
Abstract
Genotype I of hepatitis B virus (HBV) was proposed recently following sequencing of complete HBV genomes from Vietnam and Laos. However, its long-term molecular evolution is unknown. The objectives of this study were to study the molecular evolution of this genotype from an asymptomatic HBsAg carrier from the Long An cohort over a 15-year period was studied using both NGS and clone-based sequencing. The number of complete genome sequences obtained in 2004, 2007, 2013, and 2019 are 17, 20, 19, and 10, respectively. All strains belong to subgenotype I1, except for six (five from 2007 and one from 2019) and 8 further strains from 2007 which form a cluster branching out from other subgenotype I sequences, supported by a 100% bootstrap value. Based on complete genome sequences, all of the estimated intragroup nucleotide divergence values between these strains and HBV subgenotypes I1–I2 exceed 4%. These strains are recombinants between genotype I1 and subgenotype C but the breakpoints vary. The median intrahost viral evolutionary rate in this carrier was 3.88E-4 substitutions per site per year. The Shannon entropy (Sn) ranged from 0.55 to 0.88 and the genetic diversity, D, ranged from 0.0022 to 0.0041. In conclusion, our data provide evidence of novel subgenotypes. Considering that the 8 strains disappeared after 2007, while one of the 6 strains appears again in 2019, we propose these 6 strains as a new subgenotype, provisionally designated HBV subgenotype I3 and the 8 strains as aberrant genotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatitis B virus (HBV), the prototype member of the family Hepadnaviridae, has a circular, partially double-stranded DNA genome of approximately 3200 base pairs [1]. HBV replicates via reverse transcription of an RNA intermediate catalyzed by the viral polymerase that lacks proof-reading ability. The lack of proof-reading favors the development of sequence variants during long-term HBV replication. Thus, sequence heterogeneity is a feature of HBV, which persists as a quasispecies of mixed viral strains within the host [2, 3]. However, the long-term viral evolutionary rate might be slower because of ‘reverse mutations,’ through which viral variants revert back to the original form at key immunogenic residues [4].
According to an intergroup divergence of more than 8%, based on complete genomes, 9 genotypes of HBV have been identified (designated genotypes A to I) [3, 5,6,7], with a putative 10th genotype, “J,” isolated from a single individual [8]. Based on an intragroup nucleotide difference of approximately 4–8% across the complete genome and forming an independent lineage in the phylogenetic tree with support by bootstrap values of above 75% [9, 10], genotypes A–D, F, and I have been further categorized into at least 55 subgenotypes [11, 12]. The genotype distribution showed similar patterns between countries of the same region of the world but varied considerably between different parts of the world [7].
Genotype I was first reported in 2008 by Tran et al., according to the phylogenetic analysis of the complete genome of a complex (X/C) recombinant [13], which has a high similarity to the “aberrant strains” among Vietnamese isolates reported by Hannoun et al. [14] 8 years earlier. The proposal was supported by a report of isolates from Laos, which analyzed a larger number of novel sequences and assigned them to two candidate subgenotypes, I1 and I2 [15]. This genotype is widespread and has been found subsequently in other Asian countries, such as India, Thailand, and parts of China [16,17,18].
Guangxi Zhuang Autonomous Region is one of the provinces in China with the highest prevalence of persistent HBV infection, 13.4% of the general population in 1980 [19] and 9.2% in 2012 [20]. The major HBV genotypes in this province are B, C, and I. Genotype I was firstly found with a high prevalence of about 14% in Long An county, Guangxi in 2011 [16]. However, with a cross-sectional study, we found that the distribution of this genotype within Guangxi is not even and it is highly endemic in some counties [21].
In order to understand the accumulated sequence variations in the viral genome and the observed mutation rate over a long period, as well as viral pathogenesis, we used next-generation sequencing (NGS) to determine the molecular evolution of HBV in asymptomatic HBsAg carriers from the Long An cohort over a 15-year period [22]. In that study, we found one subject with aberrant genotype. The whole-genome sequence, with direct sequencing from a sample collected from that subject in 2004, was reported in 2011 (access number: FR714502) [16]. Here, we report the findings from that subject from four time point using NGS and clone-based sequencing (CBS).
Materials and methods
Study subject and ethic statement
The study subject (TZ087) was selected from the Long An cohort [23]. Serum samples from the subject were available from 2004, 2007, 2013, and 2019. The subject was negative for human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) and was antiviral therapy naïve before and during the study.
Informed consent in writing was obtained from the study subject. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and has been approved by the Guangxi Institutional Review Board.
Serological testing, measurement of serum viral loads, PCR for HBV genomic DNA, library preparation and next-generation sequencing, NGS data preprocessing and sample genotyping, haplotype construction and diversity analysis and estimation of the intrahost HBV evolutionary rate
These methods have been reported previously [22].
Clone-based sequencing
Amplicons from the fourth round were confirmed by agarose gel electrophoresis and cloned into the vector P clone 007T (The Beijing Qingke Biotech Co. Ltd, PR China) and then subsequently transformed into competent Escherichia coli (The Beijing Qingke Biotech Co. Ltd, PR China). Thirty-one clones containing the full-length viral genome were selected. Plasmid DNA was extracted using a SK1191 UNIQ-10 kit (The Beijing Qingke Biotech Co. Ltd, PR China) and the purified DNA was sequenced using a BigDye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, USA) in The Beijing Qingke Biotech Co. Ltd, PR China.
Sequences were determined for both strands to derive robust data for comparison with the full-length sequences of the various genotypes.
Forward primers:
W803-C01 (nt43-61, 5′-GGGGCCTGTATTTTCCTGCT-3′),
W798-A02 (nt254-273, 5′-TGTCAACAATTTGTGGGCCC-3′),
W807-F05 (nt505-524, 5′-ATTCCTATGGGAGTGGGCCT-3′),
W803-A01 (nt810-829, 5′-ACCAATCGGCAGTCAGGAAG-3′),
W811-C09 (nt1252-1271, 5′-GCTCCTCTGCCGATCCATAC-3′).
W798-B02 (nt1633-1652, 5′-TGTGAACAATTTGTGGGCCC-3′),
W798-E01 (nt2472-2491, 5′-GTGGGAAACTTTACCGGGCT-3′), and
W803-B01 (nt3061-3080, 5′-GGAGGTCTTTTGGGGTGGAG-3′).
Reverse primers:
W807-A01 (nt60-41, 5′-GCAGGAAAATACAGGCCCCT-3′),
W807-B01 (nt353-334, 5′-GGACAGGAGGTTGGTGAGTG-3′),
W807-C01 (nt893-874, 5′-CCCCAATCCTCGCGAAGATT-3′).
W798-C02 (nt1237-1218, 5′-CCACAAAGGTTCCACGCATG-3′),
W803-D01 (nt1543-1524, 5′-GAGGCCCACTCCCATAGGTA-3′),
W803-B03 (nt2325-2306, 5′-AGGCCCACTCCCATAGGAAT-3′), and
W798-F01 (nt2640-2621, 5′-GTATGGATCGGCAGAGGAGC-3′).
HBV genotyping
HBV genotypes were determined using phylogenies reconstructed on the basis of the complete genome and preS/S regions of the viruses. The sequences were aligned to 48 HBV sequences of all known genotypes retrieved from GenBank using Clustal W and visually confirmed with the sequence editor BioEdit [24]. The reference sequences were A1-AB116082-Japan, A2-EU859908-Belgium, A3-AB194952-Japan, B1-AB602818-Japan, B2-EU939638-China, B2-AB981582-Japan, B3-AB976562-Indonesia, B4-AB115551-Cambodia, B5-GQ924645-Malaysia, B6-KP659253-Canada, B7-GQ358143-Indonesia, B8-GQ358146-Indonesia, B9-GQ358152-Indonesia, B10-MN689123-China, C1-AF458664-China, C2-AF223960-Malaysia, C3-X75656-Polynesia, C4-AB048705-Australia, C5-JN827414-Thailand, C5-AB241111-Philippines, C6-AB493843-Indonesia, C7-EU670263-Philippines, C8-AP011106-Indonesia, C9-AP011108-Indonesia, C10-AB540583-Indonesia, C11-AB554020-Indonesia, C12-AB554025-Indonesia, C13-AB644281-Indonesia, C14-AB644284-Indonesia, C15-AB644286-Indonesia, C16-AB644287-Indonesia, C17-MG826140-China, D1-AB188244-Japan, D2-JF754621-UK, D3-AB188243.2-Japan, D4-FJ692533.2-Africa, D5-GQ205388-India, D6-FJ904397-UK, D7-KP322604.2-Tunisia, D8-FN594768-Niger, D9-JN664942-India, D10-KX357633-Ethiopia, D11-MK052961-China, E-AB032431-Liberia, F-DQ823094-Argentina, G-AF405706-Germany, H-AY090460-USA, I1-AB231908-Vietnam, I1-FJ023659-Laos, I2-FJ023664-Laos, I2-FJ023670-Laos, and I1-FR714490-China. Neighbor-Joining trees were reconstructed under the Kimura 2-parameter substitution model with the program MEGA [25]. The reliability of clusters was evaluated using interior branch test with 1000 replicates and the internal nodes with over 75% support were considered reliable.
Intragroup genetic distance between new subgenotypes and other subgenotype I
Intragroup genetic distances (mean ± SD) were calculated by pairwise comparison of nucleotide sequences using the Kimura 2-parameter method of Molecular Evolutionary Genetics Analysis (MEGA) v7.0.18 [26, 27].
Detection of recombination
Analysis of recombination was performed using the Simplot program and boot scanning analysis [28]. The complete HBV sequence was compared to consensus sequences generated using complete GenBank references for HBV genotypes A-I to search the “parenta” sequences for bootscanning analysis. In bootscanning analysis, four sequences were considered at a time: the putative recombinant sequence, reference sequences of the original genotypes, FR714490 and MZ439308, and a known outgroup, AY226578. Each informative site supported by one of three possible phylogenetic relationships among the four taxa; contiguous sites suggesting a single phylogeny were inferred to represent regions between recombination breakpoints.
Results
General characteristics and genotypes
The study subject was male and aged 34 in 2004. His alanine aminotransferase (ALT) levels were normal (6-40 U/L). Full-length HBV genomes were successfully amplified from the sera from four sampling times and analyzed successfully using NGS. On average, 800,000 reads were maintained for each sample after quality filtering, corresponding to a mean coverage of 80,000 fold at each nucleotide site. The numbers of complete genome sequences obtained in 2004, 2007, 2013, and 2019 are 17, 20, 19, and 10 (6 haplotypes were obtained by next-generation sequencing and the remainder by clone-based sequencing), respectively (Table 1).
HBV genotyping based on complete genome sequences
Because the number of strains from the 2019 sample is small, compared to the other samples, we carried out clone-based sequencing of that sample. Complete genome sequences were obtained from four clones. Consensus HBV sequences were constructed for all samples. A phylogenetic tree was constructed on the basis of the complete genomes of all strains from each sample. As shown in Fig. 1, a maximum likelihood tree of these consensus sequences indicates that the subject was infected by genotype I. All strains of 2004 and 2013 samples belong to subgenotype I1, while only 7 of 20 and 9 of 10 strains of 2007 and 2019 belong to subgenotype I1. Six strains (five obtained by NGS from the 2007 sample and one obtained by CBS from the 2019 sample) and 8 further strains from 2007 form a cluster, branching out from other subgenotype I sequences, supported by a 100% bootstrap value.

Neighbor-Joining tree. The tree was constructed using complete viral sequences under the Kimura 2-parameter substitution model with the program MEGA (Tamura et al., 2013). The branch lengths represent the number of substitutions per site. The reliability of clusters was evaluated using the interior branch test with 1000 replicates and the internal nodes with over 75% support are considered reliable
Calculation of phylogenetic distances reveals that almost all of the estimated intragroup nucleotide divergence [mean ± SD] over the complete genome sequences, by pairwise analysis between our isolates and the other HBV I1-I2 subgenotypes, exceed 4% (Table 2), suggesting that the cluster of six deviating strains from 2007 and the second cluster with eight deviating strains from 2007 belong to a different subgenotype of I. Considering that the 8 strains disappeared after 2007, while the 6 strains appear again in 2019, we propose these 6 strains as a new subgenotype, provisionally designated HBV subgenotype I3 and the 8 strains as aberrant genotype.
Phylogenetic analysis of the four ORFs of subgenotype I3
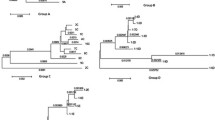
Phylogenetic trees were constructed based on the sequences of PreS/S, P, PreC/C, and X ORFs of the novel subgenotype I3. All sequences of the PreS/S ORF formed a distinct clade, supported by a 61% bootstrap value. All sequences of the P ORF formed a distinct clade, supported by a 100 % bootstrap value. All sequences of the PreC/C ORF, together with subgenotype I1, form a distinct clade, supported by a 100% bootstrap value. Except for one sequence obtained by CBS, 5 of 6 sequences of the X ORF formed a distinct clade, supported by an 80% bootstrap value. These findings suggest that the sequences of the P ORF may be more specific to subgenotype I3. The PreS/S sequences of subgenotype I3 could not be used for genotyping (Fig. 2a–d).

Neighbor-Joining trees based on open reading frames. The trees were reconstructed for the ORF PreS/S (2a), P (2b), PreC/C (2c), and X (2d) sequences of the viruses under the Kimura 2-parameter substitution model with the program MEGA (Tamura et al., 2013). The branch lengths represent the number of substitutions per site. The reliability of clusters was evaluated using the interior branch test with 1000 replicates and the internal nodes with over 75% support are considered reliable
Time series quasispecies, diversity, and intrahost HBV viral evolutionary rates
After realignment of the sequences from the samples with genotype-specific reference sequences, 62 haplotypes with an abundance greater than 1% were obtained (Table 1). The Shannon entropy (Sn) ranged from 0.55 to 0.88 and the genetic diversity, D, ranged from 0.0022 to 0.0041. The mean pairwise genetic diversity of the subjects did not change dramatically, except for that in 2019. The Sn values increased in 2007, indicating expansion of the quasispecies. However, the Sn values declined in 2013 and 2019, indicating centralization of the quasispecies.
The predominant strains in 2004 were subgenotype I1, while other minor strains expanded and lead to mixed predominant strains of I1, I3and the aberrant genotype in 2007. However, all strains of subgenotype I3 and the aberrant genotype disappeared and subgenotype I1 became predominant strains again in 2013 and 2019. This change corresponded to the value of mean genetic distance (Table 1). The subject exhibited much high intrahost viral evolutionary rates, with median values of 3.88E-4 substitutions per site per year.
Nucleotide and amino acid characteristics of subgenotype I3 and the aberrant genotype
Compared to subgenotypes I1 and I2, three amino acids (Pro\(^{179}\), Lys\(^{190}\), and Val\(^{637}\) in P gene) were found to be unique to all strains of subgenotype I3. All strains of subgenotype I3 have a length of 3215 nt. The lengths of the four ORFs, preS/S, P, preC/C, and X are 1200, 2529, 636, and 462 nt, respectively. The length of the genome and four ORFs of the aberrant genotype are the same as subgenotype I3. There are 24 amino acids that are unique to all eight strains of the aberrant genotype. These unique amino acids are distributed in three ORFs: Ala\(^{91}\), Thr\(^{120}\), Leu\(^{132}\), Lys\(^{137}\), Ala\(^{151}\), Thr\(^{157}\), and Pro\(^{160}\) in the S protein, Leu\(^{46}\), Asp\(^{81}\), Lys\(^{85}\), Lys\(^{190}\), Ser\(^{268}\), Gly\(^{271}\), Gln\(^{283}\), Tyr\(^{286}\), Pro\(^{296}\), Ser\(^{308}\), Ser\(^{309}\), Lys\(^{317}\), Leu\(^{321}\), Val\(^{343}\), Leu\(^{346}\), and Iso\(^{437}\) in the polymerase, and Asp\(^{96}\) in the core protein.
Recombinant genomes and identification of putative recombination sites
Evidence of recombination was detected in all sequences using Simplot software. The bootscanning result of subgenotype I3 showed that parts of the genome (nt 1 to 341 and nt 1832 to 3215) were more similar to subgenotype I1 than to the other genotypes. The remaining part (estimated to be from nt 341 to 1832) was more similar to genotype C. These recombination events were supported by high bootstrap values (\(P <0.05\)), suggesting that subgenotype I3 is a recombinant between genotype C and subgenotype I1 (Fig. 3a–c). The bootscanning result of the aberrant genotype showed that parts of the genome (nt 1 to 671 and nt 853 to 3215) were more similar to genotype C, while the remaining part (nt 671 to 1853) was more similar to subgenotype I1, suggesting that the aberrant genotype also is a recombinant between genotype C and subgenotype I1 (Fig. 4a–c). Clearly, both subgenotype I3 and the aberrant genotype are recombinants between genotype I1 and subgenotype C but the breakpoints differ (Fig. 5).

Simplot analysis of the recombination of strains TZ087-2007-NGS-7 (subgenotype I3) sequences. 3a and 3b show similarity for each position. 3c shows the percentage of permuted trees (BootScan). GenBank accession numbers of subgenotype C5, I1, and woolly monkey reference sequences are MZ439308, FR714490, and AY226578, respectively. P, C, S, and X indicate the polymerase, core, surface, and X genes, respectively

Simplot analysis of the recombination of strain TZ087-2007-NGS-20 (aberrant genotype) sequences. 4a and 4b show similarity for each position. 4c shows the percentage of permuted trees (BootScan). GenBank accession numbers of subgenotype C5, I1, and woolly monkey reference sequences are MZ439308, FR714490, and AY226578, respectively. P, C, S, and X indicate the polymerase, core, surface, and X genes, respectively

The breakpoints of subgenotype I3 and the aberrant genotype. Both subgenotype I3 and the aberrant genotype are recombinants between subgenotype C5 and I1. The blue portion comes from C5 and the red portion from I1
Discussion
To our knowledge, this study is the first to report a novel subgenotype of HBV, I3. The genome of subgenotype I3 comprises 3215 nucleotides. Compared to subgenotypes I1 and I2, there is one amino acid unique to subgenotype I3 and twenty-four amino acids were found to be unique to the aberrant genotype. An atypical genotyping pattern also was found in this study. These aberrant sequences clustered on a separate branch from other subgenotype I sequences in the phylogenetic tree based on whole genome. However, these disappeared after 2007 while subgenotype I3 reappeared in 2019. Both the novel subgenotype I3 and aberrant genotype are recombinants between subgenotype C5 and subgenotype I1. The appearance of minor strains varies with the Sn values. The strength of this study is that we have serial serum samples from long-term follow-up, which allows us to determine the change of various genotypes. The weakness is that subgenotype I3 has been found in only one individual and the prevalence of this novel subgenotype remains unknown.
Genotype I is found in Asia and has a distinct geographic distribution [16, 18, 29]. It has been further categorized into two subgenotypes; subgenotype I1 is found in Vietnam, Thailand, Laos, and parts of China, while subgenotype I2 is found in Laos [15]. Arankalle et al. found that the Indian strains formed a distinct branch of the cluster with 100% posterior support. The percentage divergence between the Indian strains and the strains belonging to subgenotypes I1 and I2 was \(3.5 \pm 0.29\) and \(3.1 \pm 0.3\), respectively. They thought that it should constitute a distinct subgenotype I3, although they finally classified it as a distinct cluster within genotype I and not as subgenotype [17]. In this study, we found that the percentage divergence between our strains and I1 and I2 strains was \(>4\%\), suggesting a novel subgenotype I3 circulating in China. We also found that the PreS/S sequences of subgenotype I3 could not be used for genotyping and tried to identify the HBsAg subtypes of the I3 subgenotype and of the aberrant strains according to the rules described by others [30, 31]. However, they could not be assigned.
HBV genotypes are associated with the clinical outcome of HBV infection as well as the response to antiviral therapy [32]. For example, liver inflammation activity was higher in patients with genotype C than those with genotype B and more patients with genotype C tended to have a high viral load than those with genotype B [33]. Genotype A has a greater hepatocarcinogenic potential than non-A genotypes and this is entirely attributable to subgenotype A1 [34]. Patients infected with HBV subgenotype D1 progressed from CHB to HCC through liver cirrhosis more frequently than those with subgenotype D3, suggesting HBV/D1 might have greater oncogenic potential than HBV/D3 [35]. HBV subgenotype C1 is associated with better antiviral response to nucleoside analogs in HBsAg-positive patients than B2 and C2 subgenotypes [36]. Therefore, our findings in this study have important clinical implications when considering disease prognosis and antiviral therapy among chronically infected individuals.
Eight sequences of the aberrant genotype were identified in this study. In phylogenetic tree based on whole genome, they clustered on a separate branch out from other subgenotype I sequences, supported by a 100% bootstrap value. The estimated intragroup nucleotide divergence over the complete genome sequences between these isolates and the other HBV I1-I2 subgenotypes exceed 4%. However, in the phylogenetic tree based on PreS/S sequences, they did not cluster together. This aberrant genotype is a recombinant. If every new recombinant is assigned to a new genotype, we would soon be running out of alphabet letters [37]. Furthermore, unlike subgenotype I3, this aberrant genotype disappeared in 2013 and did not appear again in 2019. Therefore, this aberrant genotype was not assigned a new subgenotype. However, recombination potentially may play a large, underestimated role in both the origins of currently known genotypes of HBV found in human and nonhuman primate populations and in future evolution of their pathogenicity and transmissibility [38].
HBV genotype shifting is common during long-term infection. It could be seen in treatment-naive patients [39], during antiviral therapy [40] and during HBeAg/anti-HBe seroconversion [41]. In this study, subgenotype I1 was found in 2004, 2007, 2013 and 2019. However, the aberrant genotype was found in 2007 only. Subgenotype I3 was found in 2007 but disappeared in 2013 and then appeared in 2019. The mechanism of genotype shifting remains obscured. The high rate of HBV replication and an error-prone RT-based replication cycle results in the generation of a diverse pool of closely related but distinct genetic variants (quasispecies) [42]. Furthermore, co-infection with one or more genotypes/subgenotypes of HBV is common [43]. Drug and immune pressure may play a key role in the selection of genotype [32]. It has been reported that pre-existing minor HCV genotypes can be selected rapidly during antiviral treatment and become transiently or permanently predominant [44]. In this study, genotype shifting may be due to co-infection with subgenotype I1 and I3. However that I3 may potentially have evolved from I1 could not be excluded. This needs to be investigated.
Both recombination and the mutation rate of the error-prone viral polymerase are important for HBV evolution [45]. Recombination may be associated with expansion of viral host ranges, emergence of new viruses, increase in virulence and pathogenesis, evasion of host immunity, and resistance to antivirals [46]. Recombination may occur between and within genotypes [38]. Subgenotype I1 is a recombinant between genotype C, G, and an unknown genotype (X) [16]. In this study, we found that subgenotype I3 is a recombinant between subgenotype C5 and subgenotype I1, suggesting that genotypes in that area are complex and more genotype/recombinants may be detected with further analysis of recombinants.
In conclusions, we were the first to analyze the long-term molecular evolution of genotype I, HBV. A novel subgenotype I3 and an aberrant genotype were identified, which appeared as minor strains during infection. The appearance of minor strains varies with the Sn values. Both the novel subgenotype I3 and aberrant genotype are recombinants between subgenotype C5 and subgenotype I1. Our study highlights the importance of using next-generation sequencing or cloned-based sequencing in finding novel quasispecies.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Magnius LO, Norder H (1995) Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology 38:24–34
Harrison TJ (2006) Hepatitis B virus: molecular virology and common mutants. Sem Liver Dis 26:87–96
Kramvis A, Kostaki EG, Hatzakis A, Paraskevis D (2018) Immunomodulatory function of HBeAg related to short-sighted evolution, transmissibility, and clinical manifestation of hepatitis B virus. Front Microbiol 9:2521
Revill PA, Tu T, Netter HJ, Yuen LKW, Locarnini SA, Littlejohn M (2020) The evolution and clinical impact of hepatitis B virus genome diversity. Nat Rev Gastroenterol Hepatol 17(10):618–634
Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai M, Miyakawa Y, Mayumi M (1988) Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol 69:2575–2583
Norder H, Courouce AM, Magnius LO (1994) Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 98:489–503
Velkov S, Ott JJ, Protzer U, Michler T (2018) The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes (Basel) 9(10):495
Tatematsu K, Tanaka Y, Kurbanov F, Sugauchi F, Mano S, Maeshiro T, Nakayoshi T, Wakuta M, Miyakawa Y, Mizokami M (2009) A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J Virol 83:10538–10547
Shi W, Zhang Z, Ling C, Zheng W, Zhu C, Carr MJ, Higgins DG (2013) Hepatitis B virus subgenotyping: history, effects of recombination, misclassifications, and corrections. Infect Genet Evol 16:355–361
Kramvis A, (2014) Genotypes and genetic variability of hepatitis B virus. Intervirology 57(3–4):141–150
Liu Y, Feng Y, Li Y, Ma J, Jia Y, Feng YM (2020) Characterization of a novel hepatitis B virus subgenotype B10 among chronic hepatitis B patients in Yunnan, China. Infect Genet Evol 83:104322
Thijssen M, Trovão NS, Mina T, Maes P, Pourkarim MR (2020) Novel hepatitis B virus subgenotype A8 and quasi-subgenotype D12 in African-Belgian chronic carriers. Int J Infect Dis 93:98–101
Tran TT, Trinh TN, Abe K (2008) New complex recombinant genotype of hepatitis B virus identified in Vietnam. J Virol 82:5657–5663
Hannoun C, Norder H, Lindh M (2000) An aberrant genotype revealed in recombinant hepatitis B virus strains from Vietnam. J Gen Virol 81:2267–2272
Olinger CM, Jutavijittum P, Hübschen JM, Yousukh A, Samountry B, Thammavong T, Toriyama K, Muller CP (2008) Possible new hepatitis B virus genotype, southeast Asia. Emerg Infect Dis 14:1777–1780
Fang ZL, Hué S, Sabin CA, Li GJ, Yang JY, Chen QY, Fang KX, Huang J, Wang XY, Harrison TJ (2011) A complex hepatitis B virus (X/C) recombinant is common in Long An county, Guangxi and may have originated in southern China. J Gen Virol 92:402–411
Arankalle VA, Gandhe SS, Borkakoty BJ, Walimbe AM, Biswas D, Mahanta J (2010) A novel HBV recombinant (genotype I) similar to Vietnam/Laos in a primitive tribe in eastern India. J Viral Hepat 17:501–510
Holzmayer V, Hance R, Defechereux P, Grant R, Kuhns MC, Cloherty G, Rodgers MA (2019) Identification of hepatitis B virus genotype I in Thailand. J Med Virol 91:717–721
Ding ZR (1982) Distribution of viral hepatitis B infection in Guangsi Province. Zhonghua Liu Xing Bing Xue Za Zhi 3:84–87
Fang ZL, Harrison TJ, Yang JY, Chen QY, Wang XY, Mo JJ (2012) Prevalence of hepatitis B virus infection in a highly endemic area of southern China after catch-up immunization. J Med Virol 84:878–884
Li GJ, Hue S, Harrison TJ, Yang JY, Chen QY, Wang XY, Fang ZL (2013) Hepatitis B virus candidate subgenotype I1 varies in distribution throughout Guangxi, China and may have originated in Long An county, Guangxi. J Med Virol 85:799–807
Chen QY, Jia HH, Wang XY, Shi YL, Zhang LJ, Hu LP, Wang C, He X, Harrison TJ, Jackson JB, Wu L, Fang ZL (2021) Analysis of entire hepatitis B virus genomes reveals reversion of mutations to wild type in natural infection, a 15 year follow-up study. Infect Genet Evol 97:105184
Fang ZL, Sabin CA, Dong BQ, Ge LY, Wei SC, Chen QY, Fang KX, Yang JY, Wang XY, Harrison TJ (2008) HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: a prospective study. Am J Gastroenterol. 103(9):2254–2262
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41:95–98
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160
Haldipur BP, Walimbe AM, Arankalle VA (2014) Circulation of genotype-I hepatitis B virus in the primitive tribes of Arunachal Pradesh in early sixties and molecular evolution of genotype-I. Infect Genet Evol 27:366–374
Norder H, Couroucé AM, Coursaget P, Echevarria JM, Lee SD, Mushahwar IK, Robertson BH, Locarnini S, Magnius LO (2004) Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 47(6):289–309
Gerlich WH, Glebe D, Kramvis A, Magnius LO (2020) Peculiarities in the designations of hepatitis B virus genes, their products, and their antigenic specificities: a potential source of misunderstandings. Virus Genes 56(2):109–119
Kmet Lunaček N, Poljak M, Matičič M (2018) Distribution of hepatitis B virus genotypes in Europe and clinical implications: a review. Acta Dermatovenerol Alp Pannonica Adriat 27:141–146
Wu JC, Huang YH, Chau GY, Su CW, Lai CR, Lee PC, Huo TI, Sheen IJ, Lee SD, Lui WY (2009) Risk factors for early and late recurrence in hepatitis B-related hepatocellular carcinoma. J Hepatol 51:890–897
Kew MC, Kramvis A, Yu MC, Arakawa K, Hodkinson J (2005) Increased hepatocarcinogenic potential of hepatitis B virus genotype A in Bantu-speaking sub-saharan Africans. J Med Virol 75(4):513–521
Datta S, Dasgupta D, Ghosh A, Ghosh S, Manna A, Datta S, Chatterjee M, Chowdhury A, Soma Banerjee S (2018) Oncogenic potential of hepatitis B virus subgenotype D1 surpasses D3: significance in the development of hepatocellular carcinoma. Carcinogenesis 39:283–292
Shen S, Liang X, Hamed K, Tanaka Y, Omagari K, Fan R, Xie Q, Tan D, Zhou B, Jia JD, Hou J, Sun J (2018) Effect of hepatitis B virus subgenotype on antiviral response in nucleoside-treated hepatitis B envelope antigen-positive patients. Hepatol Res 48:134–143
Kurbanov F, Tanaka Y, Kramvis A, Simmonds P, Mizokami M (2008) When should I consider a new hepatitis B virus genotype? J Virol 82:8241–8242
Simmonds P, Midgley S (2005) Recombination in the genesis and evolution of hepatitis B virus genotypes. J Virol 79:15467–15476
Yeh CT, Liang KH, Chang ML, Hsu CW, Chen YC, Lin CL, Lin WR, Lai MW (2017) Phenotypic and genotypic shifts in hepatitis B virus in treatment-Naive patients, Taiwan, 2008–2012. Emerg Infect Dis 23:820–821
Wang Y, Shan X, Liang Z, Shan Y, Huang W, Zhang D, Zen A, Zhou X, Zhao Y, Gong X, Xu G, Zhang X, Chen J, Huang A (2015) Deep sequencing analysis of HBV genotype shift and correlation with antiviral efficiency during adefovir dipivoxil therapy. PLoS One 10:e0131337
Wirth S, Bortolotti F, Brunert C, Postberg J, Hensel K, Jenke A (2013) Hepatitis B virus genotype change in children is closely related to HBeAg/anti-HBe seroconversion. J Pediatr Gastroenterol Nutr 57:363–366
Rodriguez-Frias F, Buti M, Tabernero D, Homs M (2013) Quasispecies structure, cornerstone of hepatitis B virus infection: mass sequencing approach. World J Gastroenterol 19:6995–7023
Hu LP, Liu DP, Chen QY, Harrison TJ, He X, Wang XY, Li H, Tan C, Yang QL, Li KW, Fang ZL (2015) Occult HBV infection may be transmitted through close contact and manifest as an overt infection. PLoS One 10(10):e0138552
Iwona BO, Karol P, Kamila CC, Pollak A, Hanna B, Agnieszka P, Andrzej H, Kosińska J, Płoski R, Tomasz L, Marek R (2018) Next-generation sequencing analysis of new genotypes appearing during antiviral treatment of chronic hepatitis C reveals that these are selected from pre-existing minor strains. J Gen Virol 99:1633–1642
Locarnini SA, Littlejohn M, Yuen LKW (2021) Origins and evolution of the primate hepatitis B virus. Front Microbiol 12:653684
Araujo NM (2015) Hepatitis B virus intergenotypic recombinants worldwide: an overview. Infect Genet Evol 36:500–510
Acknowledgements
We are indebted to staff members of Long An Center for Disease Prevention and Control and local hospitals in Long An county, Guangxi, who assisted in recruiting the study subjects and sample collection.
Funding
This study was supported by The Global Health Research Seed Grant from the Carver College of Medicine at the University of Iowa, Guangxi Key Research and Development Project (Grant No. 2018AB59002) and the National Natural Science Foundation of China (Grant No. 81860595).
Author information
Authors and Affiliations
Contributions
Conceptualization: Wu L, Fang ZL, Harrison TJ, and Jackson BJ. Data curation: Jia HH, Chen QY, Wang XY. Formal analysis: Wang XY, He X. Investigation: Jia HH, Chen QY, Jiang ZH, and Zhang WJ. Methodology: He X and Wang XY. Supervision: Wu L and Fang ZL. Writing of the original draft: Fang ZL. Writing, reviewing, & editing of the manuscript: Wu L, Fang ZL, Harrison TJ, and Jackson BJ.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no financial interests.
Ethical approval
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and has been approved by the Guangxi Institutional Review Board.
Consent to participate
Informed consent in writing was obtained from the study subject.
Additional information
Edited by Hartmut Hengel.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jia, HH., Chen, QY., Jiang, ZH. et al. A novel subgenotype I3 of hepatitis B virus in Guangxi, China: a 15-year follow-up study. Virus Genes 59, 359–369 (2023). https://doi.org/10.1007/s11262-023-01980-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-023-01980-6