
Abstract
Hepatitis B virus (HBV) poses a significant threat to blood transfusion safety in sub-Saharan Africa (SSA) where allogeneic blood donations are screened serologically, and more sensitive nucleic acid tests (NATs) are utilized infrequently. HBV strains circulating among blood donors in Botswana are not yet characterized. We designed a cross-sectional study to determine the HBV sub-genotypes and prevalence of hepatitis B surface antigen (HBsAg) among blood donors between November 2014 and October 2015. A total of 12,575 blood donations were screened for HBsAg and 50 consecutive plasma samples were selected for genotyping from confirmed HBsAg+ donations. Overlapping Pol and complete S (Pol/S) open reading frames (ORFs) were sequenced from extracted HBV DNA. To identify any signature amino acids, mutations were compared to sequences from a cohort of chronic HBV patients co-infected with HIV and were treatment naïve. The prevalence of HBsAg+ blood donors was 1.02% (95% CI 0.9–1.2%), and the circulating sub-genotypes were A1 serotype adw2 (36.1%), D2 serotype ayw2 (2.9%), and D3 serotypes ayw 1/2 (58.3%). Prevalence of escape mutations was 14% from HBV isolates of blood donors and 15% from isolates of HBV/HIV co-infected patients (p = 0.6926). The escape mutations sP120L, sG130R, sY134H, and sD144A were identified predominantly among HBV isolates from blood donors. These escape mutations have been associated with accelerated HBV sequelae [e.g., liver cirrhosis (LC) and hepatocellular carcinoma (HCC)], failure to detect HBsAg, inability to respond to immunoglobulin (Ig) therapy, and HBV vaccine escape. Characterizing the HBV burden, circulating sub-genotypes, and clinically relevant mutations among blood donors in Botswana is important to elucidate the efficacy of currently available vaccines, predicting HBV-transmission patterns, understanding the cohort’s risk to HBV-related complications, and to developing prevention strategies and effective genotype-based antiretroviral therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatitis B virus (HBV) infection is a significant global health problem with approximately 2 billion people with positive hepatitis B core antibody [anti-HBc+] confirming exposure at some point in their lives. One-eighth (257 million) of these have a chronic infection, and only a fraction is aware of their serostatus [1]. With an HBV prevalence of ≥ 8%, sub-Saharan Africa (SSA) is considered a region of high HBV endemicity [2,3,4]. HBV is transmitted through contact with infected blood or body fluids, including horizontal transfer (e.g., from sexual intercourse) and vertical transfer (e.g., from mother to child). Preventative strategies for HBV infection include testing of blood for active HBV infection and vaccination against HBV. Despite stringent guidelines on blood safety to exclude potential donors at high risk of transfusion-transmitted infection (TTIs), post transfusion-transmitted HBV infection is a major concern in resource-limited countries. In SSA, the median risk of being infected with HBV from a blood transfusion has been estimated at 4.3 infections per 1000 units, and 28,595 HBV infections per year are estimated to occur [5,6,7]. The transmission risk of HBV remains even after HBV NAT has been performed on each donated blood unit [8].
Botswana is one of the 46 African countries that serologically tests all blood donations for the four mandatory TTIs [human immunodeficiency virus (HIV)], HBV, hepatitis C virus (HCV), and syphilis [9]. In the 2000/2004 and 2010/2011 reports by the Centers for Disease Control and Prevention (CDC), the HBV prevalence among blood donors in Botswana was 4.21% and 2.21%, respectively [10]. Therefore, it is crucial to conduct routine epidemiological surveillance to obtain current data on HBV to help policy-makers improve HBV elimination strategies toward the 2030 HBV elimination goal.
HBV is a circular DNA virus belonging to the genus Orthohepadnavirus of the family Hepadnaviridae. It has four partial overlapping open reading frames (ORFs)—P, C, X, and S—that encode for seven proteins, including polymerase, precore, core, X protein, and three (L, M, and S) surface proteins [11]. The HBV surface antigen (HBsAg) (226 aa) includes the ‘a’ determinant region (aa 124–147) within the major hydrophilic region (MHR) which is targeted by antibody-based assays to screen for chronic HBV and elicits antibodies which confer protection against HBV infection.
Major mutations within the S region result in viral escape from vaccination, immunotherapy, and diagnosis. Since the P ORF overlaps the S ORF, variability within P can affect the aa sequence of the HBsAg and vice versa. Major mutations in the polymerase may lead to drug resistance [12, 13].
HBV consists of multiple genotypes (A–J) with genetic variability that differ by > 8%. Some genotypes can be further divided into sub-genotypes that differ by > 4% [13, 14]. Genotypes frequently occur in specific geographic locations and represent an important determinant of treatment response and disease progression [11]. Despite the high endemicity of HBV infection in Botswana, few studies have reported the burden of HBV among HIV co-infected patients [15,16,17]; however, there are no data on distribution of circulating sub-genotypes of the virus in HBV mono-infected including blood donors. Moreover, there is still a gap in understanding the mutation profiles of HBV isolates of HBV mono-infected compared to HBV/HIV co-infected patients. This study sought to determine the burden of HBsAg positivity among blood donors in Botswana and to identify circulating HBV sub-genotypes and clinically relevant mutations.
Methods and materials
Study design and study population
This is a cross-sectional prospective study utilizing 12,575 specimens from blood donors collected by Botswana National Blood Transfusion Service (NBTS) in Gaborone, between November 2014 and October 2015 as part of the routine blood donation screening. A subset of 50 anonymous first-time blood donors were consecutively ELISA-screened using Murex HBsAg version 3 kit [Murex Biotech, Dartford, UK] and further confirmed using Monolisa HBsAg ULTRA [Bio-Rad, France]. ELISA tests were performed on individual (rather than pooled) blood donations. The HBsAg+ confirmed samples were evaluated for HBV sub-genotypes and clinically relevant mutations. Demographic data including age and gender were obtained. Additionally, data on HIV, HCV, and syphilis seropositive samples were available for investigation. The study was approved by the University of Botswana Institute Review Board and the Health Research Development Division (HRDD) at the Botswana Ministry of Health and Wellness.
Polymerase chain reaction (PCR) and sequencing
Total nucleic acid was extracted from 1000 µl of subjects’ sera using the Qiagen UltraSens kit (QIAGEN, Hilden, Germany). An elution volume of 50 µl was used and extracts were stored at − 80 °C until use. For HBV DNA amplification, the master mix was prepared from the Superscript III Platinum One-Step kit (Invitrogen, USA) with a modification of the number of cycles to 35 and template volume of 10 µl per each reaction. We amplified a 2100 bp fragment covering nucleotides 2400–1150 according to the numbering of the EcoRI that included the full S gene overlapping with part of Pol using HBV primers Core-F and Werle-AS as previously described [15]. The PCR conditions included preheating at 95 °C for 2 min, denaturing at 95 °C for 30 s, annealing at 62.5 °C for 30 s, and extension at 72 °C for 4 min. Amplicons were confirmed on 1% agarose gel and purified using QIAquick (Qiagen, Hilden, Germany). The BigDye Terminator v3.0 kit (Applied Biosystems; Foster City, CA, USA) was used for sequencing. Six overlapping primers were used to prepare separate master-mixes for PCR. The thermocycling conditions were denaturing at 96 °C for 10 s, annealing at 50 °C for 5 s, and final extension at 60 °C for 4 min for 25 cycles. Direct sequencing was performed using the automated Sequencer (ABI PRISM 3130xl; Applied Biosystems).
Phylogenetic analysis
Electropherograms were manually edited using Sequencher v5.0 software (Gene Codes Corp., Ann Arbor, MI, USA) [18]. Alignments were performed using Clustal X v.2.1 [19] and representative references of genotypes A–H retrieved from GenBank. Additional phylogenetic inference was performed using the Bayesian Markov chain Monte Carlo (MCMC) approach implemented in the Bayesian Evolutionary Analysis with the Sampling Trees software (BEAST v1.8.4) [20] with an uncorrelated log-normal relaxed molecular clock, General Time Reversible (GTR) substitution model, and gamma site heterogeneity. The MCMC was set at a chain length of 100,000,000 with parameters logged every 10,000. The tree was visualized in FigTree v1.4.3 after a 10% burn-in using Tree Annotator v1.8.4. Posterior probabilities 0.90 and above were noted as statistically significant. The Stanford Drug Resistant Database, Geno2pheno available at http://hbv.geno2pheno.org/, and the small genome tools available at http://hvdr.bioinf.wits.ac.za/SmallGenomeTools/ were used to confirm the coverage of amplified (Pol, PreS1, PreS2, and S) genes, the quality of sequences (QA), genotypes and sub-genotypes, serotypes, drug resistance and escape mutations, and translated ORFs, respectively. The sequences from this study of blood donors are available in GenBank under accession numbers MF979142-MF979176.
Recombination and serotype analysis
All sequences in the study were assessed for recombination using the jumping profile Hidden Markov Model (jpHMM) tool available at http://jphmm.gobics.de/submission_hbv [21] and HBV serotypes were predicted based on the S gene aa positions: Lys/Arg → 122, Arg/Lys → 160, 127 → Pro/Thr/Ile-Leu, Ala → 159 or not Ala, and Ser → 140 or not Ser using the web-based tool [22] available at http://hvdr.bioinf.wits.ac.za/serotyper/.
Mutation analysis and signature amino acids
For mutation analysis, aligned sequences were compared per genotype and ORF: PreS1 genotype A versus D, PreS2 (A vs. D), S (A vs. A), and Pol (A vs. D), respectively. Comparisons were done at aa level in order to remove any synonymous mutations that have no effect on the HBV phenotype. Profiles and frequency of significant (non-synonymous) mutations obtained in the study were compared to those of HBV isolates from chronic HBV patients co-infected with HIV (HBV/HIV) and were treatment naïve. The cohort of HBV/HIV co-infected treatment naïve patients has been previously reported in Botswana [15]. The comparison was done to determine genetic diversity and any signature mutations of HBV isolates that might be found in chronic HBV/HIV co-infection patients versus HBV mono-infection. Signature mutations associated with HIV/HBV co-infection were assessed using the viral epidemiology signature pattern analysis (VESPA) tool [23].
Statistical analysis
Stata version 14 (Stata Corp, College Station, TX, USA) was used for statistical analysis. The non-parametric Wilcoxon’s test was used to analyze different mutation frequencies in the Pol/S regions. Fischer’s exact test was used for categorical data.
Results
Patient characteristics
During the period of November 2014 and October 2015, 128 of 12,575 (1.02%) (95% CI 0.9–1.2%) blood units donated to the blood bank were HBsAg+ and 50 (39.1%) specimens from the HBsAg+ were selected randomly for additional analysis. The majority of blood donors were males (67.7%), and the median age was 32 (Q1; Q3:28; 35) years. Males were significantly older (p < 0.01) than females with median age of 28 (Q1, Q3:26, 37) years (Fig. 1).

Age–sex index graph showing frequency distributions for the study participants. The male sex–age index is normally distributed and age range 30–34 years has highest HBV burden
Prevalence of HBsAg among blood donors and other TTIs
Relative to other TTIs which had the prevalence of 2.06% (258/12,548; 95% CI 1.81–2.32%) for syphilis; 1.58% (201/12,719; 95% CI 1.37–1.81%) for HIV; and 0.41% (52/15,744; 95% CI 0.30–0.53%) for HCV, HBV was ranked the second lowest at (1.02%).
Genotypes and recombination
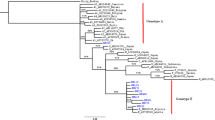
HBV DNA could be amplified from 36 of the 50 (72%) HBsAg+ samples. The phylogenetic tree showed that Botswana HBV sequences clustered with either genotype A or D regional reference sequences as shown in Fig. 2. A total of 13 of 36 (36.1%) HBV sequences from blood donors belonged to sub-genotype A1, 21 (58.3%) were sub-genotype D3, and 1 (2.9%) was sub-genotype D2. Sequence WC25 was excluded from the analysis as it was shorter than other sequences. The sequence WC5 was confirmed as a recombinant using online database but on the phylogenetic tree it clustered with high support to other genotype A sequences. This is because sequences used to construct the phylogenetic tree were trimmed to same size of 1650 base pairs, and that resulted in deletion of large fragment corresponding to genotype D. The recombination of WC5 was A1/D3 and had breakpoint at nt 531.5 ± 11.5 EcoRI shown in Fig. 3.

Phylogenetic Tree of HBV isolate sequences from blood donors supported with posterior probabilities > 0.90 shown. The HBV sequences from this study are indicted by “WC” (red for genotype A and blue for genotype D), while references are indicated by sub-genotype–accession number–country

Circular representation of WC5 showing a recombinant sequence, A1/D3, and a breakpoint at position nt 531.5 ± 11.5 EcoRI in the S protein. The graph was constructed using online database jumping profile Hidden Markov Model (jpHMM)
The distribution of genotypes between HBV sequences from HIV/HBV co-infected group and blood donors was comparable. HBV sub-genotype D3 was identified in majority of HBV sequences from blood donors 21(60%), whereas sub-genotype A1 was identified in 56 of 81 (69.1%) (p < 0.05) in the HIV/HBV co-infected group. All HBV genotype A1 sequences were serotype adw2. Apart from WC19, which was serotype HBV-ayw1, all HBV genotype D3 sequences were serotype ayw2.
Functional mutations and genetic variability
The comparison for percentage proportions for genetic diversity, and total aa differences stratified by sub-genotypes and protein of HBV sequences from HBV/HIV co-infected patients and HBV mono-infected blood donors are shown in Table 1. We compared the mutations in S gene between sequences obtained from HBV/HIV co-infected patients [15] and blood donors. Twelve mutations including diagnostic escape mutations (sY100C, sR122K, sT123A, sC124R, sM133T), vaccine escape mutations (sT126N, sQ129R, sM133L, sF134V), and immunoglobulin therapy failure (sG119R, sG130N, sT140S) were exclusive to isolates obtained from HBV/HIV patients versus n = 4 including (sP120L, sG130R, sY134H, and sD144A) (Fig. 4) obtained from some isolates from blood donors. 3 of 5 (60%) escape mutations isolated from sequences from blood donors resided within the ‘a’ determinant’s first loop (aa 124–137), 1 (20%) within the second loop (aa 138–147), and P120L was present in the mini loop outside the range (aa 124–147).

Showing consensus sequences for HBsAg per genotype; a Left: genotype A and b Right: genotype D. Escape mutations indicated for the two groups; (HIV/HBV) co-infected significant variants are shown in green and for (HBV) mono-infected variants are shown in red
Similarly, 58.3% of mutations identified in the sequences from HBV/HIV co-infected group were within the first loop. Except for the substitution at position 130 (G → R) in HBV sequences from blood donors and (G → N) in HBV/HIV co-infected individuals, there were no mutations at aa level that distinguished the two groups. There were no drug resistance mutations identified in either groups.
Discussion
This is the first study to characterize sub-genotypes and mutations in Pol/S genes among HBV mono-infected blood donors in Botswana. Our study showed that the prevalence of HBsAg+ among blood donors in Botswana decreased to 1.02% in 2014–2015 from 2.21% in 2010–2011 [24]. This decrease is expected and results from many factors including the implementation by the NBTS of stringent interventions recommended by WHO to exclude high-risk potential blood donors and HBV/HIV treatment programs. Similar trends have been observed in neighboring countries [25, 26]. However, these data may not be applicable to the general population as they excluded persons vaccinated at birth who were less than 16 years during the time of study and high-risk populations who could not donate blood [27]. HBV sub-genotypes have been associated with varied clinical outcomes and treatment efficacy [11, 28, 29]. Most HBV genotyping studies have been done among HIV/HBV co-infected cohorts and have shown that A, D, and E circulate in southern Africa, with genotype A being the most common and genotype E the least common [15,16,17, 30, 31].
This study observed that sub-genotypes A1, D2, and D3 circulate among blood donors in Botswana in agreement with other studies conducted in the country among non-blood donor cohorts [15,16,17]. To our knowledge, this is the first description of D2 and recombinant A1/D3 in our setting. Distribution of genotype D2 has been reviewed and shown to circulate in Northern Africa (Morocco, Tunisia), Europe, Asia, and the Middle East [11, 32,33,34]. It is not surprising to observe this degree of variability in genetic epidemiology in Botswana since the blood donor cohort includes diverse ethnic groups. Furthermore, this pilot study shows the current HBV burden in Botswana might be underrepresented as no study has reported HBV molecular epidemiology among HBV mono-infected including blood donors. This information may enlighten decisions on choice of regimens to be used in HBV risk groups in Botswana since HBV genotypes influence treatment responses, and this information may inspire new interventions to screen for HBV especially among blood donors. A proportion of samples could not be amplified, likely due to low viral loads [< 20 IU/ml] as previously seen in previous studies [35]. In this study, we had limited sample volumes available for HBV DNA quantification and analysis of other genomic regions. The presence of immune escape variants is of high clinical significance, as these mutants may facilitate HBV reactivation even in anti-HBs+ patients, transmission despite proper active/passive vaccination strategies, and missed diagnosis when commercial HBsAg diagnostic kits fail to detect the escape variants [36]. sD144A was the predominant mutation among genotype D sequences isolated from blood donors. Previous studies found that this mutation was associated with impaired HBsAg antigenicity, immune escape, (thereby decreasing the sensitivity of commercial HBsAg immunoassays), and escape from vaccine and immunoglobulin therapy [37]. Although it was found exclusively among HBV isolates from blood donors, the change from Asp to Glu at position s144 (D144E) has been described also in HBV isolates from HBV/HIV-infected patients [38]. Additionally, WC33 had the dual secondary escape mutations sY134H and sD144A associated with HBV reactivation. Together with sD144A they reside in the immunogenic segment (aa 139–149) and have been extensively discussed by Verheyen et al. in correlation with reactivation [38, 39]. sY134H causes a secondary co-variation with rtV142A in RT domain and is associated with failure to respond to immunoglobulin therapy in addition to HBV reactivation [39, 40]. sP120L mutation in genotype D sequences has been functionally characterized and linked with occult HBV (OBI) [41]. However, in the current study, sP120L was observed in HBV mono-infected as well. Furthermore, mutations at position 120 of the S (P120) have also been reported in several studies of HBV isolates from blood donors and other chronic HBV patients of different risk groups [42, 43]. The impact of different escape mutations found in the isolates from the HBV/HIV co-infected group has been discussed [15]. Although several studies have confirmed that there is no association between HBV infection and HIV status [17, 44], our findings suggest that the genetic diversity may differ between HBV isolates from these two groups. Except for the aa substitutions at position 130 in the S gene that overlapped in two groups, the sample size and clinical information required to establish statistical inferences of any signature mutations that may be associated with multi-virulence (HIV/HBV) compared to mono-infection were limited. The escape mutations between variants of sub-genotype A1 and D3 were not statistically significant across the S region for both cohorts (Table 1).
This pilot study focused on the volunteer blood donors and gives a more representative spectrum of the immune-pathological pattern of HBV infection in the general population than patients’ samples that would bias the results toward a specific variant of the disease, e.g., HIV/HBV co-infected [45]. In conclusion, this study shows evolutionary diversity between different HBV cohorts and highlights the need to establish mutations that correlate with HBV/HIV co-infection as opposed to HBV mono-infection. Additionally, HBV is still a significant burden in Botswana among other TTIs and the HBV molecular epidemiology might be more diverse in the general population than currently reported. Currently, Botswana is still using only serological assays to screen allogeneic blood donations. Use of NATs in addition to serological screening should be introduced at NBTS and utilized as the standard screening technique to prevent TTIs transmission through blood transfusion services. As well, routine epidemiological studies of HBV among different cohorts should be conducted to provide more robust estimates of HBV prevalence.
References
Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ (2015) Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386(10003):1546–1555. https://doi.org/10.1016/S0140-6736(15)61412-X
Spearman CW, Afihene M, Ally R, Apica B, Awuku Y, Cunha L, Dusheiko G, Gogela N, Kassianides C, Kew M, Lam P, Lesi O, Lohoues-Kouacou MJ, Mbaye PS, Musabeyezu E, Musau B, Ojo O, Rwegasha J, Scholz B, Shewaye AB, Tzeuton C, Sonderup MW, Gastroenterology, Hepatology Association of sub-Saharan A (2017) Hepatitis B in sub-Saharan Africa: strategies to achieve the 2030 elimination targets. Lancet Gastroenterol Hepatol 2(12):900–909. https://doi.org/10.1016/S2468-1253(17)30295-9
Franco E, Bagnato B, Marino MG, Meleleo C, Serino L, Zaratti L (2012) Hepatitis B: epidemiology and prevention in developing countries. World J Hepatol 4(3):74–80. https://doi.org/10.4254/wjh.v4.i3.74
Beguelin C, Fall F, Seydi M, Wandeler G (2018) The current situation and challenges of screening for and treating hepatitis B in sub-Saharan Africa. Expert Rev Gastroenterol Hepatol 12(6):537–546. https://doi.org/10.1080/17474124.2018.1474097
Jayaraman S, Chalabi Z, Perel P, Guerriero C, Roberts I (2010) The risk of transfusion-transmitted infections in sub-Saharan Africa. Transfusion 50(2):433–442. https://doi.org/10.1111/j.1537-2995.2009.002402.x
WHO (2012) Blood donor selection: guidelines on assessing donor suitability for blood donation. World Health Organization, Geneva.
WHO (2017) Blood safety and donation World Health Organisation http://www.who.int/mediacentre/factsheets/fs279/en/index.html. Accessed 04 March 2018
Vermeulen M, Swanevelder R, Chowdhury D, Ingram C, Reddy R, Bloch EM, Custer BS, Murphy EL (2017) Use of blood donor screening to monitor prevalence of HIV and hepatitis B and C viruses, South Africa. Emerg Infect Dis 23(9):1560–1563. https://doi.org/10.3201/eid2309.161594
WHO (2017) Current status on blood safety and availability in the WHO African Region—report of the 2013 survey. WHO Regional Office for Africa, Brazzaville
Apata IW, Averhoff F, Pitman J, Bjork A, Yu J, Amin NA, Dhingra N, Kolwaite A, Marfin A, Centers for Disease C, Prevention (2014) Progress toward prevention of transfusion-transmitted hepatitis B and hepatitis C infection–sub-Saharan Africa, 2000–2011. MMWR Morb Mortal Wkly Rep 63(29):613–619
Sunbul M (2014) Hepatitis B virus genotypes: global distribution and clinical importance. World J Gastroenterol 20(18):5427–5434. https://doi.org/10.3748/wjg.v20.i18.5427
Coppola N, Onorato L, Minichini C, Di Caprio G, Starace M, Sagnelli C, Sagnelli E (2015) Clinical significance of hepatitis B surface antigen mutants. World J Hepatol 7(27):2729–2739. https://doi.org/10.4254/wjh.v7.i27.2729
Locarnini SA (1998) Hepatitis B virus surface antigen and polymerase gene variants: potential virological and clinical significance. Hepatology 27(1):294–297. https://doi.org/10.1002/hep.510270144
Kramvis A (2014) Genotypes and genetic variability of hepatitis B virus. Intervirology 57(3–4):141–150. https://doi.org/10.1159/000360947
Anderson M, Gaseitsiwe S, Moyo S, Wessels MJ, Mohammed T, Sebunya TK, Powell EA, Makhema J, Blackard JT, Marlink R, Essex M, Musonda RM (2015) Molecular characterisation of hepatitis B virus in HIV-1 subtype C infected patients in Botswana. BMC Infect Dis 15:335. https://doi.org/10.1186/s12879-015-1096-4
Matthews PC, Beloukas A, Malik A, Carlson JM, Jooste P, Ogwu A, Shapiro R, Riddell L, Chen F, Luzzi G, Jaggernath M, Jesuthasan G, Jeffery K, Ndung’u T, Goulder PJ, Geretti AM, Klenerman P (2015) Prevalence and characteristics of hepatitis B virus (HBV) coinfection among HIV-positive women in South Africa and Botswana. PLoS ONE 10(7):e0134037. https://doi.org/10.1371/journal.pone.0134037
Mbangiwa T, Kasvosve I, Anderson M, Thami PK, Choga WT, Needleman A, Phinius BB, Moyo S, Leteane M, Leidner J, Blackard JT, Mayondi G, Kammerer B, Musonda RM, Essex M, Lockman S, Gaseitsiwe S (2018) Chronic and occult hepatitis B virus infection in pregnant women in Botswana. Genes (Basel). https://doi.org/10.3390/genes9050259
Sequencher® DNA Sequence Analysis Software. http://www.genecodes.com. Accessed 10 Oct 2017
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973. https://doi.org/10.1093/molbev/mss075
Schultz AK, Bulla I, Abdou-Chekaraou M, Gordien E, Morgenstern B, Zoaulim F, Deny P, Stanke M (2012) jpHMM: recombination analysis in viruses with circular genomes such as the hepatitis B virus. Nucleic Acids Res 40(Web Server issue):W193–W198. https://doi.org/10.1093/nar/gks414
Bell TG, Kramvis A (2015) Bioinformatics tools for small genomes, such as hepatitis B virus. Viruses 7(2):781–797. https://doi.org/10.3390/v7020781
Korber B, Myers G (1992) Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retrovir 8(9):1549–1560. https://doi.org/10.1089/aid.1992.8.1549
Chevalier MS, Kuehnert M, Basavaraju SV, Bjork A, Pitman JP (2016) Progress toward strengthening national blood transfusion services—14 Countries, 2011–2014. MMWR Morb Mortal Wkly Rep 65(5):115–119. https://doi.org/10.15585/mmwr.mm6505a4
Vermeulen M, Swanevelder R, Chowdhury D, Ingram C, Reddy R, Bloch EM, Custer BS, Murphy EL, Epidemiology NR, Donor evaluation Study IIIIC (2017) Use of blood donor screening to monitor prevalence of HIV and hepatitis B and C viruses, South Africa. Emerg Infect Dis 23(9):1560–1563. https://doi.org/10.3201/eid2309.161594
Mavenyengwa RT, Mukesi M, Chipare I, Shoombe E (2014) Prevalence of human immunodeficiency virus, syphilis, hepatitis B and C in blood donations in Namibia. BMC Public Health 14:424. https://doi.org/10.1186/1471-2458-14-424
Aydin OA, Karaosmanoglu HK, Sayan M, Ince ER, Nazlican O (2015) Seroprevalence and risk factors of syphilis among HIV/AIDS patients in Istanbul, Turkey. Cent Eur J Public Health 23(1):65–68. https://doi.org/10.21101/cejph.a4001
Kao JH, Chen PJ, Lai MY, Chen DS (2000) Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118(3):554–559
Orito E, Ichida T, Sakugawa H, Sata M, Horiike N, Hino K, Okita K, Okanoue T, Iino S, Tanaka E, Suzuki K, Watanabe H, Hige S, Mizokami M (2001) Geographic distribution of hepatitis B virus (HBV) genotype in patients with chronic HBV infection in Japan. Hepatology 34(3):590–594. https://doi.org/10.1053/jhep.2001.27221
Makondo E, Bell TG, Kramvis A (2012) Genotyping and molecular characterization of hepatitis B virus from human immunodeficiency virus-infected individuals in southern Africa. PLoS ONE 7(9):e46345. https://doi.org/10.1371/journal.pone.0046345
Hubschen JM, Andernach IE, Muller CP (2008) Hepatitis B virus genotype E variability in Africa. J Clin Virol 43(4):376–380. https://doi.org/10.1016/j.jcv.2008.08.018
Hou J, Liu Z, Gu F (2005) Epidemiology and prevention of hepatitis B virus infection. Int J Med Sci 2(1):50–57
Hannachi N, Bahri O, Ben Fredj N, Boukadida J, Triki H (2010) Risk of vertical transmission of hepatitis B virus in Tunisia. Arch Inst Pasteur Tunis 87(1–2):17–24
Baha W, Ennaji MM, Lazar F, Melloul M, El Fahime E, El Malki A, Bennani A (2012) HBV genotypes prevalence, precore and basal core mutants in Morocco. Infect Genet Evol 12(6):1157–1162. https://doi.org/10.1016/j.meegid.2012.04.026
Zhu HL, Li X, Li J, Zhang ZH (2016) Genetic variation of occult hepatitis B virus infection. World J Gastroenterol 22(13):3531–3546. https://doi.org/10.3748/wjg.v22.i13.3531
Amini-Bavil-Olyaee S, Vucur M, Luedde T, Trautwein C, Tacke F (2010) Differential impact of immune escape mutations G145R and P120T on the replication of lamivudine-resistant hepatitis B virus e antigen-positive and -negative strains. J Virol 84(2):1026–1033. https://doi.org/10.1128/JVI.01796-09
Lazarevic I (2014) Clinical implications of hepatitis B virus mutations: recent advances. World J Gastroenterol 20(24):7653–7664. https://doi.org/10.3748/wjg.v20.i24.7653
Coppola N, Loquercio G, Tonziello G, Azzaro R, Pisaturo M, Di Costanzo G, Starace M, Pasquale G, Cacciapuoti C, Petruzziello A (2013) HBV transmission from an occult carrier with five mutations in the major hydrophilic region of HBsAg to an immunosuppressed plasma recipient. J Clin Virol 58(1):315–317. https://doi.org/10.1016/j.jcv.2013.06.020
Salpini R, Colagrossi L, Bellocchi MC, Surdo M, Becker C, Alteri C, Aragri M, Ricciardi A, Armenia D, Pollicita M, Di Santo F, Carioti L, Louzoun Y, Mastroianni CM, Lichtner M, Paoloni M, Esposito M, D’Amore C, Marrone A, Marignani M, Sarrecchia C, Sarmati L, Andreoni M, Angelico M, Verheyen J, Perno CF, Svicher V (2015) Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology 61(3):823–833. https://doi.org/10.1002/hep.27604
Zaaijer HL, Torres P, Ontanon A, Ponte LG, Koppelman MH, Lelie PN, Hemert FJ, Boot HJ (2008) Multiple surface antigen mutations in five blood donors with occult hepatitis B virus infection. J Med Virol 80(8):1344–1349. https://doi.org/10.1002/jmv.21233
Svicher V, Cento V, Bernassola M, Neumann-Fraune M, Van Hemert F, Chen M, Salpini R, Liu C, Longo R, Visca M, Romano S, Micheli V, Bertoli A, Gori C, Ceccherini-Silberstein F, Sarrecchia C, Andreoni M, Angelico M, Ursitti A, Spano A, Zhang JM, Verheyen J, Cappiello G, Perno CF (2012) Novel HBsAg markers tightly correlate with occult HBV infection and strongly affect HBsAg detection. Antiviral Res 93(1):86–93. https://doi.org/10.1016/j.antiviral.2011.10.022
Meldal BH, Bon AH, Prati D, Ayob Y, Allain JP (2011) Diversity of hepatitis B virus infecting Malaysian candidate blood donors is driven by viral and host factors. J Viral Hepat 18(2):91–101. https://doi.org/10.1111/j.1365-2893.2010.01282.x
El Chaar M, El Jisr T, Allain JP (2012) Hepatitis B virus DNA splicing in Lebanese blood donors and genotype A to E strains: implications for hepatitis B virus DNA quantification and infectivity. J Clin Microbiol 50(10):3159–3167. https://doi.org/10.1128/JCM.01251-12
Greer AE, Ou SS, Wilson E, Piwowar-Manning E, Forman MS, McCauley M, Gamble T, Ruangyuttikarn C, Hosseinipour MC, Kumarasamy N, Nyirenda M, Grinsztejn B, Pilotto JH, Kosashunhanan N, Goncalves de Melo M, Makhema J, Akelo V, Panchia R, Badal-Faesen S, Chen YQ, Cohen MS, Eshleman SH, Thio CL, Valsamakis A (2017) Comparison of hepatitis B virus infection in HIV-infected and HIV-uninfected participants enrolled in a multinational clinical trial: HPTN 052. J Acquir Immune Defic Syndr 76(4):388–393. https://doi.org/10.1097/QAI.0000000000001511
Garmiri P, Rezvan H, Abolghasemi H, Allain JP (2011) Full genome characterization of hepatitis B virus strains from blood donors in Iran. J Med Virol 83(6):948–952. https://doi.org/10.1002/jmv.21772
Acknowledgements
The authors would like to acknowledge the support and collaborations from National Blood Transfusions Services of Botswana. We also acknowledge the Ministry of Health and Wellness of Botswana for granting the permissions to conduct this pioneer study for molecular epidemiology of HBV in blood donors in Botswana. We would like to also extend our acknowledgements to the National University of Science and Technology, Zimbabwe; University of Botswana; and the Botswana-Harvard HIV Reference Laboratory for their sponsorship, support, and contribution to the success of the study.
Funding
This work was supported through the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE), a DELTAS Africa Initiative [Grant No. DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [Grant No. 107752/Z/15/Z] and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of AAS, NEPAD Agency, Wellcome Trust, or the UK government.
Author information
Authors and Affiliations
Contributions
WTC wrote the first draft of the manuscript. WTC and MA collaborated in the lab work, primary data analysis. SM and PM were involved in statistical analysis. TM and BBP were involved in conducting some of the lab work and manuscript editing before submission. EZ, TKS, and IK supervised the project and edited the manuscript prior to submission and data evaluation. MKK provided with samples and their demographics, and also edited the manuscript before submission. JTB developed the secondary analysis plan and performed the phylogenetics of the study. ME and RMM edited the manuscript prior to submission and complemented it with contextual data. SG developed the study, main directions of the analysis plan, overall interpretation of results and edited manuscript before submission. All co-authors read and authorized the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The participants’ samples used in the study were residual from blood donors, obtained already de-identified and anonymous. These were a sub-population of HBV + screened samples from National Blood Transfusion services in Botswana (NBTS). The study was approved by the University of Botswana Institute Review Board and the Health Research Development Division (HRDD) at the Botswana Ministry of Health and Wellness (HPDME 13/18/1).
Additional information
Edited by Wolfram Gerlich.
Rights and permissions
About this article

Cite this article
Choga, W.T., Anderson, M., Zumbika, E. et al. Molecular characterization of hepatitis B virus in blood donors in Botswana. Virus Genes 55, 33–42 (2019). https://doi.org/10.1007/s11262-018-1610-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1610-z