
Abstract
The UL24 homologous genes are conserved in alphaherpesviruses. However, the proximity of the UL24 gene and the UL23 gene encoding for thymidine kinase (TK) in the genome of suid herpesvirus 1 (SuHV-1) makes it difficult to mutate UL24 without affecting the expression of the TK gene, and thus functional studies of the UL24 gene have lagged behind. In this study, CRISPR/Cas9 and homologous recombination were adopted to generate UL24 and TK mutant viruses. Deletion of either the UL24 or the TK gene resulted in significantly reduced SuHV-1 replication and spread capacity in Vero cells. However, UL24-deleted virus still maintained a certain degree of lethality in mice, while TK-deleted viruses completely lost their lethality in mice. Similarly, neurovirulence of UL24-deleted virus in mice was not significantly affected compared to parental virus. In comparison, infection with the TK-deleted viruses resulted in significantly reduced neurovirulence and complete loss of lethality. In addition, and for the first time, viral UL24 protein was found to be expressed late during SuHV-1 infection; enhanced green fluorescence protein (eGFP) labeled UL24 protein was shown to be localized in the nucleus via heterologous expression. In conclusion, the UL24 gene of SuHV-1 encodes a nuclear-localized viral protein and acts as a minor virulence-associated factor compared to the TK gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Suid herpesvirus 1 (SuHV-1) is the causative agent of Aujeszky’s disease, and belongs to Alphaherpesvirinae subfamily under the Herpesviridae family [1]. Its infection results in severe nervous system disorders and high mortality in newborn piglets and reproductive failure in sows [2]. Since late 2011 in China, SuHV-1 variants emerged in Bartha-K61 vaccinated pig farms, showed enhanced virulence to pigs, and thus caused huge economic losses [3,4,5,6].
The genome of SuHV-1 is composed of a linear double-stranded DNA molecule, that is approximately 145 kb in length encoding more than 70 genes [7], and all identified genes are conserved among herpesviruses and have their homologs in one or more related alphaherpesviruses [8]. Among them the UL24 gene has been identified in all known Herpesvirales genomes with except for the channel catfish virus [9]. The high conservation suggests that UL24 gene may play an important role in the life cycle of herpesviruses.
Previous reports have demonstrated that UL24 is an important virulence-associated protein in several herpesviruses. For example, the UL24-deficient mutants of herpes simplex virus 1 (HSV-1) replicated less efficiently than wild-type in vitro and in vivo, and was severely impaired for reactivation from latency in a mouse model of ocular infection [10]; meanwhile UL24-deficient HSV-1 exhibited a syncytial plaque phenotype in cell culture, which was especially prominent at elevated temperature (39 °C) [11]. Study in equine herpesvirus 1 (EHV-1) indicated that the UL24 homolog is a neuropathogenicity determinant of EHV-1 in the mouse encephalitis model [12]. Other research with herpes simplex virus 2 has shown that UL24 gene is a virulence determinant in murine and guinea pig disease models [13]. However, understanding the role of SuHV-1 UL24 gene in viral pathogenesis has lagged behind the similar studies performed on other herpesvirus counterparts.
As a novel genome editing tool, the CRISPR/Cas9 system was first applied to knockout target genes in cells and also animals [14, 15]. Recently, the CRISPR/Cas9 system has been further applied to engineering the genomes of different viruses, such as human immunodeficiency virus [16], HSV-1 [17], and SuHV-1 [18, 19], and has shown to be very simple and efficient method for generating mutant virus.
Here, we generated a series of SuHV-1 UL24 mutants by CRISPR/Cas9 system and homologous recombination for the first time. We identified that UL24 protein was expressed late during SuHV-1 infection and strictly located in nucleus via heterologous expression. Moreover, either UL24 or TK deletion could significantly impair virus replication and spread capacity in Vero cells. In vivo studies showed that deletion of UL24 leaded to a delay of lethal time in mice but maintained a certain content of lethality in mice, by contrast TK-deleted viruses completely lost their lethality to mice. Further neurovirulence of UL24-deleted virus was not significantly affected compared to the parental virus. In comparison, infection with the TK-deleted viruses resulted in significantly reduced neurovirulence. Finally, we conclude that UL24 is a nuclear-localized viral protein and acts as a minor virulence-associated protein compared to the TK gene.
Materials and methods
Viruses and cells
African green monkey kidney (Vero) cells and HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. Cells infected with virus were grown in DMEM containing 2% fetal bovine serum. SuHV-1 JS-2012 (GenBank Accession No. KP257591) is a variant suid herpesvirus 1 isolated in China and maintained in our laboratory [20], which was propagated and titered on Vero cells and used as the parental virus for this study.
Construction of SuHV-1 mutants
As shown in Fig. 1a, for construction of SuHV-1 ΔUL24 virus, firstly the upstream and downstream regions flanking the UL24 gene and the enhanced green fluorescence protein (eGFP) cassette were PCR amplified from SuHV-1 JS-2012 genomic DNA and pEGFP-C3 vector using primers Up-UL24-F&R, Low-UL24-F&R and GFP-UL24-F&R (Table 1) respectively, and cloned into the pBluescript II SK (+) vector through Gibson assembly. Then the above recombinant plasmid (2 μg) and JS-2012 genomic DNA (3 μg) were co-transfected into Vero cells. At 3 days post transfection, the supernatants of transfected cells were harvested and prepared for viral plaque screening. The presence of recombinant virus was confirmed by the appearance of cytopathic effect (CPE) with expression of eGFP, and plaque purified three times from Vero cells and stored until required.

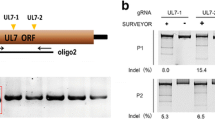
Schematic diagram of the strategies for the construction of each SuHV-1 mutant. a An overview of the construction of SuHV-1 ΔUL24 and UL24-HA viruses by homologous recombination and homologous recombination/Cas9 respectively. b Diagram showing the strategy of CRISPR/Cas9-induced deletion in UL24 gene (ΔUL24:122), TK gene (ΔTK:247) and both of them (ΔUL24:TK)
To obtain the SuHV-1 UL24-HA virus, the sequence containing UL24 ORF with an in-frame HA tag and UL24 upstream and downstream region were PCR-amplified using primers UL24-upstream-HA-F&R and UL24-downstream-HA-F&R, and further cloned into pBluescript II SK (+) by Gibson assembly, which was then co-transfected with the genome of SuHV-1 ΔUL24 virus and CRISPR/Cas9 vector targeting eGFP designed as previously described [21]. Plaques exhibiting no fluorescence were picked under a fluorescence microscope, and further validated by PCR and sequencing.
To construct the SuHV-1 ΔUL24:122 virus, we performed viral genome editing using the CRISPR/Cas9 system (Fig. 1b). The oligos UL24-gRNA-1-F&R and UL24-gRNA-2-F&R for generating guide RNAs (gRNA) were designed by Jack Lin’s CRISPR/Cas9 gRNA finder (http://spot.colorado.edu/~slin/cas9.html) and shown in Table 1. Then the oligos UL24-gRNA-1-F&R and UL24-gRNA-2-F&R were annealed and phosphorylated and then cloned into BsmBI digested plentiCRISPRv1 vector respectively. Vero cells were transfected with the above dual Cas9/gRNA plasmids and JS-2012 genomic DNA. At 3 days post-transfection, the supernatants of transfected cells were harvested and added to Vero cells for plaque screening. Then virus plaques were selected randomly and inoculated into newly plated Vero cells. One day later, viral genomic DNA was extracted by using TIANamp Virus DNA/RNA Kit (Tiangen Biotech, Beijing, China). Subsequently, PCR validation was performed using specific primers UL24-F&R in Table 1. The validated UL24 mutant was then plaque purified five times on Vero cells and stored until required.
To obtain the SuHV-1 ΔTK:247 strain, the oligos sgRNA-TK-1-F&R and sgRNA-TK-2-F&R shown in Table 1 were designed as previous studies [18, 22], and generated two sgRNAs for cleaving TK gene via the CRISPR/Cas9 system (Fig. 1b). Then the constructed pLentiCRISPRv1-gRNA plasmids were co-transfected with JS-2012 DNA into Vero cells. The rescued viruses then were harvested and added to Vero cells for plaque screening. Viral genomic DNA of each plaque was extracted as above. PCR validation was performed using specific primers TK-F&R in Table 1. The validated TK mutant was then plaque purified five times on Vero cells and stored until required. For double deletion of UL24 and TK from SuHV-1 JS-2012, the Cas9/gRNA plasmids for constructing SuHV-1 ΔUL24:122 virus were co-transfected with the genome of SuHV-1 ΔTK:247 (Fig. 1b), and UL24 and TK double deletion SuHV-1 (ΔUL24:TK) were obtained by plaque screening and PCR validation as described above.
Then all above constructed viruses and JS-2012 were inoculated into Vero cells at MOI = 1. After 12 h post infection (hpi), cells were harvested and lysed in RIPA lysis buffer (Beyotime, China) with 1 × protease inhibitor cocktail for 30 min on ice. The protein supernatants were then subjected to SDS–PAGE and Western blot. And the UL24 protein was detected by a mouse polyclonal antibody against UL24 protein, the viral VP5 protein and cellular β-actin were detected by a mouse polyclonal antibody against VP5 and a mouse monoclonal antibody (Sigma-Aldrich, Saint Louis, MO) against β-actin respectively and used for loading controls.
TK activity examination
The confluent monolayer of Vero cells was treated with 10 μM brivudin. Then 1 h later cells were infected with each virus at MOI = 1. Cell supernatants were collected at 12 and 24 hpi for virus titer determination.
Animal experiments
To study the contribution of UL24 to the pathogenicity of SuHV-1 in mice, a total of 90 6-week-old female BALB/c mice were randomly divided into nine groups in average. In group 1–3 mice were inoculated intramuscularly with 100 μl 104 50% tissue culture infective dose (TCID50) of SuHV-1 JS-2012, ΔUL24:122, and DMEM (Control group) respectively. While in group 4–8 mice were inoculated intranasally with 10 μl 104 TCID50 of SuHV-1 JS-2012, UL24-HA, ΔUL24:122, ΔTK:247 and ΔUL24:TK under general ether anesthesia, respectively; mice in group 9 were inoculated with an equivalent volume of DMEM under anesthesia as above and constituted the control group. Clinical signs of mice and number of deaths of above groups were monitored daily after infection.
To analyze the viral replication and spread in the process of acute infection, mice were inoculated intranasally with 10 μl 104 TCID50 of SuHV-1 JS-2012, ΔUL24:122, ΔTK:247 and ΔUL24:TK under anesthesia as above. Then mice were sacrificed at 3 days post infection. Mice nasal mucosa, trigeminal ganglia, brain stem, olfactory bulb, cerebrum, and cerebel were harvested and frozen at − 80 °C. Tissues were then thawed, homogenized, and frozen again. Then homogenates were thawed, sonicated, centrifuged, and titered for infectious virus by TCID50 assay on Vero cell monolayers.
Meanwhile, viral DNA from the above homogenized trigeminal ganglion of each mice was extracted as above method and used for conventional PCR and qPCR detection. The primers IE-F&R in Table 1 were adopted here for conventional PCR detection. PCR reaction conditions were pre-denaturation at 95 °C for 2 min, denaturation at 95 °C for 15 s, and annealing at 60 °C for 30 s for 35 cycles. And the primers gB-F&R in Table 1 were used for SYBR green qPCR. The reaction conditions were pre-denaturation at 95 °C for 2 min, denaturation at 95 °C for 5 s, annealing at 60 °C for 30 s, and collection of fluorescence signals for 40 cycles. Trigeminal ganglia from uninfected mice were used for DNA extraction and then PCR, and served as negative control.
UL24 protein detection and sub-cellular location
To detect the expression of UL24 protein during SuHV-1 infection, Vero cells were infected with SuHV-1 UL24-HA at MOI = 1 and the infected cells were lysed using RIPA lysate and protease inhibitor cocktail as above at 4, 8, and 12 hpi. After shaking for 30 min on ice, the cell lysate was centrifuged at 4 °C 10, 000 rpm/min for 5 min. Protein supernatants were collected for Western blot assay. The UL24-HA protein was detected by an anti-HA tag mouse monoclonal antibody (Sigma-Aldrich, Saint Louis, MO), gE was detected by a mouse monoclonal antibody in our lab, and β-actin was also detected by a mouse anti-β-actin monoclonal antibody (Sigma-Aldrich, Saint Louis, MO).
To investigate the sub-cellular localization of UL24, the UL24 gene was amplified using primers XhoI-UL24-F and Sal-UL24-R in Table 1 and ligated into the pEGFP-C3 vector to obtain a plasmid expressing UL24 protein. The recombinant plasmid and pEGFP-C3 empty vector were transfected into HeLa cells for 24 h. After dyeing the nucleus of transfected HeLa cells with DAPI, the sub-cellular location of UL24 protein and eGFP protein in the transfected cells was observed under a confocal microscope.
Results
Construction and identification of UL24-related mutant viruses
To obtain UL24 and TK deletion strains, we designed 4 specific gRNAs to cleave the UL24 and TK genes as shown in Fig. 2a. After the cleavage of Cas9/gRNA targeting UL24 or TK, SuHV-1 JS-2012 was deleted 122 bp within UL24 gene or 247 bp within TK gene (Fig. 2b, c), thereby generating the SuHV-1 ΔUL24:122, ΔTK:247 and ΔUL24:TK virus, meanwhile RFLP analysis indicated that the RFLP patterns of the CRISPR/Cas9-generated viruses were similar to JS-2012 (Fig. 2d). In addition, the SuHV-1 ΔUL24 virus and SuHV-1 UL24-HA virus were confirmed by observing the presence or absence of green fluorescence in CPE produced by the viruses (Fig. 2e), and further the specific PCR assay (Fig. 2f). Moreover, Western blot analyses demonstrated that the UL24 protein could not be detected in the extracts from cells infected with SuHV-1 ΔUL24:122 or ΔTK:247 and ΔUL24:TK (Fig. 2g).

CRISPR/Cas9 and homologous recombination mediated mutation of the corresponding genes in SuHV-1 JS-2012 genome. a The binding sites (Sequences in red box) of sgRNAs in the SuHV-1 genome (GenBank accession No. of SuHV-1 JS-2012: KP257591.1). b The genomic DNA of JS-2012, UL24- or TK-deleted strains were extracted and analyzed by PCR for the identification of the deletion of UL24 or TK respectively. c Sequence flanking the cleavage sites of SuHV-1 genome. The loci flanking the deleted sequence region was pointed by arrow and annotated with its location in the SuHV-1 genome. dBamHI-based RFLP analysis of SuHV-1 JS-2012, ΔUL24:122, ΔTK:247 and ΔUL24:TK. The genomic DNA of each virus were extracted respectively and digested with BamHI-HF (NEB) at 37 °C for 3 h. The digested mixtures were then resolved by agarose-gel electrophoresis and visualized under ultraviolet light. e Identification of SuHV-1 ΔUL24 and UL24-HA strains by visualizing CPE using a fluorescence microscope at a magnification of 200×. f Identification of SuHV-1 ΔUL24 and UL24-HA strains by specific PCR. g The identification of UL24 protein expression during each virus infection by Western blot
TK gene activity assay
Brivudin is a highly selective antiviral agent of herpesviruses. The selective activity of brivudin against herpesviruses depends on its specific phosphorylation by virus-encoded thymidine kinase (TK), and thereby exert antiviral activity [23].
Here under the treatment of brivudin the titer of SuHV-1 JS-2012 was not significantly affected at 12 hpi, but significantly inhibited at 24 hpi (Fig. 3a). The titers of SuHV-1 ΔUL24:122 and UL24-HA were also significantly suppressed at 24 hpi under treatment of the drug (Fig. 3b, c). However, the virus titer of SuHV-1 ΔTK:247 was not significant changed at either timepoint under treatment of brivudin (Fig. 3d), which proved that TK protein activity during the period of SuHV-1 ΔTK:247 infection was significantly impaired.

Effect of Brivudin against SuHV-1. Vero cells were treated with 10 μM Brivudin methanol solution and then were infected with each virus 1 h later. After 12 and 24 hpi, the supernatants were harvested and titrated. The average values of duplicates and standard deviations were shown. All statistical analyses were performed using GraphPad Prism software with Student’s t-test, *p < 0.05, ns was referred to no significance
In vitro and in vivo characteristics of UL24 and TK mutant viruses
For in vitro analysis, one-step growth curve indicated that either UL24 or TK deletion from SuHV-1 could significantly decrease virus titer in Vero cells (Fig. 4a), as indicated also by a significantly smaller plaque size (Fig. 4b). By contrast, both the virus titer and plaque size of JS-2012 and UL24-HA were almost identical in Vero cells (Fig. 4a, b). In addition, the UL24 defective strains of HSV-1 can induce Vero cells to produce syncytia-like CPE. However, UL24-deleted strain of SuHV-1-infected Vero cells can not induce syncytia-like CPE either at higher temperature (39 °C) or at lower temperature (37 °C). Instead of syncytia, it produced the same CPE as JS-2012 (Fig. 4c). For in vivo analysis, the survival curve of mice in intramuscular inoculation model showed that the mortality (4/10) of ΔUL24:122 inoculated group was significantly reduced, decreasing by 60% compared to 100% (10/10) in the JS-2012 group (Fig. 4d). However in the intranasal inoculation model all the mice infected with SuHV-1 JS-2012, UL24-HA, and ΔUL24:122 died within 5 days of infection, with only the time to death of the ΔUL24:122-infected mice being delayed by 1 day. In contrast the mice infected with SuHV-1 ΔTK:247 and ΔUL24:TK viruses survived and did not exhibited any clinical signs, indicating that UL24 is a minor virulence factor for PRV infection in mice compared to TK (Fig. 4e).

Characterization of UL24 or TK mutants in vitro and in vivo. a One-step growth curves. Vero cells were infected with each virus at MOI = 1, then cell culture supernatants were harvested at 4, 8, 12, 24, 36, and 48 hpi. Virus titers were determined by TCID50 assay. The mean titers corresponded to the averages of duplicates. Two-way ANOVA was used for analyzing the data, **p < 0.01. b Plaque sizes of SuHV-1 JS-2012, UL24-HA, ΔTK:247, ΔUL24:122, and ΔUL24:TK viruses in Vero cells cultured at 37 °C for 4 days. Relative plaque diameter from 5 randomly selected plaques of each virus was calculated and compared to those of SuHV-1 JS-2012; the average plaque diameter of SuHV-1 JS-2012 was set as 1.0. And one-way ANOVA was used for analyzing the data, **p < 0.01, ns was referred to no significance. c Plaque morphology of each virus in Vero cells. Shown were images of Vero cells infected with SuHV-1 JS-2012, ΔUL24, and ΔUL24:122 grown at 37 °C (left), 39 °C (right). d Survival percentages of mice inoculated intramuscularly with 104 TCID50 of SuHV-1 JS-2012 and ΔUL24:122 viruses. Statistical significance between survival curves was evaluated by the log-rank (Mantel-Cox) test. p value < 0.05 was considered statistically significant, ***p < 0.001. e Survival percentages of mice intranasally inoculated with 104 TCID50 of each virus
Comparison of acute infection capability of SuHV-1 JS-2012 and mutant viruses
To study the acute infection capability of each virus, viral replication in nasal mucosa and brain tissues of mice infected with 104 TCID50 of SuHV-1 JS-2012, ΔUL24:122, ΔTK:247, and ΔUL24:TK viruses were assessed. The virus titer of JS-2012 and ΔUL24:122 could be detected in nasal mucosa, trigeminal ganglion, and cerebel, and which of JS-2012 showed a little higher than that of ΔUL24:122 in all the three tissues (Fig. 5). Meanwhile the infectious virus of JS-2012 was also detected in brain stem, olfactory bulb, and cerebrum with a small amount, while infectious virus of ΔUL24:122 was not detectable in these tissues (Fig. 5). In contrast, infectious viruses of ΔTK:247 and ΔUL24:TK were not detected in all tested tissues (Fig. 5). Thus, the deletion of UL24 affects the replication of the virus in the epithelial tissue and the replication in the brain tissue, while the loss of TK can completely eliminate the ability of the virus to replicate and spread infection in animals.

Acute infection in mice via intranasal inoculation of 104 TCID50 SuHV-1 JS-2012, ΔUL24:122, ΔTK:247, and ΔUL24:TK viruses. The data represented means ± SD for five samples per data point. Statistical analyses were performed using GraphPad Prism software with Student’s t-test, ns was referred as no significance
To further explore whether SuHV-1 could enter the trigeminal ganglion after deletion of UL24 and TK genes, viral DNA of trigeminal ganglion samples infected with JS-2012, ΔUL24:122, ΔTK:247, and ΔUL24:TK were examined. It showed that all the trigeminal ganglia of mice infected with SuHV-1 JS-2012 and ΔUL24:122 were detected with large numbers of viral DNA (more than 108 viral genome copies/μl) (Fig. 6a, b), and the DNA amount of JS-2012 showed slightly larger than that of ΔUL24:122 (Fig. 6b). In addition, viral DNA was also detected in the trigeminal ganglia of mice infected with TK-deleted viruses (Fig. 6a), but their viral DNA amount (approximately 104 viral genome copies/μl) was shown quite significantly smaller than those of JS-2012 and ΔUL24:122 (Fig. 6b).

Detection of viral DNA in trigeminal ganglion by PCR. a Lanes 1–5 corresponded to 5 trigeminal ganglion samples from JS-2012-infected mice; lanes 6–10 corresponded to 5 trigeminal ganglion samples from ΔUL24:122-infected mice; lanes 11–15 corresponded to 5 trigeminal ganglion samples from ΔTK:247-infected mice; lanes 16–20 corresponded to 5 trigeminal ganglion samples from ΔUL24:TK virus-infected mice, and lane 21 corresponded to trigeminal ganglion sample from negative control mouse. b Specific absolute qPCR was performed for detection of viral genome copies/μl in the DNA extraction solution of each trigeminal ganglion. The data were analyzed by Student’s t-test, *p < 0.05, **p < 0.01
Analysis of the characteristics of UL24 protein
To investigate the characteristics of the encoded product of SuHV-1 UL24 gene, we first analyzed the expression of UL24 protein in Vero cells infected with SuHV-1. And the analysis of cellular proteins infected by UL24-HA virus showed that a low level of UL24 protein was expressed late (12 hpi) during virus infection (Fig. 7a). Meanwhile Transient transfection of plasmid expressing UL24 protein into HeLa cells indicated that UL24 protein of SuHV-1 was localized to the nucleus (Fig. 7b).

The property and function analysis of SuHV-1 UL24 protein. a The expression of UL24 protein during SuHV-1 UL24-HA infection. The UL24-HA protein was detected by the anti-HA mouse monoclonal antibody, and the viral glycoprotein gE and cellular β-actin were also detected as loading controls. b Heterologous expression of UL24 protein in HeLa cells (magnification: 20×). The fluorescent channels of eGFP alone or eGFP-UL24 fusion protein (left) and the nucleus stained by DAPI (middle) were shown, and meanwhile their merged image was shown on the right side
Discussion
Since 2011, in China SuHV-1 variants have emerged in many vaccinated pig farms and caused huge economic losses [3]. SuHV-1 encodes about 70 different genes, many of which have been studied in vitro and in vivo for their relavent functions [8]. However, the function of UL24 gene encoded by SuHV-1 has not been studied by researchers. In addition SuHV-1 UL24 gene is partially overlapped with TK gene. For analyzing function of UL24 gene, the expression and function of TK gene should not be affected, which makes the functional analysis of SuHV-1 UL24 gene much more complicated.
In this study, the specific Cas9/gRNA system was adopted to delete UL24 or TK gene from SuHV-1 JS-2012, and thus UL24 deleted SuHV-1 (ΔUL24:122), TK-deleted SuHV-1 (ΔTK:247) and double genes deleted viruses (ΔUL24:TK) were soon obtained. Like other alphaherpesvirus, after deletion of UL24 or TK gene SuHV-1 can still replicate in cells but significantly reduced the replication and spread capacity (Fig. 4a, b), indicating that UL24 is a non-essential gene of SuHV-1. However, unlike relevant studies on HSV-1, the absence of UL24 from SuHV-1 does not induce syncytia in Vero cells (Fig. 4c), by contrast deletion of which from HSV-1 often causes typical syncytia CPE in Vero cells [11]. This finding infers that the UL24 gene of SuHV-1 has lost some functional capacity compared to the HSV-1 homolog during SuHV-1 evolution.
In vivo studies showed that UL24-deleted virus exhibited a certain degree of reduced virulence in mice, but still maintained lethality to mice; by contrast after TK deletion, the virus is not pathogenic at all in mice (Fig. 4d, e). Furthermore, TK-deleted viruses could not be detected in virus-infected mice during acute infection phase (Fig. 5) and only low amounts of viral genomic DNA could be detected (Fig. 6), indicating that TK-deleted virus can infect the trigeminal ganglia but lose the ability to replicate or spread in trigeminal ganglia. In contrast, viruses with the UL24 gene deletion could infect the nasal mucosa and trigeminal ganglia, although the resulting viral titers and detected viral genome copy numbers were reduced compared to that of SuHV-1 JS-2012 (Figs. 5, 6). Therefore, the UL24 gene contributes to the virulence of the virus in mice to a certain extent; in comparison TK is the key virulence determinant of SuHV-1 in mice.
However, although the CRISPR/Cas9 system in our study is a very powerful genome-engineering tool and specificity of Cas9 is strictly controlled by gRNA and the PAM motif, it should be noted that potential off-target cleavage could still occur on viral DNA sequence with several base pair mismatches in the PAM-distal part of the gRNA-guiding sequence [24]. Therefore, strategies for minimizing off-target effects or setting of the appropriate control (revertant) group should also be considered in further studies that are related to manipulation of viral genomes by CRISPR/Cas9 system.
Ultimately, we also investigated the properties and functions of SuHV-1 UL24 protein. Western blot showed that UL24 protein can be expressed during viral infection, while the expression of which was late during SuHV-1 infection. Meanwhile heterologously expressed UL24 can be localized into the nucleus, indicating that UL24 gene of SuHV-1 can encode a viral nuclear protein.
In conclusion, SuHV-1 UL24 protein was expressed late during virus infection and localized in the nucleus. Deletion of UL24 gene resulted in significantly impaired replication and spread capacity in Vero cells but slightly reduced lethality in mice, by contrast deletion of TK gene completely loses lethality to mice. Further the neurovirulence of UL24-deleted virus was not affected significantly compared to the parental virus, while which of TK-deleted viruses were significantly affected and completely lost. Hence, SuHV-1 UL24 gene encodes a nuclear-localized viral protein and acts as a minor virulence-associated gene compared to the TK gene.
References
Pellett PE, Davison AJ, Eberle R, Ehlers B, Hayward GS, Lacoste V et al (2011) Herpesvirales. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (eds) Virus taxonomy: Ninth report of the international committee on taxonomy of viruses. Elsevier Academic Press, London, p 99–107
Pejsak ZK, Truszczyni MJ (2006) Truszczynsk: Aujeszky’s disease (pseudorabies). In: Straw BE, Zimmerman JJ, D’Allaire S, Taylor DJ (eds) Diseases of swine, 9th ed. Blackwell Publishing Ltd, Ames, p 419–433
An TQ, Peng JM, Tian ZJ, Zhao HY, Li N, Liu YM et al (2013) Pseudorabies virus variant in Bartha-K61-vaccinated pigs, China. Emerg Infect Dis 19:1749–1755
Luo Y, Li N, Cong X, Wang CH, Du M, Li L et al (2014) Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet Microbiol 174:107–115
Yu X, Zhou Z, Hu D, Zhang Q, Han T, Li X et al (2014) Pathogenic pseudorabies virus, China, 2012. Emerg Infect Dis 20:102–104
Ye C, Zhang QZ, Tian ZJ, Zheng H, Zhao K, Liu F et al (2015) Genomic characterization of emergent pseudorabies virus in China reveals marked sequence divergence: evidence for the existence of two major genotypes. Virology 483:32–43
Klupp BG, Hengartner CJ, Thomas C, Enquist LW, Mettenleiter TC (2004) Complete, annotated sequence of the pseudorabies virus genome. J Virol 78:424–440
Pomeranz LE, Reynolds AE, Hengartner CJ (2005) Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500
Bertrand L, Leiva-Torres GA, Hyjazie H, Pearson A (2010) Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J Virol 84:109–118
Jacobson JG, Chen SH, Cook WJ, Kramer MF, Coen DM (1998) Importance of the herpes simplex virus UL24 gene for productive ganglionic infection in mice. Virology 242:161–169
Jacobson JG, Martin SL, Coen DM (1989) A conserved open reading frame that overlaps the herpes simplex virus thymidine kinase gene is important for viral growth in cell culture. J Virol 63:1839–1843
Kasem S, Yu MHH, Yamada S, Kodaira A, Matsumura T, Tsujimura K et al (2010) The ORF37 (UL24) is a neuropathogenicity determinant of equine herpesvirus 1 (EHV-1) in the mouse encephalitis model. Virology 400:259–270
Blakeney S, Kowalski J, Tummolo D, DeStefano J, Cooper D, Guo M et al (2005) Herpes simplex virus type 2 UL24 gene is a virulence determinant in murine and guinea pig disease models. J Virol 79:10498–10506
Mali P, Esvelt KM, Church GM (2013) Cas9 as a versatile tool for engineering biology. Nat Methods 10:957–963
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Ebina H, Misawa N, Kanemura Y, Koyanagi Y (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep 3:2510
Bi Y, Sun L, Gao D, Ding C, Li Z, Li Y et al (2014) High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog 10:e1004090
Liang X, Sun L, Yu T, Pan Y, Wang D, Hu X et al (2016) A CRISPR/Cas9 and Cre/Lox system-based express vaccine development strategy against re-emerging Pseudorabies virus. Sci Rep 6:19176
Xu A, Qin C, Lang Y, Wang M, Lin M, Li C et al (2015) A simple and rapid approach to manipulate pseudorabies virus genome by CRISPR/Cas9 system. Biotechnol Lett 37:1265–1272
Tong W, Liu F, Zheng H, Liang C, Zhou YJ, Jiang YF et al (2015) Emergence of a pseudorabies virus variant with increased virulence to piglets. Vet Microbiol 181:236–240
Auer TO, Duroure K, Concordet JP, Del Bene F (2014) CRISPR/Cas9-mediated conversion of eGFP-into Gal4-transgenic lines in zebrafish. Nat Protoc 9:2823–2840
Tang YD, Liu JT, Wang TY, An TQ, Sun MX, Wang SJ et al (2016) Live attenuated pseudorabies virus developed using the CRISPR/Cas9 system. Virus Res 225:33–39
De Clercq E (2005) Antiviral drug discovery and development: where chemistry meets with biomedicine. Antiviral Res 67:56–75
Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4:e264
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2016YFD0500100), China Agriculture Research System (NYCYTX-009), and Shanghai Municipal Agriculture Science and Technology Key Project (2015, 1-7 and 2016, 4-2).
Author information
Authors and Affiliations
Contributions
CY and JC conducted the most experiments and drafted the manuscript. GT critically revised the manuscript. XC, SZ, SJ, JX, HZ, WT, GL helped with the experiments. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no confict of interest.
Ethical approval
All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Research involving human and animal participants
The animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals of Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China. The animal ethics approved number was SHVRI-MO-2017070588.
Additional information
Communicated by William Dundon.
Rights and permissions
About this article

Cite this article
Ye, C., Chen, J., Cheng, X. et al. Functional analysis of the UL24 protein of suid herpesvirus 1. Virus Genes 55, 76–86 (2019). https://doi.org/10.1007/s11262-018-1619-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1619-3