biotechnol 6:29–37
Chanprateep S (2010) Current trends in biodegradable polyhydroxyalkanoates. J Biosci Bioeng 110:621–632
Chardron S, Bruzaud S, Lignot B, Elain A, Sire O (2010) Characterization of bionanocomposites based on medium chain length polyhydroxyalkanoates synthesized by Pseudomonas oleovorans. Polym Test 29:966–971
Chen GQ (2003) Production and application of microbial polyhydroxyalkanoates. In: Chiellini, Solaro (eds) Biodegradable polymers and plastics. Kluwer Academic, New York, pp 155–166
Chen W, Zhang CW, Song LR, Sommerfeld M, Hu Q (2009) A high throughput Nile Red method for quantitative measurement of neutral lipids in microalgae. J Microbiol Meth 77:41–47
Corre YM, Bruzaud S, Audic JL, Grohens Y (2012) Morphology and functional properties of commercial polyhydroxyalkanoates: a comprehensive and comparative study. Polym Test 31:226–235
Degelau A, Scheper T, Bailey JE, Gushe E (1995) Fluorometric measurement of poly-b-hydroxybutyrate in Alcaligenes eutrophus by flow cytometry and spectrofluorometry. Appl Microbiol Biotechnol 42:653–657
Doan TTY, Obbard JP (2011) Improved Nile Red staining of Nannochloropsiss sp. J Appl Phycol 23:895–901
González-Garciá Y, Nungaray J, Cordova J, González-Reynoso O, Koller M, Atlic A, Braunegg G (2008) Biosynthesis and characterization of polyhydroxyalkanoates in the polysaccharide-degrading marine bacterium Saccharophagus degradans ATCC 43961(35):629–633
Gorenflo V, Steinbüchel A, Morose S, Rieseberg M, Scheper T (1999) Quantification of bacterial polyhydroxyalkanoic acids by Nile red staining. Appl Microbiol Biotechnol 51:765–772
Greenspan P, Fowler SD (2014) Spectrofluorometric studies of the lipid probe, nile red. J Lipid Res 26:781–789
Greenspan P, Mayer EP, Fowler SD (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100:965–973
Johnson K, Kleerebezem R, Van Loosdrecht MCM (2010) Influence of the C/N ratio on the performance of polyhydroxybutyrate (PHB) producing sequencing batch reactors at short SRTs. Water Res 44:2141–2152
Khanna S, Srivastava AK (2005) Recent advances in microbial polyhydroxyalkanoates. Process Biochem 40:607–619
Kim YB, Lenz RW (2001) Polyesters from microorganisms. Adv Biochem Eng Biotechnol 71:51–79
Kimura K, Yamaoka M, Kamisaka Y (2004) Rapid estimation of lipids in oleaginous fungi and yeasts using Nile red fluorescence. J Microbiol Method 56:331–338
Kitamura S, Doi Y (1994) Staining method of poly(3-hydroxyalkanoic acid) producing bacteria by Nile blue. Biotechnol Techn 8:345–350
Lee WH, Azizan MNM, Sudesh K (2004) Effects of culture conditions on the composition of poly(3-HB-co-4-HB) synthesized by Comamona acidovorans. Polym Degrad Stab 84:129–134
Legat A, Gruber C, Zangger K, Wanner G, Stan-Lotter H (2010) Identification of polyhydroxyalkanoates in Halococcus and other haloarchaeal species. Appl Microbiol Biotechnol 87:1119–1127
Müller S, Lösche A, Bley T, Scheper T (1995) A flow cytometric approach for characterization and differentiation of bacteria during microbial processes. Appl Microbiol Biotechnol 43:93–101
Murray RGE, Doetsch RN, Robinow CF (1994) Determinative and cytological light microscopy. In: Gehardt P, Murray RGE, Wood WA, Krieg NR (eds) Method for general and molecular bacteriology. American Society for Microbiology, Washington, DC, pp 21–41
Noda I, Green PR, Satkowski MM, Schechtman LA (2005) Preparation and properties of novel class of polyhydroxyalkanoate copolymers. Biomacromolecules 6:580–586
Ostle AG, Holt JG (1982) Nile blue A as a fluorescent stain for poly-b hydroxybutyrate. Appl Environ Microbiol 44:238–241
Quillaguamán J, Guzmán H, Van-Thuoc D, Hatti-Kaul R (2010) Synthesis and production of polyhydroxyalkanoates by halophiles: current potential and future prospects. Appl Microbiol Biotechnol 85:1687–1696
Ryu HW, Hahn SK, Chang YK, Chang HN (1997) Production of poly(3-hydroxybutyrate) by high cell density fed-batch culture of Alcaligenes eutrophus with phosphate limitation. Biotechnol Bioeng 55:28–32
Salgaonkar BB, Mani K, Bragança JM (2013) Accumulation of polyhydroxyalkanaotes by halophilic archae isolated from traditional solar salterns of India. Extremophiles 17:787–795
Schelegel HG, Lafferty R, Krauss I (1970) The isolation of mutantas not accumulating poly-B- hydroxybutyric acid. Arch Mikrobiol 38:209–222
Simon-Coli C, Raguenes G, Cozien J, Guezennec JG (2008) Halomonas profundus sp. Nov., a new PHA-producing bacterium isolated from deep-sea hydrothermal vent shrimp. J Appl Microbiol 104:145–1432
Spiekermann P, Rehm BHA, Kalscheuer R, Baumeister D, Steinbüchel A (1999) A sensitive, viable-colony staining method using Nile red for direct screnning of bacteria that accumulate polyhydroxyalkanoïc acids and lipid storage compounds. Arch Microbiol 171:73–80
Sudesh K, Abe H, Doi Y (2000) Synthesis, structure and properties of polyhydroxyalkanoates: biological polyesters. Prog Polym Sci 25:1503–1555
Takagi Y, Yamane T (1997) Replica technique for screening Poly(3-hydroxyalkanoïc acid)-producing bacteria by Nile Blue staining. J Ferm Bioeng 1:121–123
Van-Thuoc D, Quillaguamán J, Mamo G, Mattiasson B (2008) Utilization of agricultural residues for poly(3-hydroxybutyrate) production by Halomonas boliviensis LC1. J App Microbio 104:420–428
Wu HA, Sheu DS, Lee CY (2003) Rapid differentiation between short-chain-length and medium-chain-length polyhydroxyalkanoate-accumulating bacteria with spectrofluorometry. J Microbiol Methods 53:131–135
Acknowledgments
The authors acknowledge the regions of Bretagne and Pays de Loire for funding. We are also grateful to Breizpack and Valorial pole for their help, as well as the industrial partners of the PHApack consortium: Europlastiques, Séché Environnement, Triballat Noyal and Cap Ouest, for their contribution to this research programme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Elain, A., Le Fellic, M., Corre, YM. et al. Rapid and qualitative fluorescence-based method for the assessment of PHA production in marine bacteria during batch culture. World J Microbiol Biotechnol 31, 1555–1563 (2015). https://doi.org/10.1007/s11274-015-1904-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-015-1904-4