
Abstract
Sorption is an effective process for the remediation of mine water with low metal concentrations. To identify promising low-cost organic sorbents for nickel (Ni), adsorption and retention properties of peat, compost, brown algae, sawdust, and wood ash were compared. Batch adsorption and desorption experiments were conducted at pH 7 in 0.05 M NaNO3 solutions to simulate the ionic strength and pH of a contaminated neutral drainage. Results of adsorption kinetic experiments were best represented by the Elovich model and the fastest rates were obtained with peat (796,075 mg g−1 min−1) and compost (791 mg g−1 min−1). Results of equilibration adsorption experiments were fitted to Langmuir and Freundlich isotherms and the highest adsorption capacities were observed for peat (around 22 mg g−1) and compost (around 9 mg g−1). Desorption experiments revealed that peat and compost adsorbed more Ni and also released a lower percentage of the adsorbed metal upon exposure to Ni-free solutions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Sorption of metals onto organic materials is a rapid process that can be used in passive treatment systems to lower the metal concentrations of contaminated neutral drainage (CND) below regulatory threshold values (Calugaru et al. 2018). The financial cost of transporting these materials to remote mining sites can be substantial and, thus, studying the sorption capacity of locally available materials is of great interest. Over the past few years, the sorption of metals onto various natural materials and industrial waste products has been investigated, and numerous solid substrates with good sorption capacities have been identified (Das et al. 2008; Iakovleva and Sillanpaa 2013; Westholm et al. 2014; Zhou and Haynes 2010). Unfortunately, operational parameters such as pH, ionic strength, and sorbent:sorbate ratios vary greatly between different studies, making comparisons difficult. To establish a proper comparison, different solid substrates must be tested under identical experimental conditions. In this study, the sorption properties of five low-cost organic substrates were compared with regard to their affinity for nickel. The substrates were selected for their sorption properties and potential local availability to eastern Canadian mining sites.
The main metal(loid) contaminants of CND are Cd, Cr, Co, Cu, Fe, Hg, Mn, Ni, U, Zn, As, Sb, and Se (Calugaru et al. 2018) and their concentrations are variable and mostly contingent on the mineralogy of each particular mine site. Ni is often present in CND and, in some instances, its concentration can exceed regulatory threshold values. In the province of Quebec (Canada), the mean monthly and maximum acceptable concentrations for nickel in mining effluents are, respectively, 0.5 mg L−1 and 1 mg L−1 (MDDELCC 2012). Metal toxicity in the environment depends on speciation, itself dictated by the local geochemical conditions. The ecotoxicity of nickel is mainly attributed to its free cation, Ni2+ (Stokes 1988), which is the predominant species at circum-neutral pHs. Other soluble nickel species include hydroxides, carbonates, sulfates, nitrates, chlorides, and Ni-organic complexes. In unpolluted freshwater, an important fraction of the total nickel is bound to strong organic ligands (Xue et al. 2001). In mining effluents, competition with other cations such as Ca2+ and Mg2+ affects nickel complexation. Under these latter conditions, Ni-organic complexes tend to be more labile and the free Ni2+ fraction more important (Guthrie et al. 2005; Mandal et al. 2002). The sorption of nickel onto organic substrates takes place at functional surface sites, such as carboxylic, phenolic, and thiolic groups. The availability and structure of these sites depends on the type of organic matter. The sorption capacities of these substrates increase with pH (Stumm and Morgan 1996; Zhou and Haynes 2010) as their surface charge becomes more negative.
In the present study, three different types of organic substrates were tested: biosorbents (brown algae and maple sawdust), humified materials (peat and compost), and biochar (wood ash). Biosorbents are defined in this paper as non-degraded inert organic matter. They have regularly spaced and organized surface functional groups disposed in repeatable patterns according to their cellular structure. One disadvantage of biosorbents in their field application is their high labile organic matter content. The latter may increase the chemical and biological oxygen demands in treated waters at the detriment of aquatic species (Wan Ngah and Hanafiah 2008; Zhou and Haynes 2010). A second disadvantage is the unknown impacts of biodegradation on the sorption capacity and specific affinity of these substrates. The vast majority of information on biosorbents originates from short-term experimental studies, and their long-term behavior at the field-scale has yet to be investigated (Zhou and Haynes 2010).
Humified materials are organic substrates having undergone some degree of decomposition, polymerization, and loss of volatiles. They are characterized by the presence of complex, high molecular weight, aromatic substances, and inorganic solids (e.g., clay) or non-degraded plant material. Although highly aromatic, the structure of humic substances is poorly defined and the position of surface functional groups is heterogeneous and dependent on their chemical environment (Stumm and Morgan 1996). This complex arrangement and variable chemical properties of functional groups in humified materials can lead to multiple and possibly simultaneous sorption mechanisms such as adsorption, complexation, chelation, and co-precipitation. Metal sorption onto humified substances mostly occurs at carboxyl and phenolic functional groups (Stumm and Morgan 1996; Tan 2014). Humified substances are more recalcitrant to degradation than biosorbents and, thus, should theoretically be less affected during long-term field applications.
Biochars are a mixture of organic carbon-rich solids and ash resulting from the pyrolysis of biomass. Common by-products of biorefineries, their chemical composition depends on the nature of the parent biomass (e.g. wood residue, crop residue, manure) and production conditions (e.g. temperature, heating rate and time) (Calugaru et al. 2018; Mohan et al. 2014). They possess a high-specific surface area and porosity making them good candidates for metal adsorption and sequestration (Calugaru et al. 2018; Mohan et al. 2014; Xu et al. 2014). Sorption of metals onto biochar can take place at both organic and inorganic functional groups. The sorption of metals onto the organic fraction of biochar is attributed to the presence of oxygen-containing functional groups such as carboxyl and phenol (Tong et al. 2011; Uchimiya et al. 2011). The role of the inorganic fraction in the sorption process is linked to the presence of calcite. The dissolution of calcite increases pH, favoring either sorption or precipitation of new metal-bearing solid phases (Arán et al. 2017; Xu et al. 2014; Zhou and Haynes 2010). For many authors, the term adsorption refers strictly to 2D, non-specific, or physical adsorption and the general term sorption is often used to include both specific (chemical) and non-specific adsorption as well as surface precipitation and co-precipitation. In this paper, the terms adsorption and desorption are meant to include all surface mechanisms and exclude only true precipitation.
Passive treatment systems designed for the final rehabilitation of closed or abandoned mine sites should require minimal maintenance and sorbent materials are typically used in a single, long-term sorption cycle (years ideally) before being disposed of. Hence, substrates need to be stable over equally long periods of time and metals need to be strongly bound to their surfaces (Zhou and Haynes 2010). In this study, the Ni adsorption and retention capacity of five low-cost organic substrates (brown algae, maple sawdust, wood ash, compost, and peat) is compared by conducting short-term kinetic and equilibration experiments in the absence of competing ions. The term “equilibration experiments” is used here for experiments in which a steady state with regard to adsorption-desorption reactions is presumably reached and does not necessarily refer to the attainment of a true equilibrium. The objective of the present study are to test different materials under identical operational conditions in order to help select the best candidates for long-term treatment studies of CND and to evaluate if certain physicochemical properties of substrates could be used to predict the outcome of sorption tests.
2 Materials and Methods
2.1 Substrate, Stock Solutions, and Equipment
The cold-water brown algae Ascophyllum nodosum (ASCO-PURE kelp) were provided by Organic Ocean Inc. (Rimouski, Quebec). They were air dried and gently crushed to less than 5 mm. This substrate was used both in its original state (algae) and after being washed with hydrochloric acid (HCl-algae) to remove salts (Na, K, Mg, and Ca), as described by Fourest and Volesky (1996). Briefly, the algae were stirred in a 0.1 mol L−1 HCl solution at a 1:50 solid:liquid ratio for 24 h with a magnetic bar. The HCl solution was then removed and the algae was rinsed extensively with deionized water and dried at 60 °C. The sphagnum peat moss was purchased from a local retailer (Home Depot, Montreal, Quebec). The compost was produced from green wastes by the City of Montreal. The maple sawdust (particles < 5 mm) was provided by P.W.I. Industries (Saint-Hyacinthe, Quebec). The wood ash (particles < 5 mm) was provided by Wood Ash Industries (Kirkland Lake, Ontario). It is a mixture of bottom and fly ash from a wood and gas co-generation plant and contains a small amount of gravel. All substrates were air dried and the compost and peat moss were sieved to less than 5 mm. All substrates were kept at room temperature in plastic containers or bags until use.
Experiments were performed at pH 7.0 in 0.05 M NaNO3 solutions. The ionic strength and pH of this solution were chosen to reflect those of a nickel containing mine effluent (I ≈ 0.029 M and pH 7.31 ± 0.22) near Havre-Saint-Pierre (Quebec, Canada). Stock solutions of 0.05 M NaNO3 and 0.175 M Ni(NO3)2 were prepared in deionized water with pure chemicals (ACS grade). Initial nickel concentration in batch adsorption kinetic experiments (7.45 mg L−1) was much higher than measured in the field and was chosen as a worst-case scenario as well as to ensure analytical detection for all measurements. Nickel was analyzed by flame atomic absorption spectrometry (AAS; Perkin-Elmer AAnalyst-200). Calibration curves were built by sequential dilution of an AAS standard (~ 1000 ppm) solution (Perkin-Elmer). The detection limit was 0.01 ppm and the reproducibility of the measurements was better than 2%, based on replicate analyses of this solution. All pH measurements were carried out with a combined glass electrode (model Orion 9156BNWP, ThermoScientific), calibrated using three NIST-traceable buffer solutions (4.01, 7.00, and 10.00 at 25 °C), and a pH meter (model Orion Star A211 benchtop, ThermoScientific).
2.2 Substrate Characterization
Results of substrate characterization are presented in Table 1. The water content (water weight/humid solid weight) of each solid substrate was determined prior to the adsorption experiments by drying 5–10 g of substrate at 60 °C until constant weight was obtained. Specific surface areas (SSA) of the substrates were measured with a Micromeritics Gemini III 2375 surface analyzer using the N2-BET method BET PE2 AM-05 (Brunauer et al. 1938). The pH of a slurry of each substrate (2 g substrate: 100 mL deionized water) was measured after mixing thoroughly and letting stand for 1 h. The point of zero charge (PZC) and cation exchange capacity (CEC) of each substrate were determined respectively by the salt addition method in 0.05 M NaNO3 and by the ion adsorption method in 0.05 M NaCl at pH 4, 5, 6, 7, 8, and 9 (Bakatula et al. 2018). The total carbon (CTOT) and total organic and graphitic carbon (Corg + g) contents of the substrates were measured by combustion at 1400 °C in an induction furnace (model CS744, LECO Corporation) according to Ma. 310-CS 1.0 (CEAEQ 2013). The Corg + g samples were pre-treated by immersion of the solid for at least 1 h in a 50% v/v HCl solution to dissolve carbonate phases. Dissolved organic carbon (DOC) was analyzed on filtered (0.45 μm) extracts of the slurries (20 g substrate (wet weight):200 mL deionized water) shaken for 2 h at room temperature. Total organic carbon (TOC) of these extracts was determined at 680 °C using a DOHRMAN, model DC-190 TOC analyzer, according to Standard Method 5310 B (APHA 2005).
2.3 Adsorption and Desorption Kinetic Experiments
For these experiments, 2 g of substrate and 200 mL of the 0.05 M NaNO3 solution were placed in 500-mL Erlenmeyers and were shaken (150 rpm) at room temperature (23 ± 2 °C) on a MaxQ-2000 orbital shaker (ThermoScientific). Each experiment was conducted in duplicates and flask openings were covered by aluminum foil to limit evaporation. A third reactor without substrate was prepared in the same manner and indicated that the addition of nickel to the reaction vessels had no measurable effect on the pH of the solution. In order to prevent the pH adjustment phase and associated reactions from interfering with Ni sorption, the pH was adjusted to 7.0 24 h prior to the addition of Ni to the slurries using dilute HNO3 and NaOH solutions. Adsorption was initiated in each reactor upon the addition of 0.145 mL of a 0.175 M Ni(NO3)2 solution to obtain an initial dissolved nickel concentration of 7.45 mg L−1 (127 μmol L−1). At various time intervals over the next 24 h, nine 6-mL aliquots of the decanted liquid were collected. After 24 h, 100 mL of the liquid were decanted from the reactor. The solid and remaining liquid were transferred to a 50 mL polypropylene tube and centrifuged at 10,000×g for 10 min using a Heraeus Megafuge 8 centrifuge (ThermoScientific). Using a pipette, most of the liquid was discarded, except for the last 6 mL which were stored and later analyzed to determine the initial nickel concentration for the desorption experiment. The wet solid was transferred to an empty 500-mL Erlenmeyer flask, and its wet weight was determined by weighing the polypropylene tube before and after this transfer. The dry weight of each solid transferred to the reactors was estimated from its wet weight and a pre-determined wet:dry weight ratio (detail of the ratio determination is presented in SI). Desorption was initiated in the reactor by adding 75–200 mL of a pre-adjusted Ni-free 0.05 M NaNO3 solution to the wet solid. The added NaNO3 solution had been adjusted to a pH of 7.0 over a period of 24 h with a separate aliquot of each substrate at a solid:liquid ratio of 1:100 and filtered through a 0.45 μm Millipore MCE filter. The volume added was adjusted according to the dry weight of the solid in each reactor in order to maintain a solid:liquid ratio of 1:100, like in the adsorption experiments. At different time intervals over a 24-h period, nine 6-mL aliquots of the decanted liquid were collected. For the HCl-treated algae, 4-mL aliquots were collected because initial liquid volumes were lower. Samples collected during the adsorption and desorption kinetic experiments were immediately filtered through 0.45-μm PVDF syringe filters, stored at 4 °C, and analyzed within 14 days.
2.4 Treatment and Modeling of Experimental Kinetic Data
During adsorption experiments, the concentration of nickel adsorbed to the solids at each sampling point, \( {q}_{t_{\mathrm{ADS}}} \)(mg g−1), was calculated using Eq. 1:
where Vo and Vt are the volumes of liquid (L) in the reactor at the start of the experiment (time = 0) and at each sampling point (time = t), and m is the mass of the solid (g). \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_0} \) and \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_{\mathrm{t}}} \) are the dissolved nickel concentrations (mg L−1) at time = 0 and time = t. During the course of desorption experiments, the concentration of nickel still adsorbed to the solids at each sampling point, \( {q}_{t_{\mathrm{ADS}\left(\mathrm{des}\right)}} \)(mg g−1), was calculated using Eq. 2:
where \( {q}_{t_{\mathrm{ADS}(f)}} \) is the nickel concentration adsorbed to the solid at the end of the adsorption experiment. Vt, m, and \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_t} \) are the volumes of liquid (L), mass of solid (g), and dissolved nickel concentrations (mg L−1) in the reactor at the time of sampling (t). \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_0} \)is the initial dissolved nickel concentration in the reactor, calculated using the final dissolved nickel concentration in the adsorption experiment, the water content of the wet solid transferred to the reactor, and the volume of NaNO3 added to the reactor at the start of the desorption experiment. Vs is the volume of sample (6 mL) removed from the reactor at the previous sampling (t-1) and\( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_{t-1}} \)is the dissolved nickel concentration of this sample. Finally, the concentration of nickel desorbed from the solid during the desorption experiment, \( {q}_{t_{\mathrm{DES}}} \)(mg g−1), was obtained by subtracting the concentration of nickel still adsorbed to the solid at each sampling point, \( {q}_{t_{\mathrm{ADS}\left(\mathrm{des}\right)}} \) from the adsorbed nickel concentration at the end of the adsorption experiment,\( {q}_{t_{\mathrm{ADS}(f)}} \), using Eq. 3:
Results of the adsorption and desorption kinetic experiments were compared using three different reaction kinetics-based models: the pseudo-first-order (PFO), pseudo-second-order (PSO), and Elovich models. These empirical models were fitted to the experimental data using non-linear regressions, because linear regressions have been found to introduce a methodological bias and erroneously favor the PSO model (Tran et al. 2017; Xiao et al. 2018). Non-linear regressions were carried out using the “Solver add-in” in Microsoft Office Excel (2010 version), as described by Tran et al. (2017). The PFO model was developed by Lagergren (1898) to describe the adsorption of oxalic acid and malonic acid onto charcoal. The PSO model, often referred to as Ho’s PSO model (Ho 1995), was initially proposed by Blanchard et al. (1984) to describe the removal of heavy metals from water using zeolites. The differential and integrated, non-linear form of the PFO (Eqs. 4 and 5) and PSO (Eqs. 6 and 7) equations are:
where qe and qt are the final and time = t (min) adsorbed nickel concentrations (mg g−1) respectively, and k1 (min−1) and k2 (g mg−1 min−1) are the pseudo-first and pseudo-second-order rate constants. The Elovich equation was initially proposed by Roginsky and Zeldovich (1934) for the adsorption of carbon monoxide onto manganese dioxide. The differential and integrated, non-linear form of the Elovich equation (Eqs. 8 and 9) are:
where qt is the adsorbed nickel concentration (mg g−1) at time = t (min), α (mg g−1 min−1) is the initial rate constant, and β (mg g−1) is the desorption constant. It must be noted that the Elovich model is physically unsound as adsorbed nickel concentrations increase endlessly, never reaching a steady state. Thus, it is only suitable to describe kinetics far from equilibrium (Tan and Hameed 2017).
2.5 Equilibration Experiments
Six pre-weighted 50-mL polypropylene centrifuge tubes containing 0.4 g of substrate and 40 mL of the 0.05 M NaNO3 solution were stirred (150 rpm) horizontally at room temperature (23 ± 2 °C) on a MaxQ-2000 orbital shaker (ThermoScientific). Each experiment was conducted in triplicate and, during the first 24-h pH adjustment period, pH was adjusted to 7.0 in all tubes using dilute HNO3 and NaOH solutions. To monitor the evolution of pH in the absence of nickel, one extra blank tube was prepared in the same manner. After the pH adjustment period, adsorption was initiated in tubes by adding varying volumes of a 0.175 M Ni(NO3)2 solution to obtain initial nickel concentrations of: 0.6, 7, 15, 29, 59, and 103 mg L−1 (10, 119, 254, 494, 1000, and 1750 μmol L−1). Tubes were shaken for at least 20 h and, after allowing the solids to decant for 5 min, one 6-mL aliquot of the supernatant was collected from each tube. Subsequently, tubes were centrifuged at 10,000×g for 10 min, and all the liquid was removed using a pipette. The tubes containing the wet solids were re-weighted and wet weights of the solids were determined. The dry weight of the solids in the tubes was estimated from their wet weight and a pre-determined wet:dry weight ratio (detail of the ratio determination is presented in SI). Desorption was then initiated in the centrifuge tubes by adding 38 mL of a pre-adjusted 0.05 M NaNO3 Ni-free solution to the wet solid. The added NaNO3 solution had been adjusted to a pH of 7.0 over a period of 24 h with a separate aliquot of each substrate at a solid:liquid ratio of 1:100 and filtered through a 0.45-μm Millipore MCE filter. Tubes were shaken for at least 20 h, and subsequently, one 6 mL aliquot of the liquid was collected from each tube. Samples collected during the equilibration experiments were immediately filtered through 0.45-μm PVDF syringe filters, stored at 4 °C, and analyzed within 14 days.
2.6 Isotherm Models
The concentrations (qads) and percentages (%adsads) of nickel adsorbed to the solids at the end of adsorption equilibration experiments were calculated using Eqs. 10 and 11, respectively:
where V is the volume of liquid (L), m is the mass of solid (g), and \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_0} \) and \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_f} \) are the initial and final dissolved nickel concentrations (mg L−1), respectively. The concentrations (qdes) and percentages (%adsdes) of nickel adsorbed to the solids at the end of the desorption equilibration experiments were calculated using Eqs. 12 and 13, respectively:
where qads is the final adsorbed nickel concentration at the end of the adsorption experiment, V is the volume of liquid (L), m is the mass of solid (g), and \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_f} \) is the final dissolved nickel concentration (mg L−1). \( {\left[\mathrm{Ni}\right]}_{{\mathrm{diss}}_0} \)is the initial dissolved nickel concentration in the reactor, calculated using the final dissolved nickel concentration in the adsorption experiment, the water content of the wet solid transferred to the reactor, and the volume of NaNO3 added to the reactor at the start of the desorption experiment.
Equilibration (adsorption and desorption) results were fit to Langmuir and Freundlich isotherms. The Langmuir model was originally developed to describe the adsorption of a gas at the surface of a solid (Langmuir 1918). The underlying hypotheses for this model are that adsorption is driven by physical forces (electrostatic, London- van der Waals) and that all surface sites have identical affinities for the sorbate and adsorption is limited to a monolayer coverage of the solid surface. The general form of the Langmuir equation is:
where q is the adsorbed species concentration, qmax is the maximal concentration of surface sites, [A] is the sorbate concentration in solution, and Kads is the adsorption constant. The Langmuir parameters were derived from a linear regression of the experimental data to Eq. 15:
The Freundlich model was originally introduced to describe the logarithmic decrease in adsorption energy with increasing surface coverage of the solid (Freundlich 1907). It was later interpreted as a model describing sorption on an heterogeneous surface whose sites have a variable affinity for the sorbate (Davis et al. 2003; McLean and Bledsoe 1992). The general form of the Freundlich equation is:
where q is the adsorbed species concentration, [A] is the sorbate concentration in solution, and K and n are empirically determined constants. K is referred to as the Freundlich constant and is related to the maximum sorption capacity of the sorbent whereas n is a measure of the non-linearity of the adsorption and is related to the affinity or binding strength of adsorption sites (Davis et al. 2003; Stumm and Morgan 1996). When studying the adsorption of metals on complex substrates such as the ones evaluated in this study, the underlying hypothesis of the Langmuir model are not respected. First, complex organic substrates can form chemical bonds with metals, and second, they typically host diverse, surface functional groups (e.g., carboxylic, phenolic, and thiolic) that have varying affinities for these metals. In the present study, the Langmuir model is used empirically to evaluate and compare the sequestration potential of different sorbing materials in the neutral pH range. Hence, qmax and Kads are empirically determined constants, not to be interpreted, respectively, as the maximal concentration of surface sites and the adsorption constant originally defined in Langmuir adsorption model.
2.7 Geochemical Modeling
Geochemical modeling was conducted to obtain the equilibrium nickel speciation under the experimental conditions of this study. Speciation calculations were performed in open system conditions (pCO2 = 38.5 Pa, EH = 515 mV) at 25 °C in the absence of a solid substrate (sorbent) using the freeware, chemical equilibrium model VMINTEQ version 3.0 (Gustafsson 2016). The VMINTEQ thermodynamic database is a revised version of the original USEPA MINTEQA2 database (Allison et al. 2011) that contains updated and expanded data from the NIST Critical Stability Constant database.
3 Results and Discussion
3.1 Geochemical Modeling
We first modeled the effect of pH on nickel speciation for the Ni-NaNO3 open system solution used in the kinetic experiments (0.05 M NaNO3, 7 mg·Ni L−1). In this system, the solution reached saturation with respect to an amorphous nickel carbonate phase (NiCO3(s), Ksp = 10–11.2) at pH values above 7.33, becoming the dominant species above pH 7.5 (Fig. S1a, SI). We then modeled the effect of dissolved organic carbon on nickel speciation in the same solution at pH 7.5 using the NiCA-Donnan formulation with the default settings (Fig. S1b, SI). Under these settings, 82.5% of the DOC is composed of fulvic acids whose assumed carbon content is 50%. We chose a fixed pH value of 7.5 for this model to evaluate the effect of DOC on the solubility of NiCO3(s). As DOC increases, more Ni-DOC complexes are formed and the concentration of all other Ni species decreases. For DOC concentrations above 68 mg L−1, all nickel species are dissolved at pH 7.5. Finally, we modeled NiCO3(s) saturation as a function of pH for the range of nickel concentrations investigated in the equilibration experiments (Fig. S2, SI).
3.2 Adsorption and Desorption Kinetics
During all adsorption experiments, the dissolved nickel concentrations decreased rapidly (within minutes) (Fig. 1a). Invariant concentrations, presumably representing steady states between adsorption and desorption, were reached in all experiments with the exception of the wood ash slurry. In the presence of wood ash, dissolved nickel concentrations kept decreasing slightly even after 10 h of equilibration. During the course of the adsorption experiments, recorded pH values (6.81 to 7.48) were below NiCO3(s) saturation (7.33) for all substrates except the HCl-algae (Fig. S3a SI). For this substrate, pH increased to nearly 7.5 within the first hour of the experiment before settling back below 7.33. It is unlikely that nickel carbonate precipitated during this pH variation as the brown algae slurry contains high DOC concentrations (Table 1).

Results of the kinetic adsorption (a) and desorption (b) experiments. Dissolved nickel concentrations with time of equilibration
During desorption experiments (Fig. 1b), the dissolved nickel concentrations increased rapidly for all substrates and reached constant concentrations for the compost, wood ash, sawdust, and peat slurries after 0.5 to 4 h. In the case of algae and HCl-algae, dissolved nickel concentrations kept increasing slightly beyond 10 h of equilibration. While more nickel was adsorbed by peat and compost during adsorption experiment (Fig. 1a), desorption trials indicated that they released three to seven times less nickel than the other substrates when exposed to a nickel-free solution. In desorption experiments, recorded pH values (6.70 to 7.63) were below NiCO3(s) saturation for all substrates (Fig. S3b, SI). The saturation pH was higher during desorption (7.66) than adsorption (7.33) experiments because dissolved nickel concentrations were lower, with maximum values of 1.6 mg L−1 (27 μmol L−1).
3.3 Treatment and Modeling of Kinetic Experimental Data
As the use of data points recorded after the attainment of a steady state is likely to lead to erroneous conclusions (Tran et al. 2017), partial data sets were used to fit the experimental data (Eq. 1) to the kinetic models (Fig. 2a, Table 2) (Fig. S4, SI). The three reaction-kinetic models were compared on the basis of their coefficient of determination (R2, Eq. 17) and chi-squared test (χ2, Eq. 18). An R2 value closer to unity and a χ2 value closer to zero indicate the better fit.

Adsorbed (a) and desorbed (b) nickel concentrations (qtADS) as a function of equilibration time during HCl-algae experiment and Elovich, PSO and PFO model fits. Empty symbols identify data used for model fitting
In these equations, qt(exp) (mg g−1) is the experimental adsorbed nickel concentration at time = t, qt(cal) (mg g−1) is the adsorbed nickel concentration achieved from the model using the Solver add-in, and qt(mean) (mg g−1), the mean of the qt(exp) values.
For the adsorption experiments, the estimated qe as well as the R2 and χ2 values were always better with the PSO model; therefore, PFO results will not be discussed. Experimental steady-state values, qe(exp) (mg g−1), were estimated from adsorption experiments and values are presented in Table 2. For adsorption experiments, qe(exp) were estimated as the mean value calculated from the last (wood ash), last two (compost, sawdust, HCl-algae, and algae), or last six (peat) data points. The Elovich model provided a better fit to the selected adsorption data sets (Table 2). The steady-state adsorbed nickel concentration (qe) estimated by the PSO model was 6–12% lower than the experimental value (qe(exp)) for all substrates except for peat for which the two values were identical within one standard deviation. The rate constants derived from the Elovich and PSO models (k2 and α) indicate that the adsorption kinetics increase in the following order: peat > compost > algae > HCl-algae > sawdust > wood ash. The ability of the model constants to reproduce the experimental data (Eq. 6 and 8) was evaluated and highlighted the presence of a fast initial nickel uptake which was not well described by either model. The presence of this fast initial uptake and its influence on the Elovich model fit have been discussed elsewhere (McLintock 1967).
For the desorption experiments, data points recorded prior to reaching a steady state were used to fit the experimental data (Eq. 3) to the Elovich and PSO kinetic models (Fig. 2b, Table 3) (Fig. S5, SI). The PSO model generally provided a better fit to the selected desorption data set. The steady-state desorbed nickel concentrations (qe) estimated by the PSO model were identical, within one standard deviation, to the experimental value (qe(exp)) for all substrates but the algae. No correlation was observed between k2 and α for the desorption models which may be caused by the limited range of concentrations represented by the desorption data sets.
Kinetic-based models such as the PSO and Elovich models are empirical fits to kinetic curves that lack physical significance and combine many different models with different controlling mechanisms (Tan and Hameed 2017). Kinetic-based models are applicable when the mechanism at play is chemisorption on solids that are either non-porous or porous and exhibit high diffusion coefficients (Inglezakis et al. 2019). For porous solids (activated carbon, biochar, zeolites), diffusional mass transfer is the rate-determining step and adsorption processes may require days to weeks to reach a steady state (Inglezakis et al. 2019; Rees et al. 2014; Tran et al. 2017). Multiple adsorption mechanisms are bound to take place on the biochar, biosorbents, and humified materials used in the present study. Diffusion-based models may help identify adsorption mechanism and rate-controlling steps, but these models are more complex and can be computationally demanding and time consuming (Inglezakis et al. 2019; Tan and Hameed 2017). Nevertheless, the PSO and Elovich models still allow us to compare the performance of these materials, but the derived kinetic parameters are solely descriptive and only apply to the experimental conditions investigated in this study. Both models describe the increase in adsorbed nickel at the surface of the substrates, but they do not satisfactorily describe the attainment of a steady state or the fast initial nickel uptake.
3.4 Adsorption Equilibration Experiments and NiCO3(S) Saturation
Over the range of nickel concentrations investigated and in the absence of DOC, the pH at which NiCO3(s) saturation is reached varies from pH 6.77 ([Ni] = 103 mg L−1 or 1750 μmol L−1) to pH 7.88 ([Ni] = 0.6 mg L−1 or 10 μmol L−1) (Fig. S2 and S6, SI). These saturation pH boundaries are conservative as they apply to an open system with no DOC, whereas our samples were equilibrated in closed tubes and solutions contained various amounts of DOC. Ideally, the precipitation of a solid Ni phase should be avoided during equilibration adsorption experiments but, in the present study, field-relevant pH conditions were used and thus oversaturation with respect to NiCO3(s) could not be avoided. During the adsorption equilibration experiments, recorded pH values were below the NiCO3(s) saturation value for samples with initial nickel concentrations of 7 mg L−1 or less and above this limit for samples with initial nickel concentrations of 15 mg L−1 or more. Therefore, precipitation of a solid nickel carbonate phase may have taken place in samples with high initial nickel concentrations. During the desorption experiment, the pH values recorded were always below the NiCO3(s) saturation limit.
3.5 Adsorption and Desorption Equilibration Experiments
The final adsorbed nickel concentrations (qads, Eq. 10) were used to derive the empirical Langmuir and Freundlich isotherm parameters (Table 4). Data points from samples with initial nickel concentrations of 0.6 mg L−1 (10 μmol L−1) were ignored as they were clear outliers in the linear regression. Moreover, results from the algae experiment could not be fitted to the Langmuir model as data points show no clear curvilinear trend. The results from the HCl-algae experiment follow a more curvilinear trend but the adsorption capacity of this substrate is not significantly increased relative to the untreated algae (Fig. 4). According to the R2 coefficients, results for algae, HCl-algae, peat, and wood ash are more appropriately represented by the Freundlich isotherm while those for the compost and maple sawdust could be fitted to both models (Table 4). When compared graphically, the model that best fits the experimental data is not readily identified (Fig. 3). For example, the fit to the Langmuir model appears better for peat, but this is not reflected by the R2 coefficients. Furthermore, it should be noted that the precipitation of nickel carbonate might have occurred in samples with higher initial [Ni], reducing their final nickel concentrations and apparently increasing their adsorbed nickel concentrations, thus favoring a fit to the Freundlich model.

Compost and peat adsorption isotherms with Langmuir and Freundlich model fits
Langmuir model parameters derived from nickel adsorption studies onto organic materials similar to the ones used in this study can be found in the literature (Table S1, SI). The reported qmax values vary within one to two orders of magnitude of each other. These expected discrepancies may originate from the provenance or preparation of the tested organic materials as well as differences in experimental conditions. According to the published qmax values, brown algae display some of the highest nickel adsorption capacities. The highest values were obtained with brown algae of much smaller particle size (< 0.5 mm) than used in our experiments (< 5 mm). In contrast, under our experimental conditions, the adsorption capacities of the investigated substrates increased in the following order: sawdust/wood ash, HCl-algae, compost, and peat. This illustrates why different low-cost sorbent materials need to be compared under identical field-relevant experimental conditions.
During the adsorption experiments, pH was adjusted to 7.0 during a 24 h pH adjustment period and was then allowed to drift after the addition of nickel. It varied between the beginning (full symbols) and the end (empty symbols) of the experiments for all substrates except wood ash, for which the pH of all reactors remained stable at 7.0 (Fig. S7, SI). For the other substrates, a decrease of up to 1 pH unit was observed in most reactors. For the algae and HCl-algae, the pH also decreased in the nickel-free control vessel, likely due to an incomplete pH equilibration prior to the beginning of the experiment. For sawdust, peat, and compost, the pH decrease was proportional to the initial nickel concentration and could be interpreted as resulting from an exchange between H+ and Ni2+ ions on the solid surfaces.
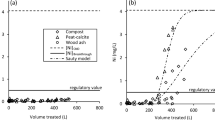
The amount of nickel released during the desorption experiments varied amongst the different substrates (Fig. 4). Desorption results show that adsorption was not reversible and that peat and compost adsorbed and retained more nickel than the other substrates. They also show that more nickel was retained by the HCl-algae than the untreated algae and that sawdust, wood ash, and HCl-algae displayed a similar hysteresis upon desorption yielding linear isotherms.

Adsorption and desorption isotherms. Results are presented as average values from triplicate reactors, with error bars representing one standard deviation from the mean
3.6 Organic Materials Properties and Adsorption
In terms of nickel adsorption capacity, retention capacity, and adsorption kinetics, the performance of humified materials surpassed that of the other substrates tested in this study. None of the physicochemical properties of the five investigated organic substrates could have alone allowed us to predict this outcome (Table 1). Humified materials have lower specific surface areas than wood ash, and yet they adsorb more nickel because of their organic surface functional groups. Furthermore, their cation exchange capacities (CEC) are not the highest, implying that a material’s capacity to adsorb and release sodium ions cannot predict its behavior with regard to nickel ions. This being said, peat does possess the second highest CEC (124 meq 100 g−1) after brown algae, and the CEC measurement for brown algae could be biased by the presence of sodium alginate, a water soluble component in the algae’s cellular membrane. The point of zero charge (PZC) of peat (3.42) is the lowest suggesting a negative surface charge at circum-neutral pH, supporting the highest Ni adsorption capacity. Regarding compost, despite having a high PZC (7.68) and, therefore, a mean positive surface charge at circum-neutral pH, it displays a strong Ni adsorption capacity. This may be interpreted as evidence of chemical interactions with Ni, rather than electrostatic binding, most likely bidentate binding to carboxylic groups, consistent with the proton exchange (decrease in pH upon adsorption).
Leaching of dissolved organic carbon also plays a role in the substrate’s capacity to adsorb nickel. DOC forms soluble complexes with divalent nickel and reduces the concentration of free nickel ions in solution (Fig. S1b, SI). Therefore, as DOC increases in a given system, the amount of charged nickel species decreases, leading to less nickel adsorption. The humified materials who performed better, release much less DOC than the other investigated biosorbents (e.g., algae, sawdust), but they release more DOC than the wood ash.
4 Conclusion
The Ni adsorption and retention capacity of five low-cost organic materials was compared by conducting short-term batch experiments at pH 7.0 in 0.05 M NaNO3. Results of adsorption and desorption kinetic experiments revealed that peat and compost adsorbed nickel more rapidly than the other substrates. A steady state was reached within 40 min in the presence of peat, whereas it took nearly 10 h with compost, sawdust, algae, and HCl-algae. For wood ash, a porous solid for which diffusional mass transfer is the expected rate-determining step, adsorption kinetics were slower and steady state was not reached within 23 h. More nickel was adsorbed by peat and compost during kinetic experiments and desorption trials indicated that they released three to seven times less nickel than the other substrates when exposed to a nickel-free solution. In equilibration experiments, peat and compost also displayed a higher adsorption capacity than the other substrates and, irrespective of the initial nickel concentrations tested, retained a higher percentage of the adsorbed nickel when exposed to nickel-free solutions. Whereas humified materials performed better than other materials under the experimental conditions of this study, other factors that affect metal sequestration need to be evaluated before moving on to long-term column experiments. These factors include the presence of competing ions, pH, and the Ni retention capacity of the substrates over periods of several weeks.
References
Allison, J., Brown, D. S., Novo-Gradac, K. J. (2011). MINTEQA2/PRODEFA2, a geochemical assesment model for environmental systems: version 3.0 user’s manual.
APHA. (2005). Standard methods for the examination of water and wastewater (21st ed.). Washington, D.C.: American Public Health Association.
Arán, D., Antelo, J., Lodeiro, P., Macías, F., & Fiol, S. (2017). Use of waste-derived biochar to remove copper from aqueous solution in a continuous-flow system. Industrial and Engineering Chemistry Research, 56, 12755–12762. https://doi.org/10.1021/acs.iecr.7b03056.
Bakatula, E. N., Richard, D., Neculita, C. M., & Zagury, G. J. (2018). Determination of point of zero charge of natural organic materials. Environmental Science and Pollution Research, 25, 7823–7833. https://doi.org/10.1007/s11356-017-1115-7.
Blanchard, G., Maunaye, M., & Martin, G. (1984). Removal of heavy metals from waters by means of natural zeolites. Water Research, 18, 1501–1507.
Brunauer, S., Emmett, P. H., & Teller, E. (1938). Adsorption of gases in multimolecular layers. Journal of the American Chemical Society, 60, 309–319.
Calugaru, I. L., Neculita, C. M., Genty, T., & Zagury, G. J. (2018). Metals and metalloids treatment in contaminated neutral effluents using modified materials. Journal of Environmental Management, 212, 142–159. https://doi.org/10.1016/j.jenvman.2018.02.002.
CEAEQ (2013). Détermination du carbone et du soufre: méthode par combustion et dosage par spectrophotométrie infrarouge, Méthode MA.310-CS 1.0. http://www.ceaeq.gouv.qc.ca/methodes/pdf/MA310CS10.pdf. Accessed 2019-06-18 2019.
Das, N., Vimala, R., & Karthika, P. (2008). Biosorption of heavy metals - an overview. Indian Journal of Biotechnology, 7, 159–169.
Davis, T. A., Volesky, B., & Mucci, A. (2003). A review of the biochemistry of heavy metal biosorption by brown algae. Water Research, 37, 4311–4330. https://doi.org/10.1016/S0043-1354(03)00293-8.
Fourest, E., & Volesky, B. (1996). Contribution of sulfonate groups and alginate to heavy metal biosorption by the dry biomass of Sargassum fluitans. Environmental Science and Technology, 30, 277–282. https://doi.org/10.1021/es950315s.
Freundlich, H. (1907). Über die adsorption in lösungen Zeitschrift für physikalische. Chemie, 57, 385–470.
Gustafsson, J. P. (2016). Visual MINTEQ ver. 3.0. https://vminteq.lwr.kth.se/. Accessed 2018-01-15 2018.
Guthrie, J. W., et al. (2005). Complexation of Ni, Cu, Zn, and Cd by DOC in some metal-impacted freshwater lakes: a comparison of approaches using electrochemical determination of free-metal-ion and labile complexes and a computer speciation model, WHAM V and VI. Analytica Chimica Acta, 528, 205–218. https://doi.org/10.1016/j.aca.2004.10.003.
Ho, Y. S. (1995). Adsorption of heavy metals from waste streams by peat. Ph. D. Thesis, University of Brimingham, Brimingham (UK).
Iakovleva, E., & Sillanpaa, M. (2013). The use of low-cost adsorbents for wastewater purification in mining industries. Environmental Science and Pollution Research, 20, 7878–7899. https://doi.org/10.1007/s11356-013-1546-8.
Inglezakis, V. J., Fyrillas, M. M., & Park, J. (2019). Variable diffusivity homogeneous surface diffusion model and analysis of merits and fallacies of simplified adsorption kinetics equations. Journal of Hazardous Materials, 367, 224–245. https://doi.org/10.1016/j.jhazmat.2018.12.023.
Lagergren, S. (1898). About the theory of so-called adsorption of soluble substances. Sven Vetenskapsakad Handingarl, 24, 1–39.
Langmuir, I. (1918). The adsorption of gases on plane surfaces of glass, mica and platinum. Journal of the American Chemical Society, 40, 1361–1403.
Mandal, R., Hassan, N. M., Murimboh, J., Chakrabarti, C. L., Back, M. H., Rahayu, U., & Lean, D. R. S. (2002). Chemical speciation and toxicity of nickel species in natural waters from the Sudbury area (Canada). Environmental Science and Technology, 36, 1477–1484. https://doi.org/10.1021/es015622e.
McLean, J. E., & Bledsoe, B. E. (1992). Behavior of metals in soils. Washington, DC: United States Environmental Protection Agency.
McLintock, I. S. (1967). The Elovich equation in chemisorption kinetics. Nature, 216, 1204–1205.
MDDELCC (2012) Directive 019 sur l’industrie minière. Ministère du Développement Durable, Environnement et Lutte contre les Changements Climatiques du Québec. http://www.mddelcc.gouv.qc.ca/milieu_ind/directive019/. Accessed 2019-06-18.
Mohan, D., Sarswat, A., Ok, Y. S., & Pittman Jr., C. U. (2014). Organic and inorganic contaminants removal from water with biochar, a renewable, low cost and sustainable adsorbent–a critical review. Bioresource Technology, 160, 191–202. https://doi.org/10.1016/j.biortech.2014.01.120.
Rees, F., Simonnot, M. O., & Morel, J. L. (2014). Short-term effects of biochar on soil heavy metal mobility are controlled by intra-particle diffusion and soil pH increase. European Journal of Soil Science, 65, 149–161. https://doi.org/10.1111/ejss.12107.
Roginsky, S. Z., & Zeldovich, J. (1934). The catalytic oxidation of carbon monoxide on manganese dioxide. Acta Physicochimica USSR, 1, 554.
Stokes, P. (1988). Nickel in aquatic systems. In H. Sigel & A. Sigel (Eds.), Metal ions in biological systems: nickel and its role in biology, vol 23 (pp. 31–46). New York: Marcel Dekker.
Stumm, W., & Morgan, J. (1996). Aquatic chemistry : chemical equilibria and rates in natural waters. Environmental Science and Technology (3rd ed.). New York: Wiley.
Tan, K. H. (2014). Humic matter in soil and the environment: principles and controversies (2nd ed.). Boca Raton: CRC Press, Taylor & Francis Group.
Tan, K. L., & Hameed, B. H. (2017). Insight into the adsorption kinetics models for the removal of contaminants from aqueous solutions. Journal of the Taiwan Institute of Chemical Engineers, 74, 25–48. https://doi.org/10.1016/j.jtice.2017.01.024.
Tong, X.-j., Li, J.-y., Yuan, J.-h., & Xu, R.-k. (2011). Adsorption of Cu(II) by biochars generated from three crop straws. Chemical Engineering Journal, 172, 828–834. https://doi.org/10.1016/j.cej.2011.06.069.
Tran, H. N., You, S.-J., Hosseini-Bandegharaei, A., & Chao, H.-P. (2017). Mistakes and inconsistencies regarding adsorption of contaminants from aqueous solutions: a critical review. Water Research, 120, 88–116. https://doi.org/10.1016/j.watres.2017.04.014.
Uchimiya, M., Wartelle, L. H., Klasson, K. T., Fortier, C. A., & Lima, I. M. (2011). Influence of pyrolysis temperature on biochar property and function as a heavy metal sorbent in soil. Journal of Agricultural and Food Chemistry, 59, 2501–2510. https://doi.org/10.1021/jf104206c.
Wan Ngah, W. S., & Hanafiah, M. A. K. M. (2008). Removal of heavy metal ions from wastewater by chemically modified plant wastes as adsorbents: a review. Bioresource Technology, 99, 3935–3948. https://doi.org/10.1016/j.biortech.2007.06.011.
Westholm, L. J., Repo, E., & Sillanpaa, M. (2014). Filter materials for metal removal from mine drainage-a review. Environmental Science and Pollution Research, 21, 9109–9128. https://doi.org/10.1007/s11356-014-2903-y.
Xiao, Y., Azaiez, J., & Hill, J. M. (2018). Erroneous application of pseudo-second-order adsorption kinetics model: ignored assumptions and spurious correlations. Industrial and Engineering Chemistry Research, 57, 2705–2709. https://doi.org/10.1021/acs.iecr.7b04724.
Xu, X., Cao, X., Zhao, L., Zhou, H., & Luo, Q. (2014). Interaction of organic and inorganic fractions of biochar with Pb(II) ion: further elucidation of mechanisms for Pb(II) removal by biochar. RSC Advances, 4, 44930–44937. https://doi.org/10.1039/c4ra07303g.
Xue, H. B., Jansen, S., Prasch, A., & Sigg, L. (2001). Nickel speciation and complexation kinetics in freshwater by ligand exchange and DPCSV. Environmental Science and Technology, 35, 539–546. https://doi.org/10.1021/es0014638.
Zhou, Y. F., & Haynes, R. J. (2010). Sorption of heavy metals by inorganic and organic components of solid wastes: significance to use of wastes as low-cost adsorbents and immobilizing agents. Critical Reviews in Environmental Science and Technology, 40, 909–977. https://doi.org/10.1080/10643380802586857.
Funding
This study was funded by the NSERC (Natural Sciences and Engineering Research Council of Canada), grant no. 469489-14, and the industrial partners of the RIME UQAT Polytechnique Montreal, including Agnico Eagle, Mine Canadian Malartic, Iamgold, Raglan Mine Glencore, and Rio Tinto. D.R., also wishes to acknowledge support from the “Fonds de recherche Nature et technologies” (FQRNT) graduate scholarship program and from the “Fondation et alumni de Polytechnique Montréal” (Bourse Banque de Montréal).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 91 kb)
Rights and permissions
About this article

Cite this article
Richard, D., Mucci, A., Neculita, C.M. et al. Comparison of Organic Materials for the Passive Treatment of Synthetic Neutral Mine Drainage Contaminated by Nickel: Adsorption and Desorption Kinetics and Isotherms. Water Air Soil Pollut 231, 556 (2020). https://doi.org/10.1007/s11270-020-04917-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-020-04917-z