
Abstract
This work describes the experimental determination of the major degradation and sorption parameters of polycyclic aromatic hydrocarbons (PAHs) in a sensitive marine environment, the Northern Adriatic Sea (Mediterranean Sea). The persistence and sorption of complex PAH mixtures were analysed in the laboratory in conditions mimicking open-sea and coastal sediments, which differ in grain size and organic carbon content and are characterized by different oceanographic conditions including abiotic influences. The study investigated how sediment type and texture and salinity, temperature and solar irradiation conditions affect the partitioning of low molecular weight (LMW) and high molecular weight (HMW) PAHs into aqueous and solid phase, which critically influence their behaviour. With regard to the change from offshore to coastal conditions, HMW PAHs were found to be more sensitive to the increased salinity (from 21 to < 37 parts per thousand), whereas LMW PHAs were more sensitive to the temperature increase (from 11 to 22 °C). HMW PAHs were more affected by sediment type changes in terms of distribution coefficient (K d). Since the physicochemical properties of organic pollutants affect their distribution, transport, bioavailability and toxicity, analysing the accumulation and persistence of dissolved pollutants can provide useful information for environmental risk assessment and management.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) are an ubiquitous class of pollutants that tend to persist in the environment. Due to their toxicity to humans, mutagenicity and carcinogenicity, 16 PAHs are included in the US Environmental Protection Agency (EPA) priority pollutant list (Laflamme and Hites 1978; Keith and Telliard 1979; Neff 1979; Elmquist et al. 2007; Honda et al. 2007; Parinos et al. 2013). These organic compounds are found in all environments. In marine water, they are found especially near the areas involved by intense human activity such as harbours, industrial site urban areas and close to river estuaries (Yunker et al. 2002; Kannan et al. 2005; Lucialli et al. 2007; Botsou and Hatzianestis 2012; Manzetti 2013). Along their route from their petrogenic or pyrogenic sources to the water column and coastal and open-sea sediments, PAHs undergo chemical and biological processes that affect their toxicity (e.g. photo-oxidation) and turn them into more reactive products that are more subject to bioaccumulation (Shor et al. 2004; Bihari et al. 2006; Parinos et al. 2013). The main processes are sorption and degradation (Neff et al. 2005; Celis et al. 2006; Sehili and Lammel 2007; Haritash and Kaushik 2009; Wu and Sun 2010). Particle-water interactions are among the most important mechanisms affecting the distribution and movement of hydrophobic organic chemicals in aquatic environments (Trevisan et al. 1995; Zhou et al. 1999; Xia and Wang 2008). Aqueous-phase PAHs are more available and therefore have a greater toxic potential, than solid-phase PAHs. Owing to their highly hydrophobic nature, PAHs show strong sorption to sediment organic matter and to fine combustion particles (like soot and/or char black carbon), which protect them from degradation during transport in water (Dachs et al. 2002; Yunker et al. 2002; Ravaioli et al. 2003; Neff et al. 2005; Parinos et al. 2013). The partitioning of organic compounds into solid and aqueous phase can be expressed by the distribution coefficient (K d), a physicochemical parameter describing the ratio of solid phase (C s, ng g−1 d.w.) to aqueous phase (C w, ng ml−1), where C is the concentration of the contaminant (Karickhoff et al. 1979; Trevisan et al. 1995; Yu et al. 2009). Moreover, the organic carbon (OC)/water partition coefficient (K oc) can help select appropriate test parameters for sorption studies based on the equation K oc = K d/f oc, where f oc is the total OC (TOC, %) fraction of sediment. K oc is similar to the octanol/water partitioning coefficient (K ow), which is frequently used in modelling non-polar organic compound bioconcentrations (Neff et al. 2005). Substance persistence in marine sediment is usually defined by its half-life (t 1/2), i.e. the loss of 50 % of the initial concentration of the contaminant (Marini and Frapiccini 2013).
This study complements previous PAH research conducted by the authors in the Northern Adriatic Sea (Marini and Frapiccini 2013). In the present work, three key degradation and sorption parameters, t 1/2, K d and K oc, are calculated experimentally and compared in conditions mimicking coastal and open sea, to gain insights into PAH bioavailability in the marine environment and provide data on the mobility of solid- and aqueous-phase PAHs.
2 Materials and Methods
2.1 Study Area
The Northern Adriatic Sea receives important PAH inputs from the Po Valley, a major industrial and agricultural area with a dense population (Stracquadanio et al. 2007; Masiol et al. 2013). Several Alpine and Apennine rivers running through it discharge organic contaminants including PAHs into the Northern Adriatic Sea (Castellarin et al. 2011). Freshwater inputs generally form a coastal layer characterized by cyclonic circulation; since most of this water is discharged by the Po River, the layer is found mainly south of the Po River delta (Campanelli et al. 2004, 2011; Marini et al. 2008). In a previous study, we examined persistence in deep Northern Adriatic Sea sediment (Marini and Frapiccini 2013). The present study compared PAH persistence and sorption in coastal and open-sea sediment. Sediment samples were collected at the same sites as in the previous study: site A, a shallow water area close to Ravenna harbour, in the Po Valley (Fig. 1) and site B, an open-sea area along the Adriatic Sea midline, 50 km off Ancona (Fig. 1).

Map of the Adriatic Sea and geographical localization of the two sampling sites
2.2 Sediment Collection and Characterization
Sediment samples (0–2 cm), two from site A and five from site B, were collected using a box corer (17 × 10 × 24.5 cm). After removal of gross material and debris, samples were stored in transparent glass jars at −18 °C until analysis. Particle size was classified according to Shepard (1954). TOC was measured in silver capsules using a FISONS NA2000 Element Analyzer (Tesi et al. 2006) after dissolving the sample in 1.5 N HCl to remove the carbonate fraction. Bottom water samples (one per site) were analysed for salinity and temperature by the CTD multiparameter probe (Campanelli et al. 2011). Sediment and water data are reported in Table 1.
2.3 Laboratory Conditions and PAH Subdivision
The characteristics of coastal (site A) and open-sea (site B) sediment were reproduced in the laboratory according to Artegiani et al. (1997) based on the laboratory conditions reported by Marini and Frapiccini (2013): 18–22 °C and sunlight for site A and <11 °C and darkness for site B. Salt concentrations, prepared with synthetic seawater, were 21–33 and >37 ppt (part per thousand, 1 ppt = 1‰) for site A and site B, respectively.
t 1/2, K d and K oc were measured in PAH compounds and averaged on the basis of their molecular weight. PAHs with two and three aromatic rings (naphthalene, acenaphthylene, acenaphthene, fluorene, phenanthrene and anthracene) were considered as low molecular weight (LMW) PAHs (Bajt 2012) and those with four, five and six rings (fluoranthene, pyrene, chrysene, benz[a]anthracene, benzo[b]fluoranthene, benzo[k]fluoranthene, benz[a]pyrene, dibenz[a,h]anthracene, indeno[1,2,3-cd]pyrene and benzo[ghi]perylene) were considered as high molecular weight (HMW) PAHs.
2.4 Chemicals and PAH Extraction
The chemicals used were dichloromethane, acetonitrile for high-performance liquid chromatography (HPLC) gradient grade (VWR International, Fontenay-sous-Bois, France), and formaldehyde 40 % (Sigma-Aldrich, St. Louis, MO, USA). A standard PAH solution with dichloromethane (1 ml) containing the 16 priority pollutants (EPA 610 PAH Mix, Supelco, Bellafonte, PA, USA) was used as the inoculum for batch tests. PAH compounds were extracted from wet sediment samples (10 g) in an ultrasonic bath (Sun et al. 1998) (three 15 min cycles) using dichloromethane (20 ml). The solvent extracted was centrifuged, and the suspended fraction was gradually reduced by rotary evaporation (28 ± 2 °C). The dry residue was recovered with acetonitrile (0.5 ml). In the sorption study, PAH was extracted only from the aqueous phase by liquid-liquid separation using dichloromethane (25 ml).
2.5 PAH Quantification and Recovery Test
PAH quantification was carried out by HPLC (Ultimate 3000, Thermo Scientific, Waltham, MA, USA) with Thermo Scientific fluorescence and diode array detectors. The 16 LMW and HMW PAHs were separated with a gradient programme (1.5 ml min−1) on an analytical reverse-phase column C16 (4.6 × 150 mm, 3 μm, 120 Å). A mixture of acetonitrile and water (Mill-Q system, Millipore, Billerica, MA, USA) was used as the mobile phase. A calibration solution was prepared from the standard PAH solution by serial dilutions (1:50, 1:100 and 1:200). The wet weight of each sediment sample was corrected to dry weight, after determination of percent sediment humidity. Concentrations were expressed on a dry weight basis. The detection limit of the method was 0.04–0.4 ng g−1, estimated as three times the background noise.
The methods employed were validated by the recovery test, which was carried out using certified sediments for PAHs (IAEA/MEL/65 1998; IAEA/MEL/67 1999) and wet marine sediments. Recovery rates were 65 % (LMW PAHs) and 82 % (HMW PAHs) in certified sediment and 59 % (LMW PAHs) and 100 % (HMW PAHs) in wet marine sediment.
2.6 Determination of the Degradation Parameter (t 1/2)
The degradation parameters were calculated by tests carried out on all sediment samples from both areas in laboratory conditions reproducing the environmental conditions of coastal and open-sea sediments. The degradation parameters were determined according to Marini and Frapiccini (2013). A known quantity of standard PAH solution (dil. 1:10) was used to spike the samples (2 ml in 200 g). The total PAH concentration of the inoculum for each sediment sample was 597 μl g−1. After an equilibration time of 24/48 h, PAH degradation rates were studied as described above for 50 days, to detect microbial degradation (Shuttleworth and Cerniglia 1995). A first-order degradation model with two fitting parameters was applied to fit the concentration data. The first-order rate constant (K) was obtained from the equation C = C 0 exp(−kt), where C was the concentration of a target compound at time t and C 0 was the average initial concentration (Hinga 2003; Chen et al. 2014). t 1/2 was calculated using the formula ln2/r = 0.693/r, where r is the mean slope of the degradation curve over the period of observation (Viñas et al. 2009).
2.7 Determination of Sorption Parameters (K d and K oc)
The sorption parameters were established by batch tests carried out to build the equilibrium sorption isotherms using five different initial concentrations of standard PAH solution (EPA 610, 1:10, 1:25, 1:50, 1:100 and 1:200) and dichloromethane as the solvent. Dry sediment (1 g) was used in each batch test with a final solution volume of 10 ml, with 0.4 % formalin to inhibit microbial activity (Means 1995; Marini and Frapiccini 2014). Samples were placed into glass centrifuge tubes, sealed, and then equilibrated in a shaking table (1000 rpm) in the dark at a temperature of 25 ± 0.5 °C. Equilibrium was achieved in about 24 h (Means 1980; Barret et al. 2010; Soares et al. 2013). After equilibration, the solutions were centrifuged at 3000 rpm at a controlled temperature (25 ± 0.5 °C) for 30 min, to separate the solid and aqueous equilibrium phases, then PAH were extracted as described above. Blank samples to calculate the PAH concentrations sorbed to the solid phase were prepared in the same way as the sediment samples, but without sediment. The PAH concentrations sorbed to the solid phase were calculated as the difference between blank and sediment samples (Kohl and Rice 1999).
3 Results and Discussions
3.1 Degradation Parameter (t 1/2)
The data regarding the degradation parameter (t 1/2) studied in the sediments mimicking coastal and open-sea environmental conditions are reported in Fig. 2a, b.

a Half-lives of LMW and HMW PAHs studied in sediment reproducing site A (coastal) and B (open sea) salinity conditions. b Half-lives of LMW and HMW PAHs studied in sediment reproducing site A (coastal) and B (open sea) temperature conditions
The experiments showed that the PAH mixtures degraded rapidly in 1–2 weeks in both conditions, as also reported in the previous study (Marini and Frapiccini 2013). A clear relationship was found between degradation rate and molecular weight, since HMW PAHs showed a longer t 1/2, owing to their lower aqueous solubility and greater incorporation to the organic fraction of sediment, which limited the access of degradation microorganisms (Lei et al. 2005; Achten et al. 2011).
It is well established that an increase in salinity affects the solubility of chemicals in water (Ni and Yalkowsky 2003). This was demonstrated by a slight difference in t 1/2 values between the two sites. The t 1/2 values of PAHs in sediment reproducing coastal salinity conditions were 3.8 and 4.6 days for LMW and HMW PAHs, respectively, whereas those of PAHs analysed in sediment mimicking open-sea salinity were respectively 4.8 and 6.7 days (Fig. 2a). The latter values were therefore slightly higher (by 1 and 2.1 day, respectively, for LMW and HMW PAHs) due to the higher salinity of open-sea water, which reduces non-polar PAH water solubility (Tremblay et al. 2005; Shukla et al. 2007). Oh et al. (2013) found that PAH water solubility decreased as salinity increased from 0 to 30 ppt in coastal sediments, resulting in enhanced PAH sorption. Our findings confirm their data, especially with regard to open-sea sediments, where salinity was higher (30 ppt). Thus, salinity increased the PAH sorption capacity of sediment and reduced PAH degradation rates. As demonstrated in our previous study (Marini and Frapiccini 2013), HMW PAHs were more sensitive to the salinity change (from 21 to <37 ppt) than LMW PAHs, resulting in a degradation rate of 30 % in presence of the higher ionic concentrations.
The t 1/2 values of the PAHs studied in conditions reproducing coastal temperature and sunlight were 6.3 and 13.3 days for LMW and HMW PAHs, respectively, whereas those of the PAHs studied in sediment mimicking open-sea temperature and darkness conditions were 12.2 and 15.6 days, respectively (Fig. 2b). As regards the influence of temperature and solar irradiation at the two sites, t 1/2 values were markedly higher in open sea compared with coastal sediments, due to lower temperature (<11 °C) and darkness. Enell et al. (2005) have demonstrated that temperature reduction affects PAH release and reduces desorption rates. In the present study, the temperature decrease (from 22 to 11 °C) reduced degradation rates; in particular, LMW PAH persistence increased by 48 % in conditions of low temperature and lack of solar irradiation. Temperature thus plays a significant role in controlling PAH bioavailability and toxicity, as also reported by Vieira and Gudhermino (2012) and as described by Marini and Frapiccini (2013) in laboratory conditions.
3.2 Sorption Parameters (K d and K oc)
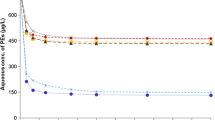
K d values were determined by construction of linear adsorption isotherms, to estimate the sediment/water partitioning of LMW and HMW PAHs in marine sediments with different grain size and TOC. The K d values were averaged on the basis of molecular weight and are reported in Fig. 3. K d values in the sandier coastal sediments, which are poor in OC, were 10.3 and 16.7 for LMW and HMW PAHs, respectively, whereas in the clayey and OC-rich open-sea sediments, they were respectively 9.8 and 31.5. As expected, HMW PAH showed a greater solid-phase fraction than LMW PAHs. The K d difference between site A and site B was particularly marked for HMW PAHs, whereas they were similar for LMW PAHs. This can be attributed to the fact that HMW PAHs, being more tightly bound to sediment particles due to their physicochemical properties, were more influenced in terms of K d by sediment-type variations compared with LMW PAHs. HMW PAHs were therefore associated with the clayey site (57.2 %) with high OC content (3.4 %), i.e. open-sea sediment.

Average K d values based on PAH molecular weight at sites A and B
Calculation of K oc, where PAH partitioning was normalized with f oc, demonstrated that PAH distribution was affected by geographical site as well as sediment grain size. Log K oc values were higher at site A than that at site B for all 16 PAHs. This work, by comparing two very different sites, lends support to the data reported by Neff et al. (2005), who often found higher log K oc values in urban estuaries or harbour sites. Moreover, the log K oc values determined experimentally in our laboratory (Table 2) were comparable to those predicted by Chu and Chan (2000) and to those calculated experimentally in situ by Yu et al. (2009). Our finding that LMW PAH log K oc values correlated linearly with log K ow values at both sites are in line with the data of Karickoff et al. (1979), who showed a strong correlation between log K oc and log K ow. These correlations were significant both for site A (r = 0.936, p < 0.01) and for site B (r = 0.846, p < 0.05). The lack of a correlation between log K oc and log K ow for HMW PAHs—not significant at site A (r = 0.629, p > 0.05) and negative at site B (r = −0.298)—may be explained by their very low aqueous solubility (Yu et al. 2009).
4 Conclusions
The present study examined the three major degradation and sorption parameters of the 16 PAHs included in the EPA priority pollutant list. The three parameters, t 1/2, K d and K oc, were calculated in laboratory conditions mimicking coastal and open-sea sediment under different temperature, salinity and sunlight conditions. The PAHs associated with open-sea sediments exhibited a lower degradation rate, resulting in greater PAH accumulation in sediment particles also as a consequence of their finer grain size and salinity, temperature and sunlight conditions; such conditions reduced the aqueous solubility of PAHs, shifted their partitioning towards the sorbed phase, and limited their biodegradation. Partitioning of the more hydrophobic HMW PAHs involved a greater amount of solid phase and varied according to the grain size and OC content of sediment to a greater extent compared with LMW PAHs. Establishing the values of these parameters allows predicting PAH mobility and the bioavailability of PAHs contained in sediments, providing useful data for environmental risk assessment and management.
References
Achten, C., Cheng, S., Straub, K. L., & Hofmann, T. (2011). The lack of microbial degradation of polycyclic aromatic hydrocarbons from coal-rich soils. Environmental Pollution, 159, 623–629.
Artegiani, A., Bregant, D., Paschini, E., Pinardi, N., Raicich, F., & Russo, A. (1997). The Adriatic Sea general circulation Part I. Air-sea interactions and water mass structure. Journal of Physical Oceanography, 27, 1492–1514.
Bajt, O. (2012). Aliphatic and polycyclic aromatic hydrocarbons in sediments of the Slovenian coastal area (Gulf of Trieste, northern Adriatic). Environmental Monitoring and Assessment, 184, 7439–7452.
Barret, M., Patureau, D., Latrille, E., & Carrère, H. (2010). A three-compartment model for micropollutants sorption in sludge: methodological approach and insights. Water Research, 44, 616–624.
Bihari, N., Fafandel, M., Hamer, B., & Kralj-Bilen, B. (2006). PAH content, toxicity and genotoxicity of coastal marine sediments from the Rovinj area, Northern Adriatic, Croatia. Science of the Total Environment, 366, 602–611.
Botsou, F., & Hatzianestis, I. (2012). Polycyclic aromatic hydrocarbons (PAHs) in marine sediments of the Hellenic coastal zone, eastern Mediterranean: levels, sources and toxicological significance. Journal of Soils and Sediments, 12, 265–277.
Campanelli, A., Fornasiero, P., & Marini, M. (2004). Physical and chemical characterization of water column in the Piceno coastal area (Adriatic Sea). Fresenius Environmental Bulletin, 13, 430–435.
Campanelli, A., Grilli, F., Paschini, E., & Marini, M. (2011). The influence of an exceptional Po River flood on the physical and chemical oceanographic properties of the Adriatic Sea. Dynamics of Atmospheres and Oceans, 52, 284–297.
Castellarin, A., Di Baldassarre, G., & Brath, A. (2011). Floodplain management strategies for flood attenuation in the River Po. River Research and Applications, 27, 1037–1047.
Celis, R., de Jonge, H., de Jonge, L. W., Real, M., Hermosin, M. C., & Cornejo, J. (2006). The role of mineral and organic components in phenanthrene and dibenzofuran sorption by soil. European Journal of Soil Science, 57, 308–319.
Chen, F., Ying, G. G., Ma, Y. B., Chen, Z. F., Lai, H. J., & Peng, F. J. (2014). Field dissipation and risk assessment of typical personal care products TCC, TCS, AHTN and HHCB in biosolid-amended soils. Science of the Total Environment, 470/471, 1078–1086.
Chu, W., & Chan, K. H. (2000). The prediction of partitioning coefficients for causing environmental concern. Science of the Total Environment, 248, 1–10.
Dachs, J., Lohmann, R., Ockenden, W. A., Méjanelle, L., Eisenreich, S. J., & Jones, K. C. (2002). Oceanic biogeochemical controls on global dynamics of persistent organic pollutants. Environmental Science & Technology, 36, 4229–4237.
Elmquist, M., Zencak, Z., & Gustafsson, Ö. (2007). A 700 year sediment record of black carbon and polycyclic aromatic hydrocarbons near the EMEP air monitoring station in Aspvreten, Sweden. Environmental Science & Technology, 41, 6926–6932.
Enell, A., Reichenberg, F., Ewald, G., & Warfvinge, P. (2005). Desorption kinetics studies on PAH-contaminated soil under varying temperatures. Chemosphere, 61, 1529–1538.
Haritash, A. K., & Kaushik, C. P. (2009). Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): a review. Journal of Hazardous Materials, 169, 1–15.
Hinga, K. R. (2003). Degradation rates of low molecular weight PAH correlate with sediment TOC in marine subtidal sediments. Marine Pollution Bulletin, 20, 466–474.
Honda, K., Mizukami, M., Ueda, Y., Hamada, N., & Seike, N. (2007). Residue level of polycyclic aromatic hydrocarbons in Japanese paddy soils from 1959 to 2002. Chemosphere, 68, 1763–1771.
Kannan, K., Johnson-Restrepo, B., Yohn, S. S., Giesy, J. P., & Long, D. T. (2005). Spatial and temporal distribution of polycyclic aromatic hydrocarbons in sediments from Michigan Inland Lakes. Environmental Science & Technology, 39, 4700–4706.
Karickhoff, S. W., Brown, D. S., & Scott, T. A. (1979). Sorption of hydrophobic pollutants on natural sediments. Water Research, 13, 241–248.
Keith, L. H., & Telliard, W. A. (1979). Priority pollutants. I—a perspective view. Environmental Science & Technology, 13, 416–423.
Kohl, S. D., & Rice, J. A. (1999). Contribution of lipids to the nonlinear sorption of polycyclic aromatic hydrocarbons to soil organic matter. Organic Geochemistry, 30, 929–936.
Laflamme, R. E., & Hites, R. A. (1978). The global distribution of polycyclic aromatic hydrocarbons in recent sediments. Geochimica et Cosmochimica Acta, 42, 289–303.
Lei, L., Khodadoust, A. P., Suidan, M. T., & Tabak, H. H. (2005). Biodegradation of sediment-bound PAHs in field-contaminated sediment. Water Research, 39, 349–361.
Lucialli, P., Ugolini, P., & Pollini, E. (2007). Harbour of Ravenna: the contribution of harbour traffic to air quality. Atmospheric Environment, 41, 6421–6431.
Manzetti, S. (2013). Polycyclic aromatic hydrocarbons in the environment: environmental, fate and transformation. Polycyclic Aromatic Compounds, 33, 311–330.
Marini, M., & Frapiccini, E. (2013). Persistence of polycyclic aromatic hydrocarbons in sediments in the deeper area of the northern Adriatic Sea (Mediterranean Sea). Chemosphere, 90, 1839–1846.
Marini, M., & Frapiccini, E. (2014). Do lagoon area sediments act as traps for polycyclic aromatic hydrocarbons? Chemosphere, 111, 80–88.
Marini, M., Jones, B. H., Campanelli, A., Grilli, F., & Lee, C. M. (2008). Seasonal variability and Po River plume influence on biochemical properties along western Adriatic coast. Journal of Geophysical Research, 113, C05S90.
Masiol, M., Formenton, G., Pasqualetto, A., & Pavoni, B. (2013). Seasonal trends and spatial variations of PM10-bounded polycyclic aromatic hydrocarbons in Veneto Region, Northeast Italy. Atmospheric Environment, 79, 811–821.
Means, J. C. (1980). Sorption of polynuclear aromatic hydrocarbons by sediments and soils. Environmental Science & Technology, 14, 1524–1528.
Means, J. C. (1995). Influence of salinity upon sediment-water partitioning of aromatic hydrocarbons. Marine Chemistry, 51, 3–16.
Neff, J.M. (1979). Polycyclic aromatic hydrocarbons in the acquatic environment. Sources, fates and biological effects. Barking, Essex, UK: Applied Science. 262.
Neff, J. M., Stout, S. A., & Gunster, D. G. (2005). Ecological risk assessment of polycyclic aromatic hydrocarbons in sediments: identifying sources and ecological hazard. Integrated Environmental Assessment and Management, 1, 22–33.
Ni, N., & Yalkowsky, S. H. (2003). Prediction of Setschenow constants. International Journal of Pharmaceutics, 254, 167–172.
Oh, S., Wang, Q., Shin, W. S., & Song, D. (2013). Effect of salting out on the desorption-resistance of polycyclic aromatic hydrocarbons (PAHs) in coastal sediment. Chemical Engineering Journal, 225, 84–92.
Parinos, C., Gogou, A., Bouloubassi, I., Pedrosa-Pàmies, R., Hatzianestis, I., Sanchez-Vidal, A., Rousakis, G., Velaoras, D., Krokos, G., & Lykousis, V. (2013). Occurrence, sources and transport pathways of natural and anthropogenic hydrocarbons in deep-sea sediments of the eastern Mediterranean Sea. Biogeosciences, 10, 6069–6089.
Ravaioli, M., Alvisi, F., & Menegazzo Vitturi, L. (2003). Dolomite as a tracer for sediment transport and deposition on the northwestern Adriatic continental shelf (Adriatic Sea, Italy). Continental Shelf Research, 23, 1359–1377.
Sehili, A. M., & Lammel, G. (2007). Global fate and distribution of polycyclic aromatic hydrocarbons emitted from Europe and Russia. Atmospheric Environment, 41, 8301–8315.
Shepard, F. P. (1954). Nomenclature based on sand-silt-clay ratios. Journal of Sedimentary Petrology, 24, 154–158.
Shor, L. M., Kosson, D. S., Rockne, K. J., Jang, L. Y., & Taghon, G. L. (2004). Combined effects of contaminant desorption and toxicity on risk from PAH contaminated sediments. Risk Analysis, 24, 1109–1120.
Shukla, P., Gopalani, M., & Ramteke, D. S. (2007). Influence of salinity on PAH uptake from water soluble fraction of crude oil in Tilapia mossambica. Bulletin of Environment Contamination and Toxicology, 79, 601–605.
Shuttleworth, K. L., & Cerniglia, C. E. (1995). Environmental aspects of PAH biodegradation. Applied Biochemistry and Biotechnology, 54, 291–302.
Soares, A. A., Moldrop, P., Minh, L. N., Vendelboe, A. L., Schjonning, P., & De Jong, L. W. (2013). Sorption of phenanthrene on agricultural soil. Water, Air, & Soil Pollution, 224, 1519.
Stracquadanio, M., Apollo, G., & Trombini, C. (2007). A study of PM(2.5) and PM(2.5)-associated polycyclic aromatic hydrocarbons at an urban site in the Po Valley (Bologna, Italy). Water, Air, and Soil Pollution, 179, 227–237.
Sun, F. S., Littlejohn, D., & Gibson, M. D. (1998). Ultrasonication extraction and solid phase extraction clean-up for determination of US EPA 16 priority pollutant polycyclic aromatic hydrocarbons in soils by reversed-phase liquid chromatography with ultraviolet absorption detection. Analytica Chimica Acta, 364, 1–11.
Tesi, T., Miserocchi, S., Langone, L., Boni, L., & Guerrini, F. (2006). Sources, fate and distribution of organic matter on the western Adriatic continental shelf, Italy. Water, Air, & Soil Pollution: Focus, 6, 229–239.
Tremblay, L., Kohl, S. D., Rice, J. A., & Gagnè, J. (2005). Effects of temperature, salinity, and dissolved humic substances on the sorption of polycyclic aromatic hydrocarbons to estuarine particles. Marine Chemistry, 96, 21–34.
Trevisan, M., Capri, E., Delre, A. A. M., Vischetti, C., Marini, M., Businelli, M., Donnarumma, L., Conte, E., & Imbroglini, G. (1995). Evalution of pesticide leaching models using three italian data-sets. In: Proceedings of the British Crop Protection Symposium—Pesticide Movement to Water. Council Monograph Series, 62, 269–274.
Vieira, L. R., & Guilhermino, L. (2012). Multiple stress effects on marine planktonic organisms: influence of temperature on the toxicity of polycyclic aromatic hydrocarbons to Tetraselmis chuii. Journal of Sea Research, 72, 94–98.
Viñas, L., Franco, M. A., Soriano, J. A., González, J. J., Ortiz, L., Bayona, J. M., & Albaigés, J. (2009). Accumulation trends of petroleum hydrocarbons in commercial shellfish from the Galician coast (NW Spain) affected by the Prestige oil spill. Chemosphere, 75, 534–541.
Wu, W., & Sun, H. (2010). Sorption-desorption hysteresis of phenanthrene—effect of nanopores, solute concentration, and salinity. Chemosphere, 81, 961–967.
Xia, X., & Wang, R. (2008). Effect of sediment particle size on polycyclic aromatic hydrocarbon biodegradation: importance of the sediment-water interface. Environmental Toxicology and Chemistry, 27, 119–125.
Yu, Y., Xu, J., Wang, P., Sun, H., & Dai, S. (2009). Sediment-porewater partition of polycyclic aromatic hydrocarbons (PAHs) from Lanzhou Reach of Yellow River, China. Journal of Hazardous Materials, 165, 494–500.
Yunker, M. B., Macdonald, R. W., Vingarzan, R., Mitchell, R. H., Goyette, D., & Sylvestre, S. (2002). PAHs in the Fraser River basin: a critical appraisal of PAH ratios as indicators of PAH source and composition. Organic Geochemistry, 33, 489–515.
Zhou, J. L., Fileman, T. W., Evans, S., Donkin, P., Readman, J. W., Mantoura, R. F. C., & Rowland, S. (1999). The partition of fluoranthene and pyrene between suspended particles and dissolved phase in the Humber Estuary: a study of the controlling factors. Science of the Total Environment, 243/244, 305–321.
Acknowledgments
This research is supported by the Bandiera RITMARE Project—La Ricerca Italiana per il Mare—coordinated by National Research Council and financed by Italian University and Research Ministry, National Research Program: 2011–2013. We are grateful to Dr. Patrizia Giordano for her assistance in sediment sampling and to Dr. Mattia Betti for his assistance in chemical analysis. The authors acknowledge Word Designs for the language revision (www.silviamodena.com).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Frapiccini, E., Marini, M. Polycyclic Aromatic Hydrocarbon Degradation and Sorption Parameters in Coastal and Open-Sea Sediment. Water Air Soil Pollut 226, 246 (2015). https://doi.org/10.1007/s11270-015-2510-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-015-2510-7