
Abstract
The extensive mass gathering of pilgrims from all over the world, as well as the constant flow of foreign workers via country entry crossings, raises the likelihood of respiratory virus outbreaks spreading and evolving in Saudi Arabia. Here, we report the sequence and phylogenetic analysis of the human parainfluenza type-2 (HPIV-2) in nasopharyngeal aspirates (NPAs) collected from Riyadh, Saudi Arabia, from 2020/21 to 2021/22 seasons. RNA was extracted from the clinical samples and subjected to RT-PCR analysis for the detection of IAV and IBV. The full-length HN gene of HPIV-2 was amplified and sequenced. Multiple sequence alignments (both nucleotides and deduced amino acids) were aligned using Clustal W, MegAlign program of Lasergene software, and MEGA 7.0. HPIV-2 was found in (4; 2% of 200) NPAs. Sequence and phylogenetic analysis results showed that indicated a genotype shifting from G3 to G4a with 83% sequence homology 62-M786 from Japan, which was prominent throughout the winter seasons of 2008/09. Multiple amino acid sequence alignment revealed 25 sites of possible difference between G3 genotypes and G4a. A total of twenty- two of these locations were shared by the other G4a genotypes, whereas three positions, 67 V, 175 S, and 377Q, were exclusively shared by G3. Only eight conserved N-glycosylation sites were found at amino acids 6(NLS), 286(NTT), 335(NIT), 388(NNS), 498(NES), 504(NPT), 517(NTT), and 539(NGT) in four Riyadh isolates. Our findings also revealed that the G4a genotype of HPIV-2 predominated in our samples population during the winter seasons of 2020/21 and 2021/22. Further research with a larger sample size covering numerous regions of Saudi Arabia throughout different epidemic seasons is needed to achieve an improved knowledge of HPIV-2 circulation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human parainfluenza viruses (HPIV) are one of the primary causes of lower respiratory tract infections (LRTI), accounting for 13% of LRTI cases, 4–14% of LRTI hospitalizations, and 4% of LRTI mortality in children under the age of five worldwide among children younger than 5 years [1]. These viruses were first isolated from children with croup in the late 1950s and were known as croup-associated viruses [2, 3].
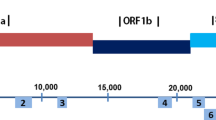
The HPIV virion contains a single-stranded negative-sense RNA genome of 15,462 nucleotides that encode six genes and eight proteins. The two surface glycoproteins that are responsible for infecting host cells are the hemagglutinin-neuraminidase (HN) and fusion (F) proteins [4]. HN is responsible for binding to sialic acid residues on the surface of host epithelial cells and F protein induces fusion of the viral envelope with the host cell membrane [5]. The HN gene has been chosen as the optimal target gene region for HPIV phylogenetic analysis due to its antigenicity and variability [6, 7]. HPIVs belong to the family Paramyxoviridae, of the order Mononegavirales [3]. HPIVs are classified into four serotypes (HPIV-1, 2, 3, and 4) in two genera: Respirovirus (HPIV-1 and HPIV-3) and Rubulavirus (HPIV-2 and HPIV-4) [8].
Of the HPIVs detected, the prevalence of HPIV-2 was ranked third behind HPIV3 and HPIV1, accounting for 2.6 to 14% of HPIV infections [1, 9, 10]. Although the fact that HPIV-3 has been identified as a major viral cause of acute respiratory infections in newborns and young children [11]. Infections with HPIV-1 and HPIV-2 were more prevalent in older children [12]. Furthermore, Villaran et al. discovered that the detection rate of HPIV-3 was four times higher in children under the age of five than in those over the age of five [13].
The epidemiology and economic effects of HPIV infections in Saudi Arabia are mostly unclear. During the winter seasons of 2003 and 2004, among 282 screened samples of NPAs obtained from infants and young children admitted to the Buraidah Maternity and Pediatric Hospital in Al-Qassim, Saudi Arabia, and clinically diagnosed with acute lower respiratory tract infections, the frequency of HPIV-1, -2, and − 3 was 9 (3.2%), 4 (1.4%), and 1 (0.4%), respectively [14]. In Riyadh, we previously show that HPV-3 was detected in 42 cases (8%) of 1429 Saudi infants hospitalized at King Khaled University Hospital with LRTIs between April 1993 and March 1996. We also discovered that HPIV-3 could be detected in all months, with outbreaks occurring from June to August when the air temperature was 40 °C [15]. Only a few surveillance studies have been conducted to detect HPIV-2 in clinical samples collected from hospitalized patients in various districts. We previously demonstrated that HPIV-2 had been observed in Riyadh over the winter-spring season. We previously showed that only one sample (0.56%) was HPIV-2 positive using nested RT-PCR among the 180 nasopharyngeal aspirates (NPAs) collected from possible infections in Riyadh [16]. This study aimed to examine the molecular characteristics of HPIV-2 isolates throughout admitted children in Riyadh during two seasons, 2020/21 and 2021/22.
Materials and methods
Sample collection
A total of 200 NPAs were collected from all children admitted to King Khalid University Hospital (KKUH) in Riyadh, Saudi Arabia (120 between October and January 2020/21 and 80 in 2021/22). After receiving informed consent from their parents, samples were collected from children’s hospitals with acute respiratory illness and respiratory infection (fever, cough, sore throat, wheezing, or apnea), and the study protocol was approved by the institutional review board (IRB) of KKUH, Riyadh (Ethics Reference No. 14/4463/IRB 03).
Virus detection
Viral RNA was extracted from samples using kits from (Qiagen, Hilden, Germany) as directed by the manufacturer. In brief, the 140 µl samples were thawed and lysed in 560 ml of lysis solution containing carrier RNA. To disperse any visible precipitate, one volume of ethanol (96–100%) was added, and QIAamp Mini column binding was performed by centrifuging the lysate at 8000 rounds per minute (rpm) for 30 s. Columns were washed with 500 µl wash buffers with ethanol at 8000 rpm for 15 s. Columns were dried, and Viral RNA was eluted in 60 µl elution buffer by centrifugation at room temperature for 2 min at 8000 rpm.
The presence of HPIV-2 RNA in extracted RNA was evaluated using the One-step Ahead RT-PCR Kit with Taq High Fidelity DNA Polymerase (Qiagen, Hilden, Germany) and the primer pair HPIV2-Cdna-F/HPIV2-R (Table 1). The PCR amplification procedure was followed exactly as specified in the kit’s instruction manual. MM Mix 2.5 × (6 µl), Reaction Mix (25X) (1 µl), RNase-free water (10 µl), forward primer (F) (0.6 M, 1.5 µl), and reverse primer (R) (0.6 M, 1.5 µl) were combined to make the master mix. Individual tubes were filled with template RNA extracts (5 µl). The template RNA extracts (5 µl) were added to the individual tubes. The runs were performed in a 25-µl reaction mixture in a GeneAmp 9700 thermal cycler (Applied Biosystems, Foster City, CA) using the manufacturer’s specified reaction conditions: 30 min of reverse transcription at 50 °C, followed by 35 cycles of 15 min at 95 °C denaturation, 30 s of annealing at 52 °C, and 2 min of extension at 72 °C. The final extension was performed for two minutes at 72 °C.
DNA sequencing
The sequences of the whole positive sample (4 isolates) from each of the two epidemic years (2020/21 and 2021/22) were designated (Riyadh 40/2020, Riyadh 74/2020, Riyadh 25/2021, and Riyadh 37/2021) used for sequencing and phylogenetic analysis of the complete HN gene. Using a GeneAmp 9700 thermal cycler, the same kit (Step Ahead RT-PCR Kit with Taq High Fidelity DNA Polymerase) was utilized to amplify HN antigenic glycoprotein. Three overlapping primer sets were utilized to get the full HN gene, as indicated in Table 1. RT-PCR products were purified from agarose gel with the Illustra GFX PCR DNA Kit (GE Healthcare) and sequenced on both strands with a commercial service (Macrogen Inc, Seoul, South Korea).
Sequence data analysis and phylogeny
The HN gene sequence data was edited and assembled for the selected strains using the Bioedit tool (Ibis Biosciences, Carlsbad, CA). Multiple sequence alignment was used to compare the acquired sequences to the sequences of 39 reference and vaccination strains from GenBank (Table 2).
Divergence analysis, mutation site identification, and amino acid change prediction were performed with the EditSeq and MegAlign programs, Lasergene software 3.18 (DNAStar Inc., Madison, WI). NetNGlyc 1.0 was used to predict probable N-linked glycosylation sites (Asn/X/Ser/Thr, where X is any amino acid other than proline) and O-linked glycosylation sites (Ser or Thr) (http://www.cbs.dtu.dk/services/NetNGlyc) [17] and NetOGlyc 3.1 (http://www.cbs.dtu.dk/services/NetOGlyc), [18], respectively. MEGA 7.0’s neighbor-joining approach [19] was used to create a phylogenetic tree based on a complete nucleotide sequence of the HN gene with bootstrap values of 1,000 replicates.
Statistical analysis
The Fisher’s Exact test was performed to identify significant differences in the prevalence of HPIV2 isolates across gender and age groups. The Z-test with Bonferroni adjustment was used for the posthoc comparison. P < 0.05 was considered significant.
Results
Prevalence of HPIV-2
HPIV-2 was found in (4; 2% of 200) NPAs collected throughout two epidemic seasons (2020/21 and 2021/22). Based on gender, HPIV-2 isolates were more prevalent (P < 0.05) in males (3; 2.83% of 106) than females (1, 1.06% of 94 cases) (P < 0.05). Infants aged 1–2 years were the most (P < 0.05) afflicted age group, accounting for (2; 2.56% of 78 cases as demonstrated in (Table 3).
Amino acid sequence analysis of the HN gene
Analysis of HN Gene amino acid residues, 571 amino acids with a TAA as a stop codon similar to the prototype HPIV-2 strain (NC_003443) with the other genotypes. Specific amino acids were assigned to G3 genotypes, including 67 V, 175 S, and 377Q (Table 4). The four Riyadh isolates shared an amino acid sequence with the 62-M786 strain except for eleven positions: 48 V, 101P, 111I, 118 S, 117T, 164 N,316 K,345Q,351 N, and 360 N.
N- and O-Glycosylation site analysis in amino acid sequences
Analyzing the N-glycosylation sites at the HN gene of four Riyadh isolates revealed only 8 conserved N-glycosylation sites at residues at amino acids 6(NLS), 286(NTT), 335(NIT), 388(NNS), 498(NES), 504(NPT), 517(NTT), and 539(NGT). Conversely, the HN Gene is extensively glycosylated with O-linked carbohydrates at serine and threonine residues. All isolates from this study had a total of 40 to 45 potential O-linked glycosylation sites, as shown in (Fig. 1).
Genetic analysis of HOPV strains
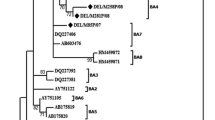
We examined the phylogenetic tree generated by the neighbor-joining method for HPIV-2 positive samples to other reference sequences from GenBank, including local and international strains. Sequence and phylogenetic analyses revealed a genotype shift from G3 to G4a genotype with sequence homology of 83% 62-M786 from Japan, which was prevalent during the winter seasons of 2008/09, as shown in (Fig. 2).

Alignment of deduced amino acid sequences of the HN gene of HPIV-2 isolates. The amino acid positions matched to amino acid positions in the prototype HPIV-2 strain (NC_003443) sequences. Various amino acids are assigned a color, and similar residues are shown by dots. Maroon rectangles surround predicted N-glycosylation sites. The blue rectangle represents amino acids assigned to G3 genotypes. Small filled circles relate to O-glycosylation sites predicted

HPIV-2 phylogenetic trees. For alignment, the HN gene was employed. The phylogenetic tree was generated using the MEGA7 program and the neighbor-joining method. The numbers at the tree’s internal nodes are the bootstrap values from 1,000 replicates. Only values greater than 60% are displayed. Isolates from seasons 2020/21 and 2021/22 are in red font, whereas isolates from seasons 2008/09 are in blue font
Discussion
HPIVs primarily impact children under the age of five, with the pathogenic range including upper and lower respiratory tract infections. They cause 30-40% of all acute respiratory tract infections in infants and children under the age of five, causing a variety of clinical symptoms such as colds, croup, bronchiolitis, and pneumonia [3]. HPIV-2 is responsible for 60% of all infections in children under the age of two and immunocompromised patients, and so cannot be ignored. However, features of this virus’s epidemiology and evolutionary dynamics are currently being challenged. As a result, the purpose of this study was to examine the seasonality, circulation pattern, genetic diversity, and evolutionary dynamics of HPIV-2 in Riyadh, Saudi Arabia over an extended period (2020/21 and 2021/22).
Only four (4; 2% of 200) clinical samples collected throughout the two epidemic seasons 2020/21 and 2021/22 were positive for HPIV-2. HPIV infections are identified at a low incidence throughout the year [20]. HPIV-1, -2, and − 3 were found in 9 (3.2%), 4 (1.4%), and 1 (0.4%) of the 282 NPAs screened from newborns and young children admitted to the Buraidah Maternity and Pediatric Hospital in Al-Qassim, Saudi Arabia, during the winter seasons of 2003 and 2004 [14]. The circulation pattern of HPIVs in Saudi patients treated at King Khaled University Hospital in Riyadh was studied in our laboratory. RT-PCR was used to screen for HPIV-2 in NPAs collected from hospitalized children during two consecutive seasons (2007/08 and 2008/09). Only one (0.56%) of the 180 samples tested positive for HPIV-2 [16]. During an early investigation in Beijing, China, from 2004 to 2012, HPIVs were found in 1675 (6.50%, 1675/25,773) individuals, including 261 (1.01%, 261/25,773) for HPIV-1, 28 (0.11%, 28/25,773) for HPIV-2, and 1388 (5.39%, 1388/25,773) for HPIV-3. [11]. In a recent study, 21 HPIV-2 HN sequences were acquired from LRTI pediatric cases in seven Chinese provinces (Henan, Jilin, Gansu, Chongqing, Shandong, Anhui, and Hebei) in children aged 1 to 12 years between 2011 and 2017 to 2021. [21].
In addition, we sequenced the complete HN gene to investigate the genetic characteristics and phylogeny of Riyadh HPIV-2 strains. Riyadh HPIV-2 isolates were belonging to the G4a genotype with sequence homology of 83% 62-M786 from Japan. Our earlier work of Phylogenetic analysis of HPIV-2 was classified in cluster G3 related to a strain reported in the US state of Oklahoma during the winter seasons of 2008/09 [16], as shown in (Fig. 2). Despite this, there are little studies on the virus’s epidemiology and evolutionary dynamics. Phylogenetic analysis indicated two groups within HPIV2 isolates, but due to the small number of isolates, the phylogenetic tree was insufficiently informative [6]. Later, a phylogenetic tree was created using more diverse international HPIV-2 isolates, which revealed the existence of four clusters (G1-4) [16]. A previous investigation of HPIV-2 genetic diversity in Croatia between 2011 and 2014 predicted that the G1a genotype will supplant the G3 genotype in this period [22]. Although the G1a genotype is currently dominant, the G3 genotype can still be identified irregularly among the isolates. [23].
For instance, an examination of the deduced amino acid sequence alignment revealed 25 possible locations of difference between G3 genotypes and G4a (Table 3). Twenty-two of these positions were shared by the other G4a genotypes, whereas three positions, 67 V, 175 S, and 377Q, were solely shared by G3. In comparison to the 62-M786 strain, the four Riyadh isolates shared an amino acid sequence with eleven points of potential variation: 48 V, 101P, 111I, 118 S, 117T, 164 N,316 K,345Q,351 N, and 360 N. The effect of these changes on the antigenic characteristics of the mature HN protein, however, could not be established because the positions of the antigenic sites for HPIV-2 have not been determined yet. The two glycoproteins HN and F proteins are the most common targets of neutralizing antibodies [23]. These two proteins were thoroughly examined. HN glycoprotein is organized as a tetramer and trimer on the surface of the viral envelope with the host cell membrane [5].
Glycosylation is an essential protein modification process that involves the enzymatic process of adding a carbohydrate chain to a protein, resulting in a posttranslational modification that modifies protein folding, antigenicity, and biological activity [24, 25]. A wide range of viruses rely on O-linked and N-linked glycosylation to carry out essential biological activities. Viruses frequently contain integral proteins that contribute to host-cell interactions such as adhesion to receptors and membrane fusion. Fusion proteins from a variety of viruses, including HPIVs, and influenza viruses, share several common characteristics that are increased by glycosylation. Each of these viruses has several glycosylation sites that must be processed and modified by the host post-translational machinery to be fusogenically active. Glycosylation is involved in the biogenesis, stability, antigenicity, and infectivity of most viruses [26]. The N-glycosylation sites at the HN gene of four Riyadh isolates revealed only eight conserved N-glycosylation sites at amino acids 6 (NLS), 286 (NTT), 335 (NIT), 388 (NNS), 498 (NES), 504 (NPT), 517 (NTT), and 539 (NGT). These findings are consistent with a recent work that found four N-glycosylation sites (N6, N115, N142, and N272) to be substantially conserved in all HN sequences of Chinese HPIV2 strains [21].
This study was limited by a small sample size, geographical and chronological distribution. Additional thorough studies with larger sample sizes encompassing various regions of Saudi Arabia throughout different epidemic seasons are needed to gain an improved knowledge of HPIV-2 circulation.
Conclusion
In conclusion, we present data on the genetic diversity of HPIV-2 in the HN protein gene in Riyadh throughout the winter seasons of 2020/21 and 2021/22. In addition, HPIV-2 was identified only in (4; 2% of 200) clinical samples. A sequencing and phylogenetic investigation of the HPIV-2 full-length HN gene indicated a genotype shifting from G3 to G4a with 83% sequence homology 62-M786 from Japan, which was prominent throughout the winter seasons of 2008/09. Multiple amino acid sequence alignment revealed 25 sites of possible difference between G3 genotypes and G4a. A total of twenty- two of these locations were shared by the other G4a genotypes, whereas three positions, 67 V, 175 S, and 377Q, were exclusively shared by G3. Only eight conserved N-glycosylation sites were found at amino acids 6(NLS), 286(NTT), 335(NIT), 388(NNS), 498(NES), 504(NPT), 517(NTT), and 539(NGT) in four Riyadh isolates. The variation in the glycosylation profile, as well as the into genotypic and intergenotypic changes across HPIV-2 isolates, may increase the virus’s ability to avoid the host immune response. Our findings also revealed that the G4a genotype of HPIV-2 predominated in Riyadh during the winter seasons of 2020/21 and 2021/22. Further research with a bigger sample size covering numerous regions of Saudi Arabia throughout different epidemic seasons is needed to achieve an improved knowledge of HPIV-2 circulation.
Data Availability
All the data is provided.
References
Wang X, Li Y, Deloria-Knoll M, Madhi SA, Cohen C, Arguelles VL, Basnet S, Bassat Q, Brooks WA, Echavarria M (2021) Global burden of acute lower Respiratory Infection associated with human parainfluenza virus in children younger than 5 years for 2018: a systematic review and meta-analysis. The Lancet Global Health 9:e1077–e1087
Henrickson KJ (2003) Parainfluenza viruses. Clin Microbiol Rev 16:242–264
Branche AR, Falsey AR (2016) Parainfluenza virus Infection. Seminars in respiratory and critical care medicine. Thieme Medical Publishers, pp 538–554
Phan MV, Arron G, GeurtsvanKessel CH, Huisman RC, Molenkamp R, Koopmans MP (2019) Cotten M.Complete genome characterization of eight human parainfluenza viruses from the Netherlands Microbiol Resource Announcements. (8): e00125–e00119
Eveno T, Dirr L, El-Deeb IM, Guillon P, von Itzstein MT (2019) Human Parainfluenza Virus Type-1 Haemagglutinin-Neuraminidase with Mechanism-Based Inhibitors Viruses. (11)
Terrier O, Cartet G, Ferraris O, Morfin F, Thouvenot D, Hong S, Lina B (2008) Characterization of naturally occurring parainfluenza virus type 2 (hPIV-2) variants J Clin Virol. (43): 86–92
Mao N, Ji Y, Xie Z, Wang H, Wang H, An J, Zhang X, Zhang Y, Zhu Z (2012) Cui A.Human parainfluenza virus-associated respiratory tract infection among children and genetic analysis of HPIV-3 strains in Beijing, China
Parija S, Marrie T (2019) Human parainfluenza viruses (HPIV) and other parainfluenza viruses. Medscape. In
Liu W-K, Liu Q, Chen D-H, Liang H-X, Chen X-K, Huang W-B, Qin S, Yang Z-F, Zhou R (2013) Epidemiology and clinical presentation of the four human parainfluenza virus types BMC infectious diseases. (13): 1–8
Ng X, Zj D, Zp X, Ll Z, Sz Z, Huang H, Gao H (2016) Zhang B.Human Parainfluenza virus types 1–4 in hospitalized children with acute lower Respiratory Infections in China J Med Virol. (88): 2085–2091
Wang F, Zhao L-Q, Zhu R-N, Deng J, Sun Y, Ding Y-X, Tian R, Qian Y (2015) Parainfluenza virus types 1, 2, and 3 in pediatric patients with acute respiratory infections in Beijing during 2004 to 2012 Chinese medical journal. (128): 2726–2730
Soudani S, Mafi A, Al Mayahi Z, Al Balushi S, Dbaibo G, Al Awaidy S, Amiche AA Systematic Review of Influenza Epidemiology and Surveillance in the Eastern Mediterranean and North African Region (2022) Infectious Diseases and Therapy. (11): 15–52
Villaran MV, García J, Gomez J, Arango AE, Gonzales M, Chicaiza W, Alemán W, Lorenzana de Rivera I, Sanchez F Aguayo N.Human Parainfluenza virus in patients with influenza-like Illness from C entral and S outh a merica during 2006–2010 (2014) Influenza and other respiratory viruses. (8): 217–227
Meqdam MM, Subaih SH, Thwiny IR (2005) Rapid detection and clinical features of Influenza and parainfluenza in infants and young children hospitalized with acute lower respiratory illnesses J Trop Pediatr. (51): 160–165
Almajhdi FN (2015) Hemagglutinin-neuraminidase gene sequence-based reclassification of human parainfluenza virus 3 variants Intervirology. (58): 35–40
Almajhdi FN, Alshaman MS, Amer HM (2012) Human parainfluenza virus type 2 hemagglutinin-neuramindase gene: sequence and phylogenetic analysis of the Saudi strain Riyadh 105/2009 Virology journal. (9): 1–5
Gupta R, Jung E, Brunak S (2004) Prediction of N-glycosylation sites in human proteins
Julenius K, Mølgaard A, Gupta R, Brunak S (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites Glycobiology. (15): 153–164
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets Molecular biology and evolution. (33): 1870–1874
Farrag MA, Hamed ME, Amer HM, Almajhdi FN (2019) Epidemiology of respiratory viruses in Saudi Arabia: toward a complete picture Arch Virol. (164): 1981–1996
Feng Y, Zhu Z, Xu J, Sun L, Zhang H, Xu H, Zhang F, Wang W, Han G, Jiang J (2023) Molecular Evolution of Human Parainfluenza Virus Type 2 Based on Hemagglutinin-Neuraminidase Gene Microbiology Spectrum. e04537-04522
Šantak M, Slović A, Ljubin-Sternak S, Mlinarić Galinović G (2016) Forčić D.Genetic diversity among human parainfluenza virus type 2 isolated in Croatia between 2011 and 2014 J Med Virol. (88): 1733–1741
Šantak M, Balija ML, Galinović GM, Sternak SL, Vilibić-Čavlek T, Tabain I (2018) Genotype replacement of the human parainfluenza virus type 2 in Croatia between 2011 and 2017–the role of neutralising antibodies Epidemiol Infect. (146): 1372–1383
Cipollo JF, Parsons LM Glycomics and glycoproteomics of viruses: Mass spectrometry applications and insights toward structure–function relationships (2020) Mass spectrometry reviews. (39): 371–409
Shao N, Liu B, Xiao Y, Wang X, Ren L, Dong J, Sun L, Zhu Y, Zhang T, Yang F (2021) Genetic characteristics of human parainfluenza virus types 1–4 from patients with clinical respiratory tract Infection in China Front Microbiol. (12): 679246
Feng T, Zhang J, Chen Z, Pan W, Chen Z, Yan Y, Dai J (2022) Glycosylation of viral proteins: implication in virus–host interaction and virulence Virulence. (13): 670–683
Acknowledgements
The authors thank the Researchers Supporting Project number (RSPD2023R801), King Saud University, Riyadh, Saudi Arabia.
Funding
The authors thank the Researchers Supporting Project number (RSPD2023R801), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization A.N.A. and I.M.A.; methodology, A.N.A.; software, N.A.A. and I.M.A.; validation, A.N.A, N.A.A., I.M.A., and F.N.A.; formal analysis, A.F.A.; investigation, F.N.A.; resources, F.N.A.; data curation, A.N.A and I.M.A.; writing original draft preparation, A.N.A and I.M.A.; writing review and editing, F.N.A.; review and editing, visualization, I.M.A.; supervision, F.N.A; project administration, F.N.A.; funding acquisition, N.A.A. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by Research Ethics Committee (RES) at the King Saud University, Riyadh, Saudi Arabia (Ethics Reference No. 21/4463/IRB 03).
Informed consent
Informed consent was obtained from all subjects involved in the study.
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by Seung-Kook Choi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alsaleh, A.N., Aziz, I.M., Alkubaisi, N.A. et al. Genetic analysis of human parainfluenza type 2 virus in Riyadh, Saudi Arabia. Virus Genes 60, 1–8 (2024). https://doi.org/10.1007/s11262-023-02035-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-023-02035-6