
Abstract
The genome sequence and annotation of two novel poxviruses, NY_014 and Murmansk, are presented. Despite being isolated on different continents and from different hosts, the viruses are relatively similar, albeit distinct species. The closest known relative of the novel viruses is Yoka poxvirus. Five novel genes were found in these genomes, two of which were MHC class I homologs. Although the core of these genomes was well conserved, the terminal regions showed significant variability with large deletions and surprising evidence of recombination with orthopoxviruses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Poxviruses are large, complex viruses with linear, double-stranded DNA genomes that replicate entirely in the cytoplasm and infect insects (subfamily Entomopoxvirinae) and vertebrates (subfamily Chordopoxvirinae). Chordopoxviruses have been placed into 10 genera, i.e., Avi-, Capri-, Cervid-, Crocodylid-, Lepori-, Mollusci-, Ortho-, Para-, Sui-, and Yatapoxvirus; however, there are a number of fully sequenced viruses that are currently not assigned to a genus and are likely to require the designation of several new genera. These include salmon gill poxvirus (SGPV) isolated from salmon [1], Yoka poxvirus [2], Cotia virus and Embu virus [3] isolated from mosquitos, and Pteropoxvirus isolated from the Australian little flying fox (a bat) [4]. When further reports of novel poxviruses with only part of their genomes sequenced are taken into account, it is apparent that the numbers of Chordopoxvirus species and genera have taken a recent leap. Included in this group are a novel poxvirus isolated in big brown bats [5] that has been proposed to be designated as a new genus Chiropoxvirus, a poxvirus isolated from humans in the country of Georgia that has been classified as a novel Orthopoxvirus species [6] and two novel poxviruses from grey kangaroos (Dr. M. O’Dea, personal communication).
All of the Chordopoxviruses share a common core of about 80 genes that regulate and perform transcription, replication, and virus assembly functions [7]. However, the viral genomes are substantially varied in size, encoding between 129 and 328 genes. Thus, it is clear that there is an abundance of viral genes that are specific to various subsets of the Chordopoxviruses. Many of these genes are of “unknown function,” but are suspected to encode some type of host range or virulence factor because of their non-essential nature.
Poxviruses first assigned to a genus of the subfamily Chordopoxvirinae can be considered “low hanging fruit” discovered by virtue of their abundance and, or ability to cause observable effects in humans and animals. In contrast, those that have been discovered more recently have been isolated from relatively poorly surveyed hosts (fish, bats, mosquitos, birds, and aquatic mammals) or rare infections of humans.
Here, we report the complete genome sequences of two novel poxvirus species: one, NY_014, from an immune-suppressed patient living in the US [8] and the second, Murmansk from a root vole (Microtus oeconomus) isolated in Russia [9]. Phylogenetic analysis reveals that these two genomes are remarkably similar considering where they were isolated, and indicates that they should be placed in a new genus together with Yoka poxvirus.
Materials and methods
Viral strains
The isolation of NY_014 from a renal transplant patient with a progressive panniculitis/blistering rash in Upstate New York, USA and DNA isolation has been described previously [8]. Although not substantiated, this infection may have been acquired from a feral cat.
The Murmansk virus was isolated as follows [9]: during the period of June–July 1985, 225 rodents of different species were captured in the forest-tundra of Kolsky Peninsula, Russia, (latitude N 65°, longitude E 38°). Pooled organs (brain, liver, and spleen) from each species were analyzed for viral activity by intracerebral inoculation of suckling baby mice. From one such pool of root vole (Microtus oeconomus) organs, the LEIV-11411 Mur-Lovozero (Murmansk) viral strain was isolated. Electron microscopic study suggested that it belonged to the Poxviridae family. It had a low hemagglutinin activity, and formed hemorrhagic plaques on chorioallantoic membranes and plaques on Vero cell monolayers.
DNA extraction and PCR
To extract DNA, the Murmansk strain was propagated on Vero cells, and after 3 days cells were harvested and freeze-thawed three times. Low-speed centrifugation removed cellular debris and DNA was extracted from the supernatant using the Magna Pure apparatus (Roche, Germany). NY_014 was propagated on BSC40 cells and DNA was extracted as described by [8]. Use of a pan-orthopoxvirus real-time PCR assay [10] produced no signal with both viruses, whereas the pan-poxvirus PCR [11] method produced a specific amplicon. Sequencing of the amplicons resulted in unique sequences with no identical counterparts in the GenBank database.
Genome sequencing and de novo assembly
The purified Murmansk viral DNA was sequenced using the Illumina HiSeq 2500 platform (Illumina, Inc, San Diego, CA). The de novo assembly of the viral genome was performed using CLC genomic workbench software (CLC bio, Aarhus, Denmark) with an average coverage of over 1000×. The NY_014 assembly has an average coverage of 500×. Briefly, raw reads were imported into CLC and quality was assessed to remove duplicate reads, low-quality reads (quality score >0.01), and reads with >2 ambiguous nucleotides. After de novo assembly at the default setting, output contigs were screened against poxviruses (Taxid:10240) using BLAST. Four contigs were generated from the Murmansk DNA and three contigs were generated from NY_014. The order and orientation of the contigs were determined based on the BLAST results, and the gaps between the contigs were filled by Sanger sequencing using designed specific primers based on the assembled contigs. The gaps in the de novo assembling were due to tandem repeats and the inverted terminal repeats sequences (ITRs) at both the ends of poxvirus genome. The ITR regions usually form separate contigs and have elevated level of raw reads coverage relative to other contigs. ITR contigs were manually added to both the ends of the assembled genomes, and PCR and sequencing were used to confirm the assembly of the ITR ends.
Genome and phylogenetic analysis
The two genomes were annotated using the Genome Annotation Transfer Utility (GATU) [12] with Yoka poxvirus (YKV) and Cowpox virus strain Brighton Red (CPXV-BR) as the reference genomes. This program first transfers known gene positions to the target genomes and then identifies possible novel ORFs for further evaluation by the annotator. All predicted ORFs were searched against the NCBI nr database using BLAST programs [13]. Multiple sequence alignments (MSA) were performed with MAFFT [14] and ClustalO [15] in Base-By-Base (BBB). Phylogenetic analysis were performed using both maximum likelihood and neighbor-joining methods with MEGA v7 [16] and 500 bootstrap replicates; these methods generated similar trees. Dotplots were calculated and visualized using JDotter [17]. BBB, JDotter, and GATU are available at the Viral Bioinformatics Resource Centre (virology.uvic.ca).
Viruses and sequence accession numbers used for analysis
The following viruses were also used in the phylogenetic analysis [species/strain name (abbreviation; GenBank accession number)]: the Orthopox species, Camelpox virus CMS (CMLV; AY009089.1), Cowpox virus Brighton Red (CPXV; NC_003663.2), Ectromelia virus Moscow (ECTV; AF012825.2), Monkeypox virus Zaire-96-I-16 (MPXV; NC_003310.1), Vaccinia virus Western Reserve (VACV; NC_006998.1), Variola virus United Kingdom 1946 Harvey (VARV; DQ441444.1), Taterapox virus (TATV; NC_008291.1), Raccoonpox virus Herman (RCNV; NC_027213.1), Skunkpox virus WA (SKPV, NC_031038.1), and Volepox virus CA (VPXV, NC_031033.1); the Avipoxvirus species, Canarypox virus (CNPV; NC_005309.1), Fowlpox virus Iowa (FWPV; NC_002188.1), Penguinpox virus (PEPV; NC_024446.1), Pigeonpox virus (PIPV; NC_024447.1), and Turkeypox virus (TKPV; NC_028238.1); the Molluscipoxvirus species, Molluscum Contagiosum virus (MOCV; NC_001731.1); the Leporipoxvirus species, Myxoma virus Lausanne (MYXV; NC_001132.2) and Rabbit Fibroma virus (RFV; NC_001266.1); the Suipoxvirus species, Swinepox virus (SWPV; NC_003389.1); the Capripoxvirus species, Lumpy Skin Disease virus Neethling 2490 (LSDV; NC_003027.1), Sheeppox virus TU-V02127 (SPPV; NC_004002.1), and Goatpox virus Pellor (GTPV; NC_004003.1); the Cervidpoxvirus species, Deerpox virus W-1170-84 (DPV; NC_006967.1); the Yatapoxvirus species, Yaba-like disease virus (YLDV; NC_002642.1), Yaba monkey tumor virus (YMTV; NC_005179.1), and Tanapox virus (TANV; NC_009888.1); the Crocodylidpoxvirus species, Nile crocodilepox virus (CRV; NC_008030.1); the Parapox virus species, Bovine Papular Stomatitis virus AR02 (BPSV; NC_005337.1), Orf virus OV-SA00 (ORFV; NC_005336.1), Pseudocowpox virus VR634 (PCPV; NC_013804.1), and the unassigned viruses Cotia virus SPAn232 (COTV; NC_016924.1), Squirrel poxvirus (SQPV; NC_022563.1), and Yoka poxvirus (YKV; NC_015960.1). Accession numbers for genomic and MHC class I-like sequences used are indicated in their respective figures. The genome sequences of NY_014 and Murmansk poxviruses have been deposited in GenBank with accession numbers MF001305 and MF001304, respectively.
Results
NY_014 and Murmansk genome characteristics
The genome sequences of NY_014 and Murmansk were 200,223 bp (ITR = 1646 bp; A + T = 70.5%) and 204,055 bp (ITR = 4182 bp; A + T = 70.2%), respectively. There were only limited tandem repeat sequences in the ITRs of both NY_014 and Murmansk. 197 and 206 ORFs were annotated in the NY_014 and Murmansk genomes, respectively. We took a conservative approach to the process, annotating (1) ORFs matching previously characterized poxvirus genes, (2) unique ORFs larger than 65 codons, and (3) ORFs matching more than 50% of a characterized poxvirus gene with promoter region and initiating Met codon intact. The goal was to limit annotations to those ORFs with a high likelihood of encoding a polypeptide with some biological function. The NY_014 and Murmansk genome annotations are presented in Table 1. The lists of genes in Table 1 confirm an overall co-linear arrangement of the two new genomes with the Yoka poxvirus genome throughout the central core. The NY_014 and Murmansk genomes share 187 genes, which have an average aa identity of 94.3%; for the core region, this average increases to 98.1%. Although the core regions of these genomes have high similarity, towards the right end of the central region of the genome, Murmansk has two genes (Murmansk-156 and -157) that are absent from NY_014. These two genes, which are also absent from Yoka poxvirus, are orthologs of orthopoxvirus genes that encode a semaphorin-like protein (CPXV-BR-176) and a chemokine binding protein (CPXV-BR-178) with 50 and 34% aa identity, respectively. Table 1 also highlights the considerable variation at the left and right termini between these genomes and Yoka poxvirus, which is in part because the Yoka poxvirus genome is approximately 25 kbp shorter than the NY_014 and Murmansk genomes. To identify those genes without orthologs in Yoka poxvirus, we searched the NCBI non-redundant (nr) database using BLASTP [13]. The ITRs are unremarkable, although they are different sizes in the two viruses with one and four genes duplicated for NY_014 and Murmansk genomes, respectively. Such differences are commonly seen among the poxviruses and it is unclear whether having two copies of particular genes makes much difference to the biology of the viruses.
NY_014 and Murmansk phylogenetic analysis
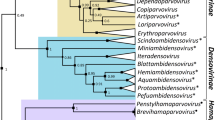
Considering the locations from which these two viruses were isolated (New York, USA, and Murmansk, close to the Russia/Sweden border), we were somewhat surprised by their high level of nucleotide similarity. Alignment of the central core genes revealed that NY_014 and Murmansk viruses are approximately 98% identical (nucleotide); thus, they are each other’s closest relative. The next closest known relative is Yoka poxvirus, which shares approximately 85% nucleotide identity with each of these two novel viruses. These relationships suggest that these three viruses should be placed in a common genus, proposed to ICTV as Centapoxvirus. In order to increase the reliability of the phylogenetic analysis, alignments were constructed using approximately 25 kbp of the most conserved core of the Chordopoxvirus genomes (equivalent to VACV-Cop-A2L to VACV-Cop-A24R). This greatly reduced the number of gaps required for the alignments, which are a common source of errors. The phylogenetic tree is shown in Fig. 1, and Table 1 provides the percent nucleotide identity for the individual genes through the aligned regions. For a frame of reference, NY_014 and Murmansk are more similar (98.2% nt identity) than variola and ectromelia viruses (97.0% nt identity) and almost as similar as variola and camelpox viruses (98.5% nt identity). Predicting dates of divergence for poxviruses is difficult, especially when recombination events are suspected, but it has been estimated that variola and camelpox viruses diverged 3–4000 years ago [18]. Within the core alignment region, for the most part, these novel viruses were syntenic with Yoka poxvirus, their closest relative, and much of this is also in common with the Orthopoxviruses. However, both ends of these genomes are much more variable than the core, not only in the order of genes, but also in their relationship to other genomes (discussed below). This emphasizes that it is essential to construct the viral phylogenetic tree using sequences that have a single common evolutionary history and not with regions that may have been targets of recombination. The variation beyond the left and right extremes of the core is best illustrated by a Dotplot [17] of the Yoka poxvirus and Murmansk genomes (Fig. 2). At the resolution provided by this dotplot, no indels can be observed within the core, but, in fact, numerous small indels are present. It is notable that the phylogenetic tree generated by the 25-kbp core is closely mirrored by a tree built using only the conserved RNA polymerase (RPO147) gene (data not shown), which supports its utility as an indicator of accurate relationships between poxviruses.

Maximum likelihood phylogenetic tree of the subfamily Chordopoxvirinae constructed using MEGA 7 with a 25-kbp segment of the genome core (VACV-Cop-A2L to VACV-Cop-A24R). All branches except five: MOCV origin branch (67), SWPV origin branch (57), DPV origin branch (72), VACV origin branch (71), and VARV/CMLV/TATV origin branch (49) had bootstrap scores of 95 or higher. These values were generated from 500 bootstrap replicates. The scale bar represents nucleotide substitutions per site

Dotplot of Murmansk (vertical) and Yoka (horizontal) poxvirus genomes. Blue and red boxes represent genes transcribed towards the left and right, respectively
Novel poxvirus genes in the NY_014 and Murmansk genomes
Our annotation of the genomes (Table 1) revealed 5 pairs of orthologs common to these 2 viruses, but absent from other poxviruses. Murmansk and NY_014 each contain three MHC class I-like homologs (Murmansk-037, -038, and -196 and NY_014-033, 034, and 186), while the Yoka genome contains a single MHC class I homolog (YKV173). All 6 of these poxvirus MHC class I-like proteins are predicted to possess a signal sequence and a C-terminal transmembrane domain and possibly function as MHC class I mimics. The Murmansk-196 and NY_014-186 pair are most similar to YKV173, suggesting that this gene was likely present in an ancestor of all three viruses. The remaining 2 pairs of MHC class I-like homologs present only in Murmansk and NY_014 are more similar to the MHC class I proteins of vertebrates than they are to any poxvirus protein. Each of the poxvirus orthologous protein pairs is >95% aa identical as expected; however, the pairs of paralogs from a single virus are only 36% aa identical. Interestingly, the NY_014-033/Murmansk-037 pair were 54–55% aa identical to rodent MHC class I proteins, whereas the NY_014-034/Murmansk-038 pairs were 37–42% aa identical to the same set of rodent proteins. A comparison of the group of rodent MHC class I proteins revealed them to be 66–80% aa identical among themselves. For the 2 sets of poxvirus genes, proteins from different rodent species were the top matches; however, a “best database match” does not imply these were the hosts from which the poxvirus genes were acquired especially since the top scores were very similar. These results suggest that a poxvirus, ancestral to Murmansk and NY_014, first acquired one MHC class I-like gene and this was subsequently duplicated a long time very much before these two viruses diverged.
BLASTP failed to match the proteins encoded by 2 of the pairs of orthologs (Murmansk-033 and -048) with proteins in the non-redundant protein database leaving these with an “unknown function” annotation. However, the final novel poxvirus gene NY_014-173/Murmansk-179 encodes a product with very low similarity to an IL-1 receptor-like protein, mostly matching to an immunoglobulin-like domain. Although a similarly annotated protein exists in some Capripoxviruses, these share only 21% aa identity to the NY_014- and Murmansk-predicted proteins. Since the host proteins that match these 2 distantly related poxvirus proteins share very few residues, within an immunoglobulin-like domain, it is unlikely that the NY_014 and Murmansk orthologs described here share a common ancestral protein within the poxvirus lineage.
The NY_014-165 and Murmansk-171 genes were found to be orthologous (47% aa identity) to RCNV-Herman-171, which previously was unique to the NAOV. Interestingly, the position of the gene is syntenic with RCNV; however, it is encoded by the opposite DNA strand in NY_014 and Murmansk (Fig. 3). Additionally, in this same region NY_014 and Murmansk genomes possess a gene (NY_014-166 and Murmansk-172), which is immediately to the right of the previously discussed gene, that they share only with Yoka poxvirus (YKV-157).

Organization of the variable region at right end of viral genomes. Direction of gene transcription is illustrated by the blue arrows representing the genes
Unusual features within the terminal regions of the NY_014 and Murmansk genomes
The left and right terminal regions are more variable than the central conserved core of the poxvirus genomes in several ways: (1) the position of orthologous genes, (2) the particular genes present, and (3) the percent nucleotide identity between orthologous genes. Usually, such lower DNA similarity is simply due to the types of proteins encoded by the genes in these regions. For example, comparing the DNA polymerase and RNA polymerase (RPO147) genes shows that the NY_014 and Murmansk orthologs are 98–99% identical (nt), whereas the large surface glycoprotein genes are only 90% identical. Therefore, it was a surprise when we examined the regions from the termini that are present in only one or the other of the NY_014 or Murmansk genomes and discovered that they have very unusual relationship patterns with other poxvirus genomes.
Left terminal region
DNA and protein alignments first revealed that some of the NY_014 proteins from the viral termini were much more similar to RCNV proteins than the conserved core proteins. It was also apparent that these NY_014 proteins were even more similar to RCNV proteins than the Yoka poxvirus orthologs contradicting the pattern of the phylogenetic tree produced with the genome cores (Fig. 1). For example, the NY_014 and Murmansk DNA polymerases are approximately 86 and 83% (aa) identical to the Yoka poxvirus and RCNV orthologs, respectively, whereas the NY_014-005 (encoding an Ankyrin-like protein (Cop-B18R)) has 94% aa identity with the RCNV ortholog. Although this switching between different viruses as “most similar ortholog” appears complex and at odds to a straightforward evolutionary trajectory, it is often simplified when the presence or absence of genes is considered. For example, if genes have been lost from Yoka poxvirus, then the closet match to NY_014 and Murmansk genes is likely to be RCNV. If the genes are also absent from RCNV, then the nearest neighbor most likely becomes another orthopoxvirus. Also since NY_014 and Murmansk are approximately evolutionarily equi-distant from the various orthopoxvirus species, then minor differences between orthologs can result in different species appearing to be the closest relative. However, a closer examination of similarity scores between these orthologs clearly reveals that some genome segments have been exchanged between viruses. Terminal genes are generally quite variable in poxviruses, for example, terminal genes NY_014-020, -021, and -022 have only 62–69% aa identity to their orthologs in Yoka poxvirus. In contrast, orthologs of the genes NY_014-002 to -006 (Fig. 4 and Supplemental Fig. 1), which are absent from the Murmansk and Yoka poxvirus genomes, are 83–96% identical (aa) to corresponding orthologs in RCNV. This high percent identity suggests a recombination event that resulted in an exchange of genes from an RCNV-like virus to an ancestor of NY_014. Using a comparison of these genes to CPXV orthologs as a control indicates that the NY_014 genome acquired the DNA from an RCNV-like ancestor, and not the other way around. For several other genes such as Murmansk-002/003, the closest ortholog is in CPXV (78 and 69% aa identity, respectively). However, given the location of the genes and the predicted rarity of recombination events compared to deletion events, this arrangement may be the result of the previously described recombination followed by loss of genes from RCNV and NY_014 ancestors (Fig. 4 and Supplemental Fig. 1).

Alignment of the left terminal regions of the NY_014 (bottom) and RCNV-Herman (top) genomes. The MAFFT alignment generated 90% nucleotide identity over aligned regions
Figure 4 illustrates the synteny between the NY_014 and RCNV genomes, but several genes are missing from the NY_014 genome, which could be achieved by 2 deletion events following the introduction of the region into the NY_014 genome. Within this region, one of the genes has also been lost from RCNV, but in this instance gene loss was due to mutation that fragmented the gene to produce RCNV-006f/007f. Both scenarios are consistent with the high variability that is observed for the terminal regions of poxvirus genomes, especially with respect to the generation of indels.
Yet another category of gene organization is represented by Murmansk genes -010 to -021, most of which are also present in NY_014 (Supplemental Fig. 1). The most similar orthologs of these genes are found in CPXV; however, the percent identity is relatively low (28–50% nt identity), suggesting that this region was acquired from a virus not represented by the currently known poxvirus species.
There are 3 pairs of adjacent genes in the Murmansk genome 010/011 (unknown function), 015/016 (IL-18 binding protein), 188/189 (interferon alpha/beta binding protein) that may be the result of gene duplication events (Supplemental Figs. 1 and 2). Although the NY_014 genome contains only one member of each pair that is likely to be functional, an alignment of the genomic sequences suggests that the gene duplications occurred before these 2 viruses diverged. One member of each of the 3 NY_014 pairs has been disrupted in a slightly different manner. The ortholog of Murmansk-010 is mostly deleted and the ortholog of Murmansk-016 has been disrupted by mutations that destroy the start of the coding region. A large deletion in NY_014 has created NY_014-181 from the Murmansk-188 and Murmansk-189 gene pair, generating a fusion of the N-terminal 2/3 of Murmansk-188 and the C-terminal 1/3 of Murmansk-189 (Supplemental Fig. 2). The nt identity between these Murmansk gene pairs is 63, 52, and 59%, respectively. Since the Murmansk proteins 010/011 have only approximately 33% aa identity with a CPXV hypothetical protein, they are unlikely to be orthologs.
Right terminal region
Examination of the right terminal regions of the NY_014 and Murmansk genomes revealed that these too have a complex relationship with each other and the orthopoxviruses. Within this region, 8 Murmansk genes are absent from NY_014 and 5 NY_014 genes are absent from Murmansk (Table 1). Furthermore, there are no Yoka poxvirus or RCNV counterparts of several of the NY_014 and Murmansk genes in the right region. This results in the most similar homolog being in the orthopoxvirus group (CPXVs in Table 1). However, the relationship between the genomes is not that simple because although some of the Murmansk and NY_014 proteins are in the order of 40–60% identical to the CPXV homologs, which would be expected for the relationship shown by the phylogenetic tree in Fig. 1, several of the Murmansk and NY_014 proteins are more than 70% identical to the CPXV protein homologs. Similar to the relationship with RCNV in the left terminal region, NY_014 has three genes with high aa identity to CPXV orthologs: NY_014-194 and C7L (Type 1 IFN inhibitor) 93% aa identity, NY_014-195 and C8L (unknown protein) 92% aa identity, and NY_014-196 and C9L (Ankyrin-like protein) 79% aa identity. These three genes are also absent from Murmansk in this region. For example, the C7L-like gene in NY_014 (194) is not syntenic with the C7L orthologs Murmansk-193 and Yoka poxvirus-171, and sequence alignment reveals that a gene syntenic with Yoka/Murmansk poxvirus C7L-like genes may have lost function in NY_014 (Supplemental Fig. 2). Phylogenetic analysis of C7L revealed obvious rearrangement compared to phylogenies created using more conserved genes, supporting the idea of horizontal gene transfer (Fig. 5).

Maximum likelihood phylogenetic tree of orthologs of NY_014-194 (Type 1 IFN inhibitor) constructed using MEGA 7. The scale bar represents nucleotide substitutions per site
Host range and virulence genes in Murmansk and NY_014
Poxviruses typically encode many genes that are known to target different aspects of the host immune response, and different clades of poxviruses tend to have somewhat distinct repertoires of host range and virulence genes simply by virtue of their evolutionary history. As might be predicted because of their position on the phylogenetic tree, the repertoire of these genes in the genomes of the Murmansk and NY_014 viruses is most similar to the orthopoxviruses, including the presence of orthologs of genes almost exclusively found in the orthopoxvirus genus such as K1L and CrmB-E. However, gene duplication and loss through mutation or out-right deletion have generated complex patterns (presence/absence) for this type of gene in poxviruses. Additionally, since the molecular mechanisms by which most of these proteins function are unknown, it is impossible to gauge the contribution that the different genes make in the different hosts.
Discussion
We have presented the genome sequences of two novel poxviruses that are likely new species within the genus typified by Yoka poxvirus. Five genes, present in each of these viruses, are otherwise unique in poxviruses. Two of the genes encode MHC class I-like proteins and another has low similarity to an IL-1 receptor-like protein. The other 2 unique genes are also likely to encode some sort of virulence protein, adding to the repertoire of processes that the poxviruses, as a family, have acquired to overcome host defense mechanisms. Although much of the genomes of the two viruses presented here show synteny with their closest relative, Yoka poxvirus, a close examination of the similarity of the genomes across the entire length revealed the results of several ancient recombination events. Based on the similarity of these exchanged regions, they appear to have been acquired from an ancestor of the North American Orthopoxvirus RCNV. This finding strengthens the notion that recombination has played an important role in the evolution of the poxviruses [19,20,21,22], including variola virus [23].
Poxvirus genomes typically encode many virulence genes that are known to target different aspects of the host immune response. The host range gene repertoire of Murmansk and NY_014 is very typical of what has been reported for orthopoxviruses, but there are a few notable similarities to clade II poxviruses. We observed several differences in host range genes between the individual isolates of the Yoka-Murmansk-NY_014 lineage including the expansion of ANK-containing proteins, the presence of two K1L orthologs in NY_014, the disruption or loss of several host range genes in Yoka poxvirus, and an expansion of TNF receptor-like genes in Murmansk. Similar differences in the host range gene repertoire have been correlated with differences in the host range of other poxviruses (for a recent review, see [24]).
One of the perennial fears regarding poxviruses is the possibility of emergence of a virus capable of creating a new smallpox-like disease. NY_014, presented here, may represent a special risk since it was isolated from an immunocompromised human. Natural processes that could lead to such a catastrophic event include (1) changes to a known zoonotic poxvirus that leads to persistence and rapid spread in humans, (2) introduction of a novel poxvirus to humans from an unknown animal reservoir, and (3) recombination among poxviruses that currently infect animals to create a variant able to infect humans. It is immensely difficult to predict the likelihood of any of these events. However, given that there has been a steady discovery of novel poxviruses and evidence supporting recombination among the various viral genomes, such possibilities are real. Furthermore, the geographic isolation of each of these three viruses on three different continents from an immunocompromised human (NY_014), vole (Murmansk), and mosquito pool (Yoka) only contributes to the question of the natural host and geographical range of these isolates. Further studies on these isolates, as well as collection and identification of more poxviruses, will help us understand their host range/evolution and potential for more serious and widespread zoonotic infections.
References
M.C. Gjessing, N. Yutin, T. Tengs, T. Senkevich, E. Koonin, H.P. Rønning, M. Alarcon, S. Ylving, K.-I. Lie, B. Saure, L. Tran, B. Moss, O.B. Dale, J Virol JVI.01174 (2015)
G. Zhao, L. Droit, R.B. Tesh, V.L. Popov, N.S. Little, C. Upton, H.W. Virgin, D. Wang, J. Virol. 85, 10230 (2011)
M.S. Ibrahim, M. Antwerpen, E. Georgi, P. Vette, G. Zoeller, H. Meyer, Genome Announc. 2, e01155 (2014)
M.A. O’Dea, S.-L. Tu, S. Pang, T. De Ridder, B. Jackson, C. Upton, J. Gen. Virol. 97, 2363 (2016)
G.L. Emerson, R. Nordhausen, M.M. Garner, J.R. Huckabee, S. Johnson, R.D. Wohrle, W.B. Davidson, K. Wilkins, Y. Li, J.B. Doty, N.F. Gallardo-Romero, M.G. Metcalfe, K.L. Karem, I.K. Damon, D.S. Carroll, Emerg. Infect. Dis. 19, 1002 (2013)
N.M. Vora, Y. Li, M. Geleishvili, G.L. Emerson, E. Khmaladze, G. Maghlakelidze, A. Navdarashvili, K. Zakhashvili, M. Kokhreidze, M. Endeladze, G. Mokverashvili, P.S. Satheshkumar, N. Gallardo-Romero, C.S. Goldsmith, M.G. Metcalfe, I. Damon, E.F. Maes, M.G. Reynolds, J. Morgan, D.S. Carroll, N. Engl. J. Med. 372, 1223 (2015)
A. Ehlers, J. Osborne, S. Slack, R.L. Roper, C. Upton, Bioinformatics 18, 1544 (2002)
N.S. Lakis, Y. Li, J.L. Abraham, C. Upton, D.C. Blair, S. Smith, H. Zhao, I.K. Damon, Clin. Infect. Dis. 61, 1543 (2015)
S.D. L’vov, V.L. Gromashevskiĭ, S.S. Marennikova, G.V. Bogoiavlenskiĭ, F.N. Baĭluk, Vopr. Virusol. 33, 92 (1988)
M.W. Eshoo, C.A. Whitehouse, A. Nalca, S. Zoll, J.A. Ecker, T.A. Hall, T.-T.D. Pennella, D.D. Duncan, A. Desai, E.K. Moradi, K. Rudnick, B. Libby, R. Ranken, R. Sampath, S.A. Hofstadler, D.J. Ecker, L.B. Blyn, PLoS ONE 4, e6342 (2009)
Y. Li, H. Meyer, H. Zhao, I.K. Damon, J. Clin. Microbiol. 48, 268 (2010)
V. Tcherepanov, A. Ehlers, C. Upton, BMC Genom. 7, 150 (2006)
S.F. Altschul, T.L. Madden, A.A. Schäffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman, Nucleic Acids Res. 25, 3389 (1997)
K. Kazutaka, D.M. Standley, Mol. Biol. Evol. 30, 772 (2013)
F. Sievers, D.G. Higgins, Methods Mol. Biol. 1079, 105 (2014)
S. Kumar, G. Stecher, K. Tamura, Mol. Biol. Evol. 33, 1870 (2016)
R. Brodie, R.L. Roper, C. Upton, Bioinformatics 20, 279 (2004)
I.N.B. Igor, V. Babkin, Viruses 7, 1100 (2015)
C. Smithson, S. Kampman, B. Hetman, C. Upton, Computation (n.d.)
D. Coulson, C. Upton, Virus Genes 42, 171 (2011)
L. Qin and D. H. Evans, J Virol JVI.00022 (2014)
K.A. Bratke, A. McLysaght, BMC Evol. Biol. 8, 67 (2008)
C. Smithson, A. Purdy, A.J. Verster, C. Upton, PLoS ONE 9, e91520 (2014)
S.L. Haller, C. Peng, G. McFadden, S. Rothenburg, Infect. Genet. Evol. 21, 15–40 (2013)
Acknowledgments
The authors thank the many University of Victoria undergraduate Co-op students who have helped develop the Virology.ca bioinformatics tools, and especially Colum McClay for modifications to Base-By-Base. This study was funded by a Natural Sciences and Engineering Research Council Discovery Grant to CU. The authors would like to acknowledge the following people for their technical support and comments on the manuscript: Miriam Laker, Richard Kline, Scott Sammons, Ginny Emerson—U. S. Centers for Disease Control and Prevention.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Author information
Authors and Affiliations
Contributions
HM, ID, and YL conceived of, or designed study; JG, HZ, and DB performed research; CS, CG, and CU analyzed data; and CS, HM, CU, and YL wrote the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
No human subjects were involved in this study.
Additional information
Edited by Joachim Jakob Bugert.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2017_1501_MOESM1_ESM.pdf
Supplemental Fig. 1: Alignment of the left terminal regions from NY_014 (465–25,217 nt) and Murmansk (277–24,579 nt) viruses. (PDF 40 kb)
11262_2017_1501_MOESM2_ESM.pdf
Supplemental Fig. 2: Alignment of the right terminal regions from NY_014 (176,994–199,759 nt) and Murmansk (177,936–203,779 nt) viruses. (PDF 51 kb)
Rights and permissions
About this article

Cite this article
Smithson, C., Meyer, H., Gigante, C.M. et al. Two novel poxviruses with unusual genome rearrangements: NY_014 and Murmansk. Virus Genes 53, 883–897 (2017). https://doi.org/10.1007/s11262-017-1501-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-017-1501-8