
Abstract
The virus that causes Marek’s disease (MD) is globally ubiquitous in chickens, continuously evolving, and poses a significant threat to the poultry industry. Although vaccines are extensively used, MD still occurs frequently and the virus has evolved increased virulence in China. Here, we report an outbreak of MD in vaccinated chickens and unvaccinated turkeys in a backyard farm in Guangdong province, China, in 2018. Phylogenetic analysis revealed two lineages of MDVs at this farm, with one lineage, containing isolates from two turkeys and five chickens, clustering with virulent Chinese strains and displays a relatively high genetic divergence from the vaccine strains. These new isolates appear to have broken through vaccine immunity, yielding this outbreak of MD in chickens and turkeys. The second lineage included four chicken isolates that clustered with the CVI988 and 814 vaccine strains. The large diversity of MDVs in this single outbreak reveals a complex circulation of MDVs in China. Poor breeding conditions and the weak application of disease prevention and control measures make backyard farms a hotbed for the evolution of viruses that cause infectious diseases. This is especially important in MDV as the MD vaccines do not provide sterilizing immunity, which allows the replication and shedding of virulent field viruses by vaccinated individuals and supporting the continuous evolution of MDVs. Hence, constant monitoring of the evolution of MDVs is necessary to understand the evolution of these field viruses and potential expansions of their host range.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Marek’s disease (MD) is highly contagious in chickens and is characterized by immunosuppression and neurological disorders, with eventual tumor formation involving the peripheral nerves, visceral organs, eye, muscle or skin [1]. The causative agent of MD is the Marek’s disease virus (MDV). MDV is a member of the genus Mardivirus and belongs to the Alphaherpesvirinae subfamily of the family Herpesviridae.
MD is widespread in chickens [6]. In China, MD vaccines are used extensively, however, they have limited effect on viral infection and transmission [7]. The vaccines protect against tumors but do not provide sterilizing immunity, thus, vaccinated chickens still support the replication and shedding of virulent field viruses. The MD vaccine program frequently fails, and occasionally virulent MDVs are isolated from vaccinated chickens in China [8], which has two major consequences. First, virulent virus shed by the vaccinated chickens is still pathogenic to the non-vaccinated [9]. Second, with the widespread use of MD vaccines, MDV strains are continuously evolving leading to the generation of strains with greater virulence [10].
MDV encodes more than 100 genes, including MDV EcoRIQ (meq), phosphorylated protein 38 kDa (pp38), virus-encoded interleukin 8(vIL8), glycoprotein B (gB), gE, and gI, with meq and pp38 being of utmost importance for the pathogenicity and tumorigenicity of MDV [11]. pp38 may be involved in immune modulation against MD and the absence of the pp38 gene can reduce lymphoproliferative lesions [12]. The meq gene is involved in the transformation of lymphocytes, and the absence of the meq gene prevents the formation of lymphomas in chickens after MDV infection [13].
Here, we report an outbreak of MD in chickens and turkeys in a backyard farm in Guangdong province, China, in 2018. Whole genome sequences of 11 MDVs were generated and analyzed to examine the evolution of MDVs in China. The aim of this study was to understand the transmission and evolution of Marek's disease virus (MDV) in poultry. Our results emphasize the importance of continuous monitoring of MDV evolution and provide a basis for the control of MD in poultry.
Materials and methods
Samples and virus detection
Vaccinated chickens and unvaccinated turkeys with severe MD-like symptoms, including paralysis of the feet and emaciation, were collected from a backyard farm in Guangdong Province, China, in April 2018. The clinical symptoms in this flock began at about day 90 after hatching. Livers and feathers from the sick birds were collected and kept at -80℃. DNA from the livers and feathers was extracted using the Universal Genomic DNA Extraction Kit Ver.3.0 (TaKaRa Biotechnology Dalian Co., Ltd.) according to the manufacturer’s instructions. PCR was performed with primer sets for MDV viral meq (F 5′-ATGTCTCAGGAGCCAGAGCC-3′; R 5′-TCAGGGTCTCCCGTCACCTG-3′) [14] and pp38 (F 5′-TTAATTTGATTCAGATTTTG-3′; R 5′-ATGGAATTCGAAGCAGAACA-3′) genes. PCR products for meq and pp38 of the anticipated sizes were isolated and purified from agarose gels using the E.Z.N.A.® Gel Extraction Kit (Omega Bio-tek, Inc., Guangzhou, China).
PCR products were cloned into the pJET1.2 vector using the conditions defined by the manufacturer (Wuhan Miaoling Bioscience & Technology Co., Ltd, Wuhan, China). All ligation products were transformed into E. coli DH5a cells and were selected on LB agar plates containing 100 mg/ml of ampicillin. Colonies were screened and those with inserts of the appropriate size for the meq or pp38 PCR products were used for DNA sequencing.
Genome sequencing
MDVs from nine chickens (designed MDV-C1, MDV-C2, MDV-C3, MDV-C4, MDV-C5, MDV-C6, MDV-C7, MDV-C8, and MDV-C9) and from two turkeys (designed MDV-T6 and MDV-T7) were selected for genome sequencing. Sequencing libraries were prepared with the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs), and sequencing was performed on the Illumina NovaSeq platform with 6000 and 150 bp paired-end reads generated.
Raw reads were cleaned to exclude adaptor and low-quality sequences using fastp (v.0.19.7) [15]. Clean reads that mapped to the host genome sequence (NCBI reference genomes: chicken, GCA_000002315.5; turkey, GCA_000146605.4) with BWA-MEM (v.0.7.17) were filtered out [16]. For each sample, the viral genome was de novo assembled using Spades (v.3.13.1) with the parameter “-k 21,33,55,77 –careful”[17]. The assembled contigs were aligned against the reference MDV genome (strain name: Md5 NCBI. Accession no. NC_002229.3) using the nucmer program from the MUMmer package [18] to generate contiguous viral genomes. Newly assembled viral genomes were deposited into GenBank (Accession numbers: OP887017- OP887027).
Phylogenetic analysis
In addition to the genomes sequenced in this study, genomes of other MDV strains were obtained from GenBank (Table S1). MAFFT (v 7.0) was used to obtain a multiple sequence alignment with the iterative refinement method (FFT-NS-i) [19]. For phylogenetic analysis, alignment gaps associated with incomplete genomic data were removed using Trimal (v1.4) [20]. IQ-TREE (v1.6.12) [21] was used to reconstruct maximum likelihood (ML) trees for the MDV genome sequences, meq gene sequences, and pp38 gene sequences. The parameters for IQ-TREE were iqtree-s input. fasta– m MFP-bb 1000.
Recombination analysis
RDP4 was used to detect potential recombination events between the sequences using seven different methods: RDP, GENCONV, BOOTSCAN, MAXCHI, CHIMERA, SISCAN, and 3SEQ [22]. Only recombination events that had significant signals from at least three different methods were considered to be potential recombinations. To further analyze the possibility of recombination, the genomes were analyzed with Simplot software (version 3.5.1) [23]. Parameters for the similarity plots are: window, 200 bp; step, 50 bp.
Results and discussion
Upon dissection, sick chickens and turkeys with symptoms of MD were found to possess multifocal nodules on their livers, with many more multifocal nodules observed in the chickens compared to the turkeys (Fig. 1). PCR amplification and sequencing of the target genes meq and pp38 confirmed that the sick turkeys and chickens were infected with MDVs. The occurrence of MDV-induced tumors in turkeys is unusual. In this case, the turkeys had been raised with chickens in a backyard farm, thus, they were likely in contact with a high density of MDV from the chickens, leading to infection and a more serious disease in these turkeys. In our study, the turkeys infected by MDV possessed tumors with fewer multifocal nodules on their livers compared to their companion chickens (Fig. 1B). Turkey herpesvirus (HVT) is reported to be ubiquitous in domestic turkeys, and is used as a vaccine against MD in chickens, thus it might also protect turkeys against natural infection by MDV [24, 25]. However, due to recent evolutionary changes in MDV, HVT no longer fully protects chickens against tumors and mortality from MDV. These evolutionary changes may also be a factor in the infection of turkeys by MDV at this farm. In addition to chickens and turkeys, MDV has been reported in other species of wild birds [26,27,28,29,30]. The expansion of the host range of MDVs requires additional attention.

Pathological lesions of MD seen in the livers of sick birds. A chickens. B turkeys. Representative livers of sick birds display multifocal nodules, with many more multifocal nodules of varying size observed in the chicken compared to the turkeys
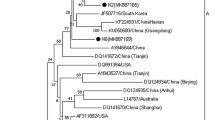
To further explore the evolution of these MDVs, MDV isolates from nine chickens and two turkeys were selected for genome sequencing. Homology analysis showed the nucleotide identity of the genome, meq and pp38 genes were 97.14–99.94%, 99.02–100%, and 98.67–100%, respectively, among the nine chicken isolates, 99.90%, 100%, and 100%, respectively, between the two turkey isolates, and 97.14–99.94%, 99.71–100%, and 98.67–100%, respectively, between the chicken and turkey isolates. meq and pp38 genes have very important functions for the pathogenicity and tumorigenicity of MDV [12, 13]. They may under more strict selection pressure than other parts of the genome. Therefore, these two genes and the full-genome were selected for further phylogenetic analyses. Phylogenetic relationships based on whole genome sequence and the meq gene showed similar topologies, where the MDVs were separated into two clusters (Fig. 2A, B). Five chicken isolates and two turkey isolates belonged to clade I and the four remaining chicken isolates belonged to clade II. The clade 2 sequences included MDV-C1 and MDV-C6, which clustered with the CVI988 vaccine sequence, and MDV-C4 and MDV-C5, which clustered with the 814 vaccine sequence. The meq gene is associated with the evolution of MDV virulence and considered to be an MDV oncogene that plays a role not only in tumor formation but also in its immunosuppressive effects [31, 32]. In this study, the nucleotide sequences for the meq gene identified in the clade 1 genome sequences (MDV-T6, MDV-T7, MDV-C2, MDV-C3, MDV-C7, and MDV-C8) had relative low nucleotide identities (99.02–99.12%, Table 1) when compared with the sequences from the genomes of the two commercial vaccine strains (CVI988 and 814). In addition, the meq gene sequences from these isolates encode amino acid substitutions K77E, D80Y, V115A, T139A, P176R, and P217A that are characteristic of MDV strains isolated from China, and isolates with these substitutions have been found to yield higher morbidity [33]. It is also worth noting that these amino acid substitutions, with the exception of K77E and V115A, were exclusive to these isolates and are not present in the vaccine sequences. Point mutations can influence the transcriptional activity of the meq protein, thereby affecting pathogenicity [34]. Notably, substitutions P176R and P217A occur at the second-position in the four proline repeat (PPPP) sequences found in the proline-rich central region of the protein, and may be associated with the increase in virulence of these strains [4, 35]. The functional consequences of other substitutions need further study. The virulence and genetic characteristics of MDV strains have been changing with the introduction of vaccines [36, 37]. In the past few years several highly virulent MDV strains have been isolated from vaccinated chickens in China [38]. Like them, the virulent strains described here appear to have broken through vaccine immunity, leading to an outbreak of MD in chickens and turkeys in this farm.

Phylogenetic analysis of MDVs estimated using maximum likelihood (ML). A whole genome, B meq gene, C pp38 gene. Phylogenetic trees were estimated with IQ-TREE (Minh et al. 2020) using the best fit substitution model and 1000 bootstrap replicates. Numbers (> 70) above branches are percentage bootstrap values for the major nodes. Scale bars depict the number of amino acid substitutions per site. Strains isolated from different hosts are in different colors, Red = chicken, Blue = turkey. Vaccine strains are marked in green
The phylogenetic tree for the pp38 gene showed a different topology compared to those for the genome sequence or the meq gene (Fig. 2C). Sequences for the MDV-C4, C5, C9, and MDV-T7 pp38 gene clustered with the 814 vaccine sequence and some Chinese virulent strain sequences such as CC/1409, JL/1404, LTS, J-1, GX0101, and LMS, but did not cluster with MDV-T6, MDV-C2, MDV-C3, MDV-C7, MDV-C1, MDV-C6, and MDV-C8. Since pp38 and meq sequences display different phylogenetic patterns, this suggests that recombination occurred within the MDV genomes. RDP4 software [22] detected evidence for 21 recombination events (Table S2) in these genomes, with one of the potential recombination events located in the region surrounding the pp38 gene. According to these results, the strain MDV-C8 was further used as the query to against strains MDV-C9, AF147806, and EU499381 in the SimPlot program [23]. The results indicated potential multiple recombination events in the region surrounding the pp38 gene (Fig. 3). These recombination events explained the different topologies between the pp38 and meq genes. Although MDVs are DNA viruses, and have relatively lower mutation rates compared with RNA viruses, recombination can facilitate their evolution and allowing them to adapt to new hosts [39]. Research has demonstrated that recombination increases the complexity of disease diagnosis, prevention, and control [40].

Identification of potential recombination events in the MDV genomes. Similarity plot of the pp38 gene of MDV-C8 against sequences from MDV-C9, AF147806, and EU499381. Parameters for the similarity plots are: window, 200 bp; step, 50 bp
Marek's disease is globally ubiquitous in chickens, necessitating a strategy of comprehensive vaccination. In China, chickens are inoculated after hatching with commercial vaccines, such as CVI988, HVT, and 814 [10, 41, 42]. Although MD vaccines have proven to be very successful in protecting chickens from tumor development and mortality, they do not provide sterilizing immunity. Some reports have shown that MDV field viruses have continuously evolved toward greater virulence and resistance to immune responses [43], leading to vaccinated chickens retaining the ability to support the replication and shedding of virulent field viruses [10]. In this study, our 11 new MDV genome sequences from chickens and turkeys show that these isolates had a deep divergence from the vaccine strains, which suggests rapid evolution of MDVs in China. Rapid evolution and large divergence may explain the outbreak of MD in these birds. Generally, HVT is ubiquitous in domestic turkeys, and protects turkeys from infection by MDVs [44]. The turkeys and chickens examined here were raised in the same backyard farm and it appears that HVT failed to provide protection to the turkeys from these newly evolved MDV strains, hence, constant monitoring of the evolution of MDV is necessary to control future outbreaks of MD in both chickens and turkeys.
Data availability
The sequences generated in the current study were deposited to the GenBank under the accession numbers OP887017-OP887027.
Abbreviations
- MD:
-
Marek’s disease
- MDV:
-
Marek’s disease virus
- HVT:
-
Turkey herpesvirus
References
Calnek BW (2001) Pathogenesis of Marek’s disease virus infection. Curr Top Microbiol Immunol 255:25–55. https://doi.org/10.1007/978-3-642-56863-3_2
Davison AJ, Eberle R, Ehlers B et al (2009) The order Herpesvirales. Arch Virol 154:171–177. https://doi.org/10.1007/s00705-008-0278-4
Witter RL (1983) Characteristics of Marek’s disease viruses isolated from vaccinated commercial chicken flocks: association of viral pathotype with lymphoma frequency. Avian Dis 27:113–132
Shamblin CE, Greene N, Arumugaswami V et al (2004) Comparative analysis of Marek’s disease virus (MDV) glycoprotein-, lytic antigen pp38- and transformation antigen Meq-encoding genes: association of meq mutations with MDVs of high virulence. Vet Microbiol 102:147–167. https://doi.org/10.1016/j.vetmic.2004.06.007
Liao Y, Lupiani B, Ai-Mahmood M et al (2021) Marek’s disease virus US3 protein kinase phosphorylates chicken HDAC 1 and 2 and regulates viral replication and pathogenesis. PLoS Pathog 17:e1009307. https://doi.org/10.1371/journal.ppat.1009307
Osterrieder N, Kamil JP, Schumacher D et al (2006) Marek’s disease virus: from miasma to model. Nat Rev Microbiol 4:283–294. https://doi.org/10.1038/nrmicro1382
Nair V (2018) Spotlight on avian pathology: Marek’s disease. Avian Pathol 47:440–442. https://doi.org/10.1080/03079457.2018.1484073
Zhuang X, Zou H, Shi H et al (2015) Outbreak of Marek’s disease in a vaccinated broiler breeding flock during its peak egg-laying period in China. BMC Vet Res 11:157. https://doi.org/10.1186/s12917-015-0493-7
Baigent SJ, Smith LP, Nair VK et al (2006) Vaccinal control of Marek’s disease: current challenges, and future strategies to maximize protection. Vet Immunol Immunopathol 112:78–86. https://doi.org/10.1016/j.vetimm.2006.03.014
Reddy SM, Izumiya Y, Lupiani B (2017) Marek’s disease vaccines: current status, and strategies for improvement and development of vector vaccines. Vet Microbiol 206:113–120. https://doi.org/10.1016/j.vetmic.2016.11.024
Sun A, Zhao X, Zhu X et al (2022) Fully attenuated meq and pp38 double gene deletion mutant virus confers superior immunological protection against highly virulent Marek’s disease virus infection. Microbiol Spectr 10:e0287122. https://doi.org/10.1128/spectrum.02871-22
Gimeno IM, Witter RL, Hunt HD et al (2005) The pp38 gene of Marek’s disease virus (MDV) is necessary for cytolytic infection of B cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of MDV. J Virol 79:4545–4549. https://doi.org/10.1128/jvi.79.7.4545-4549.2005
Lupiani B, Lee LF, Cui X et al (2004) Marek’s disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc Natl Acad Sci USA 101:11815–11820. https://doi.org/10.1073/pnas.0404508101
Shi M, Li M, Wang P et al (2021) An outbreak in three-yellow chickens with clinical tumors of high mortality caused by the coinfection of reticuloendotheliosis virus and Marek’s disease virus: a speculated reticuloendotheliosis virus contamination plays an important role in the case. Poult Sci 100:19–25. https://doi.org/10.1016/j.psj.2020.09.034
Chen S, Zhou Y, Chen Y et al (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. https://doi.org/10.1093/bioinformatics/bty560
Li H, Durbin R (2010) Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26:589–595. https://doi.org/10.1093/bioinformatics/btp698
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Kurtz S, Phillippy A, Delcher AL et al (2004) Versatile and open software for comparing large genomes. Genome Biol 5:R12. https://doi.org/10.1186/gb-2004-5-2-r12
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. https://doi.org/10.1093/bioinformatics/btp348
Nguyen LT, Schmidt HA, von Haeseler A et al (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Martin DP, Murrell B, Golden M et al (2015) RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. https://doi.org/10.1093/ve/vev003
Lole KS, Bollinger RC, Paranjape RS et al (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. https://doi.org/10.1128/jvi.73.1.152-160.1999
Davidson I, Malkinson M, Weisman Y (2002) Marek’s disease in turkeys. I. A seven-year survey of commercial flocks and experimental infection using two field isolates. Avian Dis 46:314–321. https://doi.org/10.1637/0005-2086(2002)046[0314:Msditi]2.0.Co;2
Mescolini G, Lupini C, Davidson I et al (2020) Molecular characterization of a Marek’s disease virus strain detected in tumour-bearing turkeys. Avian Pathol 49:202–207. https://doi.org/10.1080/03079457.2019.1691715
Kenzy SG, Cho BR (1969) Transmission of classical Marek’s disease by affected and carrier birds. Avian Dis 13:211–214
Haesendonck R, Garmyn A, Dorrestein GM et al (2015) Marek’s disease virus associated ocular lymphoma in Roulroul partridges (Rollulus rouloul). Avian Pathol 44:347–351. https://doi.org/10.1080/03079457.2015.1056088
Murata S, Chang KS, Yamamoto Y et al (2007) Detection of the virulent Marek’s disease virus genome from feather tips of wild geese in Japan and the Far East region of Russia. Arch Virol 152:1523–1526. https://doi.org/10.1007/s00705-007-0982-5
Lian X, Ming X, Xu J et al (2018) First molecular detection and characterization of Marek’s disease virus in red-crowned cranes (Grus japonensis): a case report. BMC Vet Res 14:122. https://doi.org/10.1186/s12917-018-1437-9
Murata S, Hayashi Y, Kato A et al (2012) Surveillance of Marek’s disease virus in migratory and sedentary birds in Hokkaido, Japan. Vet J 192:538–540. https://doi.org/10.1016/j.tvjl.2011.07.006
Murata S, Machida Y, Isezaki M et al (2020) Genetic characterization of a Marek’s disease virus strain isolated in Japan. Virol J 17:186. https://doi.org/10.1186/s12985-020-01456-1
Li Y, Sun A, Su S et al (2011) Deletion of the Meq gene significantly decreases immunosuppression in chickens caused by pathogenic Marek’s disease virus. Virol J 8:2. https://doi.org/10.1186/1743-422x-8-2
Zhang YP, Lv HC, Bao KY et al (2016) Molecular and pathogenicity characterization of Gallid herpesvirus 2 newly isolated in China from 2009 to 2013. Virus Genes 52:51–60. https://doi.org/10.1007/s11262-015-1264-z
Sato J, Murata S, Yang Z et al (2022) Effect of insertion and deletion in the meq protein encoded by highly oncogenic Marek’s disease virus on transactivation activity and virulence. Viruses. https://doi.org/10.3390/v14020382
Murata S, Okada T, Kano R et al (2011) Analysis of transcriptional activities of the Meq proteins present in highly virulent Marek’s disease virus strains, RB1B and Md5. Virus Genes 43:66–71. https://doi.org/10.1007/s11262-011-0612-x
Padhi A, Parcells MS (2016) Positive selection drives rapid evolution of the meq oncogene of Marek’s disease virus. PLoS ONE 11:e0162180. https://doi.org/10.1371/journal.pone.0162180
Trimpert J, Groenke N, Jenckel M et al (2017) A phylogenomic analysis of Marek’s disease virus reveals independent paths to virulence in Eurasia and North America. Evol Appl 10:1091–1101. https://doi.org/10.1111/eva.12515
Yu ZH, Teng M, Luo J et al (2013) Molecular characteristics and evolutionary analysis of field Marek’s disease virus prevalent in vaccinated chicken flocks in recent years in China. Virus Genes 47:282–291. https://doi.org/10.1007/s11262-013-0942-y
Li K, Yu Z, Lan X et al (2022) Complete genome analysis reveals evolutionary history and temporal dynamics of Marek’s disease virus. Front Microbiol 13:1046832. https://doi.org/10.3389/fmicb.2022.1046832
Zhang Y, Lan X, Wang Y et al (2022) Emerging natural recombinant Marek’s disease virus between vaccine and virulence strains and their pathogenicity. Transbound Emerg Dis 69:e1702–e1709. https://doi.org/10.1111/tbed.14506
Cui H, Gao H, Cui X et al (2013) Avirulent Marek’s disease virus type 1 strain 814 vectored vaccine expressing avian influenza (AI) virus H5 haemagglutinin induced better protection than turkey herpesvirus vectored AI vaccine. PLoS ONE 8:e53340. https://doi.org/10.1371/journal.pone.0053340
Reddy SK, Sharma JM, Ahmad J et al (1996) Protective efficacy of a recombinant herpesvirus of turkeys as an in ovo vaccine against Newcastle and Marek’s diseases in specific-pathogen-free chickens. Vaccine 14:469–477. https://doi.org/10.1016/0264-410x(95)00242-s
Sun GR, Zhang YP, Lv HC et al (2017) A chinese variant Marek’s disease virus strain with divergence between virulence and vaccine resistance. Viruses 9:71. https://doi.org/10.3390/v9040071
Baigent SJ, Petherbridge LJ, Smith LP et al (2006) Herpesvirus of turkey reconstituted from bacterial artificial chromosome clones induces protection against Marek’s disease. J Gen Virol 87:769–776. https://doi.org/10.1099/vir.0.81498-0
Acknowledgements
This work was supported by the Guangdong Major Project of Basic and Applied Basic Research (2020B0301030007), Laboratory of Lingnan Modern Agriculture Project (NT2021007), the Guangdong Provincial Key R&D Program, (2022B1111040001), the National Natural Science Foundation of China (31822056), the Guangdong Science and Technology Innovation Leading Talent Program (2019TX05N098), the 111 Project (D20008), the Double first-class discipline promotion project (2023B10564003) and the Department of Education of Guangdong Province (2019KZDXM004 and 2019KCXTD001).
Funding
This study was funded by Guangdong Major Project of Basic and Applied Basic Research, 2020B0301030007.
Author information
Authors and Affiliations
Contributions
YS conceptualized, designed and and wrote the manuscript. WL, HM, JP and XS performed the experiments. XL analyzed the data. DMI participated in revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Edited by Juergen Richt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, W., Meng, H., Liang, X. et al. The genome evolution of Marek’s disease viruses in chickens and turkeys in China. Virus Genes 59, 845–851 (2023). https://doi.org/10.1007/s11262-023-02034-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-023-02034-7