
Abstract
Industrial synthesis processes produce high concentration of hazardous organic pollutants into water body, which must be removed before being discharged. Advanced oxidation processes (AOPs) using heterogeneous catalysts has been widely utilized for wastewater treatment. Here, RuO2-based catalyst was synthesized by a general impregnation method and used to oxidize phenol by peroxymonosulfate (PMS) as an oxidant in aqueous solution. The properties of this supported catalyst were characterized by SEM (scanning electron microscopy), XRD (powder X-ray diffraction), and nitrogen adsorption-desorption. The mesoporous Al2O3 support had large surface area and high thermal stability. It is found that ruthenium oxide-based catalyst is highly effective to activate PMS to related sulfate radicals. The effects of catalyst loading, phenol concentration, PMS concentration, reaction temperature, and reusability of the as-prepared catalyst on phenol degradation have been investigated. Overall, our findings demonstrate that in RuO2/Al2O3 mesoporous catalyst, Oxone (PMS) is effectively activated, and 100% phenol degradation occurs in 60 min. To regenerate the deactivated catalyst and improve its catalytic properties, three different methods involving annealing in air, washing with water, and applying ultrasonics were used. The catalyst was recovered thoroughly by heating treatment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Many organic contaminants in polluted water are toxic and extremely refractory to natural degradation causing significant problems to public health and ecological system (Shukla et al. 2010b; P. R. Shukla et al. 2010; Lu et al. 2015). Several methods such as adsorption, flocculation, membrane separation, and chemical oxidation can be used to remove the organic toxins. Among these methods, advanced oxidation process (AOP) such as wet air oxidation, thermal destruction, ozonation, and homogeneous/heterogeneous oxidation can achieve complete degradation of organic contaminants in wastewater to water and carbon dioxide as final products (Wang 2008; Cao et al. 2009; Agustina et al. 2005; M Anbia and Amirmahmoodi 2011; Mansoor Anbia and Ghaffari 2009). Fenton oxidation using OH radical in the presence of Fe2+ ions is a typical AOP. However, they suffer from disadvantages such as low pH range, metal leaching, and generation of large quantity of sludge (Melero et al. 2007). In order to eliminate this drawback, another oxidant such as peroxymonosulfate has been proposed. The high oxidation potential of sulfate radicals has caused them to be suggested as an alternative recently (Anipsitakis and Dionysiou 2003; Muhammad et al. 2013). For the oxidation of organic pollutants, conjunction of heavy metals such as cobalt, ruthenium, and iron with PMS to produce sulfate radicals has been proposed. The radical generation process can be described as shown below (Anipsitakis and Dionysiou 2003).
The main issue, in the use of metal ions as catalysts, is their toxicity causing health problems to humans. Therefore, heterogeneous catalysts operating at room temperature with normal pressure and low energy have been proposed recently to overcome the disadvantages of the homogeneous process (Anipsitakis et al. 2005; Shukla et al. 2011b; Chen et al. 2008; Zhang et al. 2010). Heterogeneous catalysts are usually synthesized using cheap materials as supports which bring excellent catalyst performance and practicability (Saputra et al. 2012). Among the various supports such as metal oxides, silica, and zeolite, Al2O3 is an important one that widely used in the chemical and petrochemical industries owing to its diverse merits featuring the acid-base characteristics, having high specific surface area, high mechanical strength, excellent chemical stability, and low price (Rao et al. 2005; Gao et al. 2009; Khalil 2007; Li et al. 2014). In addition, many reports show that ruthenium is one of the best transition metal catalysts that it can be used in oxidation of organic compounds such as aniline, acetic acid, chlorophenols, and phenol (Gallezot et al. 1997; Carrier et al. 2009; Liu et al. 2010; Minh et al. 2006). However, the utilization of this heavy metal-based catalysts with peroxymonosulfate (PMS or Oxone) for phenol degradation has been less developed so far (Muhammad et al. 2012). Therefore, this paper investigates the use of ruthenium heterogeneous catalyst with PMS for chemical oxidation of phenol in aqueous solution. In order to prepare of ruthenium-based catalyst, firstly, mesoporous Al2O3 supports with different surface area were synthesized via precipitation method, and then during impregnation treatment, RuO2 was loaded on the surface of as-prepared alumina supports.
2 Materials and Methods
2.1 Mesoporous Al2O3 Preparation
At first, 27.5 g of aluminum sulfate octadecahydrate (Al2 (SO4)3, Aldrich) was dissolved in deionized water under constant stirring. Then, NH3·H2O solution (1.56 wt. %) was added dropwisely so that the pH of the solution reached 8.0; the solution was heated at 40 °C. After the completion of reaction, the white product was isolated and washed with deionized water until the precipitate became free of SO4 2− and then heated at 90 °C for 3 h by water bath method. After that, the precipitate was washed with anhydrous ethanol (Merck) to remove water and then dispersed in a solution containing 1.0 g cetyltrimethylammonium bromide (CTAB, Aldrich) and 50 mL anhydrous ethanol for 4 h. In the next step, the vessel was transferred in an oven to be heated at 120 °C for 12 h and finally calcined in the air at 600, 800, and 1000 °C for 6 h, respectively, at a heating rate of 1 °C/min.
2.2 Preparation of RuO2/Al2O3 Catalyst
A general impregnation method was used for the synthesis of RuO2/Al2O3 catalyst. At first, 5 g Al2O3 was dispersed in 90 mL of ethanol for 15 min. Next, ruthenium chloride (Aldrich) and 50 mL ethanol were added into the solution and kept stirring vigorously for 16 h, and then dried at 100 °C. The resultant solid was heated at 200 °C for 2 h, and finally calcined at 550 °C for 6 h. The loading of Ru on the support was maintained at 5 wt. %.
2.3 Characterization
The characterization of the obtained catalyst was performed by SEM (scanning electron microscopy), XRD (X-ray diffraction), and nitrogen adsorption-desorption. The mineralogy and the structural features of the support and catalyst were studied by XRD patterns. These experiments were carried out on a Philips 1830 diffractometer with Cu-Kα (λ = 0.154) radiation at a current of 30 mA and voltage of 30 kV. The textural and morphological information of the mesoporous Al2O3 support were obtained using scanning electron microscopy (SEM, Philips XL 30). The specific surface areas were identified by N2 adsorption-desorption experiments performed at 77 K on micromeritics model ASAP 2010 sorptometer. The support and catalyst were degassed at 120 °C for 12 h prior to the analysis.
2.4 Experimental Procedure
To perform the catalytic oxidative examinations, 500 mL of phenol solution with initial concentration of 50 mg/L was poured into a glass beaker and stirred constantly to obtain a homogeneous mixture at 25 °C. Next, a certain amount of catalyst was added to the mixture, and then, it was allowed to reach adsorption equilibrium between phenol molecules and the catalyst during reaction time. The next step of the experiment was to blend an amount of Oxone as an initial reactant. The reaction proceeded for 3 h. Then, during appropriate time interval namely 0 to 160 min, 50 mL of the solution was separated, and an excess of sodium nitrite as a quenching reagent was added to the sample. After that, the solution was filtered with filter paper. Next, phenol concentrations were determined using spectrophotometric measurements (UV/Vis mini 1240 Shimadzu) by standard method employed 4-aminoantipyrine at maximum wavelength of phenol i.e., 500 nm. The same procedures were performed for other initial concentration of phenol (20, 70, and 100 ppm).
3 Results and Discussion
3.1 Characterization of Ru Impregnated Al2O3 Catalyst
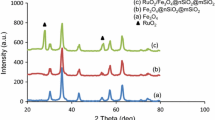
XRD patterns of mesoporous Al2O3 calcined at 600 °C (A-1), 800 °C (A-2), 1000 °C (A-3), and RuO2/Al2O3 (R-A-1) are presented in Fig. 1. It seems very week diffraction peaks in the XRD patterns of A-1, when the calcination temperature increased from 600 to 1000 °C, the diffraction peaks became stronger, which corresponded to the γ-Al2O3 phase. It can be seen that ruthenium species are found in the form of RuO2 on RuO2/Al2O3 at 2θ coordinate of 28°, 35°, and 54.3°.

XRD patterns of A-1, A-2, A-3, and R-A-1
In Fig. 2, SEM images of the synthesized Al2O3 particles are shown. The samples are composed of very tiny spherical particles. Homogeneous particle size distributions are perceived in all samples.

SEM images of A-1 (a), A-2 (b), and A-3 (c)
The N2 adsorption-desorption isotherms of A-1, A-2, A-3 as supports and R-A-1 as catalyst sample are shown in Fig. 3. The specific surface area of A-1, A-2, A-3, and R-A-1 was determined to be 222.05, 140.97, 82.1, and 143.37, respectively. Since the specific surface area of A-1 is larger than A-2 and A-3, it was utilized as the catalyst support. After the RuO2 loading on the support, the specific surface area of catalyst became lower than support. The hysteresis loop indicated the mesoporous structure of Al2O3 and RuO2/Al2O3 samples.

N2 adsorption-desorption isotherms of A-1, A-2, A-3, and R-A-1
3.2 Preliminary Study of Phenol Degradation Using Catalyst
Figure 4 shows preliminary tests of adsorption and phenol degradation in aqueous solution on A-1 and R-A-1 under various experimental conditions. Generally, phenol can adsorb on the surface of A-1 and R-A-1 even at low efficiency. The adsorption efficiency on A-1 and R-A-1 is 8 and 3 % in 2 h, respectively. The results indicate that the surface area of support decreases by loading ruthenium on the surface of the support. In oxidation tests, the Oxone without a solid catalyst could not oxidize phenol. Phenol degradation would occur when catalyst and oxidant (PMS) simultaneously were present in the solution. In RuO2/Al2O3-Oxone, phenol could be removed completely from solution in 60 min.

Phenol removal in various reaction conditions
Anipsitakis and Dionysiou (Anipsitakis and Dionysiou 2004) have reported that Co (II) and Ru (III) are the best transition metal catalysts for the activation of PMS. The reaction of Ru (III) with PMS can be described as following:
The oxidation process is proposed as below.

At first, catechol was produced in the presence of sulfate radicals and then converted to hydroquinone and benzoquinone (Anipsitakis et al. 2006; Mijangos et al. 2006; Yalfani et al. 2009). Maleic acid, oxalic acid, and acetic acid, as the latter intermediates, were finally mineralized to CO2 and H2O (Hu et al. 2011).
3.3 Effects of Reaction Parameters on Phenol Degradation
Several variables such as phenol concentration, catalyst loading, Oxone concentration, and reaction temperature can influence phenol degradation in aqueous solution. These variables were investigated here.
Figure 5 shows various concentrations of phenol (between 20 and 100 ppm) on the phenol degradation. Phenol degradation efficiency decreased with increasing phenol concentration. At low phenol concentrations (20–50 ppm), 100 % phenol degradation could be achieved within 60 min. At phenol concentration of 100 ppm, degradation efficiency was only 50 % within 140 min.

Effect of phenol concentration on phenol removal. R-A-1 RuO2/Al2O3. Reaction conditions: 1 g Oxone, 0.2 g catalyst, 25 °C
In phenol degradation, production of sulfate radicals is the main reaction, which depends on supported ruthenium catalyst and Oxone. Due to the same concentration of RuO2/Al2O3 and Oxone, sulfate radical concentration produced in solution will be the same. Thus, a high amount of phenol in solution will require more time to achieve the same removal rate, thus lowering phenol degradation efficiency.
Degradation of phenol in the solution depends on the amount of the RuO2/Al2O3 (R-A-1) catalyst (Fig. 6). Phenol oxidation efficiency is enhanced by increasing catalyst dosage in the solution. At 0.1 g RuO2/Al2O3, phenol degradation would be 88 % at 100 min, while RuO2/Al2O3 loading of 0.2 g, oxidation of phenol would reach 100 % at 60 min. It was noticed that the highest degradation efficiency was achieved at the highest Ru-based catalyst dosage, mainly because increasing catalyst dosage could increase the active sites.

Effect of catalyst loading on phenol removal. R-A-1 RuO2/Al2O3. Reaction conditions: 50 ppm, 1 g Oxone, 25 °C
Figure 7 indicates that degradation of phenol depends on the Oxone amount. It was reasonable that higher Oxone concentration led to more degradation of phenol, because Oxone was the origin of driving force for the forming of SO4 ·−. At 0.2 g Oxone, 88 % removal of phenol could be achieved in about 100 min. When concentration of oxidant component is increased to 1 g, phenol could be fully removed within 60 min. Degradation efficiency would decrease, at extra Oxone loading. It has been reported that self-quenching phenomenon of hydroxyl and sulfate ions by PMS are possible as shown below (P. Shukla et al. 2010a).

Effect of Oxone concentration on phenol removal. R-A-1 RuO2/Al2O3. Reaction conditions: 50 ppm, 0.2 g catalyst, 25 °C
The reduction potential of SO5 ·−/HSO5 − is 0.95 V at pH 7. Meanwhile, SO4 ·−/SO4 2− has an oxidation potential of [2.5–3.1 V] (Shukla et al. 2011a), which makes it possible that extra amounts of HSO5 − from Oxone would consume the active SO4 ·−, resulting in a lower degradation rate.
The reaction temperature effect on phenol degradation is shown in Fig. 8. As can be seen, the rate of reaction would increase considerably with increase in temperature owing to accelerating the formation of SO4 ·−. Complete phenol degradation at temperature of 25 °C would be achieved in 60 min. When the temperature is raised to 35 °C, 100 % degradation efficiency of phenol would be achieved in about 40 min. At 45 °C, complete removal could be achieved in about 20 min. Taking into account the practical application of the system, the optimal condition for the following experiments was ambient temperature.

Effect of temperature on phenol removal. R-A-1 RuO2/Al2O3. Reaction conditions: 50 ppm, 1 g Oxone, 0.2 g catalyst
Based on the results, it can be concluded that supported ruthenium catalyst has accelerated the phenol degradation due to physicochemical properties of catalyst. The catalyst was used once before the regeneration process, and then, it was tested after its regeneration by annealing in air, using ultrasonication and washing with water. The deactivation of catalyst occurs probably due to adsorption of intermediates and a fraction of loose ruthenium leaching from the support. One hundred percent phenol removal was achieved after 70 min for the catalyst regenerated by annealing in air. This shows that the catalytic activity of the catalyst was recovered thoroughly by heating treatment, and surface-attached intermediates were removed, whereas regeneration of the used catalyst by applying ultrasonic and washing with water partially eliminated the intermediates presented on the surface of the catalysts (Fig. 9).

Regeneration studies of the used catalyst
4 Conclusions
This study provided a feasible approach for applying the mesoporous alumina as appropriate support for synthesizing of based catalyst. Heterogeneous oxidative degradation of phenol using RuO2/Al2O3-Oxone is effective methods for waste water treatment. Investigations indicated that RuO2/Al2O3 is a good catalyst for generating sulfate radicals in the presence of Oxone to degrade phenol under atmospheric pressure and close to ambient temperature. Performance of phenol oxidation was dramatically influenced by operating parameters, such as catalyst dosage, oxidant and phenol concentration, and reaction temperature. Our findings demonstrate that in this catalyst, Oxone effectively activated, and 100 % phenol degradation occurs in 60 min. Regeneration by heat treatment could fully recover the catalyst activity.
References
Agustina, T. E., Ang, H., & Vareek, V. (2005). A review of synergistic effect of photocatalysis and ozonation on wastewater treatment. Journal of Photochemistry and Photobiology C: Photochemistry Reviews, 6(4), 264–273.
Anbia, M., & Amirmahmoodi, S. (2011). Adsorption of phenolic compounds from aqueous solutions using functionalized SBA-15 as a nano-sorbent. Scientia Iranica, 18(3), 446–452.
Anbia, M., & Ghaffari, A. (2009). Adsorption of phenolic compounds from aqueous solutions using carbon nanoporous adsorbent coated with polymer. Applied Surface Science, 255(23), 9487–9492.
Anipsitakis, G. P., & Dionysiou, D. D. (2003). Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt. Environmental Science & Technology, 37(20), 4790–4797.
Anipsitakis, G. P., & Dionysiou, D. D. (2004). Radical generation by the interaction of transition metals with common oxidants. Environmental Science & Technology, 38(13), 3705–3712.
Anipsitakis, G. P., Dionysiou, D. D., & Gonzalez, M. A. (2006). Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. Implications of chloride ions. Environmental Science & Technology, 40(3), 1000–1007.
Anipsitakis, G. P., Stathatos, E., & Dionysiou, D. D. (2005). Heterogeneous activation of oxone using Co3O4. The Journal of Physical Chemistry B, 109(27), 13052–13055.
Cao, S., Yeung, K. L., Kwan, J. K., To, P. M., & Samuel, C. (2009). An investigation of the performance of catalytic aerogel filters. Applied Catalysis B: Environmental, 86(3), 127–136.
Carrier, M., Besson, M., Guillard, C., & Gonze, E. (2009). Removal of herbicide diuron and thermal degradation products under catalytic wet air oxidation conditions. Applied Catalysis B: Environmental, 91(1), 275–283.
Chen, X., Chen, J., Qiao, X., Wang, D., & Cai, X. (2008). Performance of nano-Co3O4/peroxymonosulfate system: kinetics and mechanism study using Acid Orange 7 as a model compound. Applied Catalysis B: Environmental, 80(1), 116–121.
Gallezot, P., Chaumet, S., Perrard, A., & Isnard, P. (1997). Catalytic wet air oxidation of acetic acid on carbon-supported ruthenium catalysts. Journal of Catalysis, 168(1), 104–109.
Gao, C., Lin, Y.-J., Li, Y., Evans, D. G., & Li, D.-Q. (2009). Preparation and characterization of spherical mesoporous CeO2−Al2O3 composites with high thermal stability. Industrial & Engineering Chemistry Research, 48(14), 6544–6549.
Hu, L., Yang, X., & Dang, S. (2011). An easily recyclable Co/SBA-15 catalyst: heterogeneous activation of peroxymonosulfate for the degradation of phenol in water. Applied Catalysis B: Environmental, 102(1), 19–26.
Khalil, K. M. (2007). Synthesis and characterization of mesoporous ceria/alumina nanocomposite materials via mixing of the corresponding ceria and alumina gel precursors. Journal of Colloid and Interface Science, 307(1), 172–180.
Li, D., Wu, C., Tang, P., & Feng, Y. (2014). In situ synthesis and properties of ZSM-5/α-Al2O3 composite. Materials Letters, 133, 278–280.
Liu, W.-M., Hu, Y.-Q., & Tu, S.-T. (2010). Active carbon–ceramic sphere as support of ruthenium catalysts for catalytic wet air oxidation (CWAO) of resin effluent. Journal of Hazardous Materials, 179(1), 545–551.
Lu, M., Yao, Y., Gao, L., Mo, D., Lin, F., & Lu, S. (2015). Continuous treatment of phenol over an Fe2O3/γ-Al2O3 catalyst in a fixed-bed reactor. Water, Air, & Soil Pollution, 226(4), 1–13.
Melero, J., Calleja, G., Martínez, F., Molina, R., & Pariente, M. (2007). Nanocomposite Fe2O3/SBA-15: an efficient and stable catalyst for the catalytic wet peroxidation of phenolic aqueous solutions. Chemical Engineering Journal, 131(1), 245–256.
Mijangos, F., Varona, F., & Villota, N. (2006). Changes in solution color during phenol oxidation by Fenton reagent. Environmental Science & Technology, 40(17), 5538–5543.
Minh, D. P., Gallezot, P., & Besson, M. (2006). Degradation of olive oil mill effluents by catalytic wet air oxidation: 1. Reactivity of p-coumaric acid over Pt and Ru supported catalysts. Applied Catalysis B: Environmental, 63(1), 68–75.
Muhammad, S., Saputra, E., Sun, H., Ang, H.-M., Tadé, M. O., & Wang, S. (2013). Removal of phenol using sulphate radicals activated by natural zeolite-supported cobalt catalysts. Water, Air, & Soil Pollution, 224(12), 1–9.
Muhammad, S., Shukla, P. R., Tadé, M. O., & Wang, S. (2012). Heterogeneous activation of peroxymonosulphate by supported ruthenium catalysts for phenol degradation in water. Journal of Hazardous Materials, 215, 183–190.
Rao, P. M., Wolfson, A., Kababya, S., Vega, S., & Landau, M. (2005). Immobilization of molecular H3PW12O40 heteropolyacid catalyst in alumina-grafted silica-gel and mesostructured SBA-15 silica matrices. Journal of Catalysis, 232(1), 210–225.
Saputra, E., Muhammad, S., Sun, H., Ang, H. M., Tadé, M. O., & Wang, S. (2012). Red mud and fly ash supported Co catalysts for phenol oxidation. Catalysis Today, 190(1), 68–72.
Shukla, P., Fatimah, I., Wang, S., Ang, H., & Tadé, M. O. (2010a). Photocatalytic generation of sulphate and hydroxyl radicals using zinc oxide under low-power UV to oxidise phenolic contaminants in wastewater. Catalysis Today, 157(1), 410–414.
Shukla, P., Sun, H., Wang, S., Ang, H. M., & Tadé, M. O. (2011a). Co-SBA-15 for heterogeneous oxidation of phenol with sulfate radical for wastewater treatment. Catalysis Today, 175(1), 380–385.
Shukla, P., Sun, H., Wang, S., Ang, H. M., & Tadé, M. O. (2011b). Nanosized Co3O4/SiO2 for heterogeneous oxidation of phenolic contaminants in waste water. Separation and Purification Technology, 77(2), 230–236.
Shukla, P., Wang, S., Singh, K., Ang, H., & Tadé, M. O. (2010b). Cobalt exchanged zeolites for heterogeneous catalytic oxidation of phenol in the presence of peroxymonosulphate. Applied Catalysis B: Environmental, 99(1), 163–169.
Shukla, P. R., Wang, S., Sun, H., Ang, H. M., & Tadé, M. (2010c). Activated carbon supported cobalt catalysts for advanced oxidation of organic contaminants in aqueous solution. Applied Catalysis B: Environmental, 100(3), 529–534.
Wang, S. (2008). A comparative study of Fenton and Fenton-like reaction kinetics in decolourisation of wastewater. Dyes and Pigments, 76(3), 714–720.
Yalfani, M. S., Contreras, S., Medina, F., & Sueiras, J. (2009). Phenol degradation by Fenton’s process using catalytic in situ generated hydrogen peroxide. Applied Catalysis B: Environmental, 89(3), 519–526.
Zhang, W., Tay, H. L., Lim, S. S., Wang, Y., Zhong, Z., & Xu, R. (2010). Supported cobalt oxide on MgO: highly efficient catalysts for degradation of organic dyes in dilute solutions. Applied Catalysis B: Environmental, 95(1), 93–99.
Acknowledgments
The authors are thankful to Research Council of Iran University of Science and Technology (Tehran) and Iran National Science Foundation (INSF) for the financial support to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Anbia, M., Rezaie, M. Synthesis of Supported Ruthenium Catalyst for Phenol Degradation in the Presence of Peroxymonosulfate. Water Air Soil Pollut 227, 349 (2016). https://doi.org/10.1007/s11270-016-3047-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-016-3047-0