
Abstract
H9N2 avian influenza viruses (AIVs) have been recorded in Eurasian for several years. Since 2004–2005, the disease has become endemic in Iraq, causing serious economic losses in the poultry industry. The hemagglutinin (HA) and neuraminidase (NA), two out of eight protein-coding genes, play an important role during the early stage of infection and hinder virus assembling. Little is known about the genetic information of the H9N2 viruses currently circulating in Iraq; thus, gene sequences of six AIVS of the H9N2 subtype have been detected and analyzed in the period of 2014–2015 from different outbreaks of broiler flocks in five provinces situated in the middle and southern parts of Iraq. Genetic comparison of the partial sequences of HA gene indicated that all Iraqi viruses are related to each other and could be divided into two subgroups. Viruses of the first and the second subgroups demonstrated a high similar identity with Pakistani and Iranian viruses, respectively. The nucleotide sequences of the NA protein of the all studied Iraqi viruses were very similar (95.2–100% identity), and shared high nucleotide sequence identity with Iranian, Pakistani, and Lebanese strains. All six recent viruses possessed histidine, alanine, and leucine at positions 183, 190, and 226, respectively, which are the key residues in receptor-binding sites. The Iraqi viruses were closely related to viruses of G1-like lineage isolated from poultry flocks of Iran and Pakistan, suggesting that possible epidemiological links could be derived from a common origin. Further investigations are required and should include the viral isolation and full-length molecular characterization of H9N2 AIVs in this area.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
H9N2 avian influenza (AI) virus has been reported widely around the world since its first detection in the USA in 1966 and is classified in type A influenza virus [1]. Type A influenza viruses are members of the Orthomyxoviridae family, being capable of causing infection in a variety of hosts, including birds and mammals [2]. The negative-sense RNA genome of influenza A virus consists of eight segments which encode 10 to 12 proteins, including two surface glycoproteins, hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), three polymerase proteins (PA, PB1, PB2), two matrix (M1, M2), and non-structural proteins (NS1, NS2) [3, 4]. There are 18 HA and 11 NA subtypes, and the viruses are classified into subtypes based on their two surface glycoproteins: HA and NA. Sixteen HA (H1–H16) and nine NA (N1–N9) subtypes have been isolated from wild birds, in particular, the aquatic ones, which are the major natural reservoir [5, 6]. H5, H7, and H9 subtypes are considered as the most common avian subtypes [7]. The HA surface glycoprotein of influenza viruses plays an indispensable role in the initial stages of infection and is responsible for the virus binding to the cell receptor named sialic acid; sialic acid is present on the host cell surface and promotes fusion of viral and endosomal membranes and eventually facilitates viral entry into the host cell [8]. The NA surface glycoprotein of influenza viruses hinders virus assembling by cleaving the α-ketosodic linkage between sialic acid and the adjacent sugar residue [9]. Two distinct lineages of H9N2 influenza viruses exist: North American and Eurasian lineages. The Eurasian lineage is composed of at least three sub-lineages. This lineage is divided into three major sub-lineages represented by their prototype strains: A/chicken/Korea/38349-p96323/96 (Korean-like), A/duck/Hong Kong/Y280/97 (Y280-like), and A/quail/Hong Kong/G1/97 (G1-like) [10]. Throughout the second half of the 1990s, outbreaks resulting from H9N2 subtypes were reported in many countries, namely Germany, Italy, Ireland, South Africa, USA, Korea, China, and the Middle Eastern countries such as Saudi Arabia and Iran [5]. The H9N2 subtype is classified as low pathogenic AI (LPAI), and generally, the affected chickens show mild to severe respiratory signs, edema of the head and the face, and declines in egg production accompanied with soft-shelled or misshapen eggs. The high morbidity and mortality rates in poultry flocks have been reported, particularly in combination with other respiratory pathogens [11]. Furthermore, the H9N2 viruses have led to occasional respiratory illnesses in humans and have been accounted for as a global concern in human societies [2]. AIV infection in Iraq has brought about remarkable economic losses to the poultry industry, especially in broiler flocks during the past decade, and is still continuing [12]. The genetic and antigenic evolution of H9N2 viruses in some countries, particularly those neighboring Iraq, has been well documented, and Iraq is located in a critical region of H9 AI outbreaks. However, information is limited on the genetic properties of H9N2 viruses now circulating in Iraq; therefore, gene sequences of six AIVS of the H9N2 subtype have been detected and analyzed in the course of 2014–2015 from different outbreaks of broiler flocks in five provinces situated in the middle and southern parts of Iraq. The current research is aimed at describing the changes having occurred in avian (H9N2) viruses in recent years and preparing beneficial guidance on the control of H9N2 AIVs and vaccine candidate selection.
Materials and methods
Sampling
From September 2014 to June 2015, Samples were collected from 80 broiler chicken farms in five provinces representing central and southern parts of Iraq (Al-Basra, Wasit, An-Najaf, Al-Muthanna, and Al-Qadisyia; Fig. 1). Each farm had one flock with 10,000 birds. Five tracheas(pooled together) per flock were collected in a sterile phosphate buffered saline from three to 4 weeks old broiler flocks suffered from respiratory syndrome such as coughing, gasping, rales, and excessive lacrimation in all of the affected broiler flocks. In addition, non-characteristic generalized clinical signs, namely ruffled feathers, huddling, depression, decreased activity, and diarrhea were observed. All the infected flocks have been vaccinated with oily inactivated vaccine of AI subtype H9N2.

Area of origin of H9N2 AIVs from broiler flocks in Iraq during 2014–2015. The names of the provinces are indicated in boldface and the names of H9N2 AIVs (QMG-1 to QMG-6) are indicated in non-boldface and 10, 20, 21, 15, and 14 denote the numbers of tested flocks in Wasit, Al- Qadisyia, An-Najaf, Al-Muthanna, and Al-Basra, respectively
RNA extraction and cDNA synthesis
Total RNA was extracted directly from tracheas, using CinnaPure RNA extraction kit (SinaClon, Iran) according to the manufacturer’s instructions. The cDNA was synthesized by the reverse transcription (RT) reaction that was performed with a mixture of 5 µL of extracted RNA and 1 µL of influenza universal primer, Uni12: 5′-AGC AAA AGC AGG-3′ and incubated at 65 °C for 5 min and 5 °C for 1 min. Then, 14 µL of master mix consisting of 7.25 µL of DEPC-treated water (SinaClon, Iran), 4 µL Buffer 5×, 2 µL dNTP mix (SinaClon, Iran), 0.5 µL Revert Aid Reverse Transcriptase (Thermo Fisher Scientific, USA), and 0.25 µL Ribolock Rnase inhibitor (Thermo Fisher Scientific, USA) was added to each tube. And the mixture was heated at 25 °C for 5 min, at 42 °C for 60 min, and at 95 °C for 5 min. Finally, it cooled down to 4 °C for 1 min and was stored at −20 °C until it was used. The preliminary RNA extraction and cDNA synthesis were performed in Iraq, and the other molecular characterizations were conducted at the central lab of the Veterinary Faculty of Tehran University, Iran.
Real-time PCR
Real-time PCR was performed as a screening test to detect the presence of influenza virus genome in the pooled samples as the first step for primary screening, and in the next step, the positives tested separately for a precise finding of positive samples. The amplification was performed using an amplification kit (Bioneer, South Korea) with the forward primer 5′-ATGGGGTTTGCTGCC-3′, reverse primer 5′-TTATATACAAATGTTGCAYCTG-3′, and Taqman probe FAM -5′-TTCTGGGCCATGTCCAATGG-3′-TAMRA as described by Monne et al. [13].
PCR reaction for amplification of HA and NA gene
A part of 776 bps and 314 bps of the HA and NA genes was subsequently amplified by RT-PCR, using one pair of specific primers for each gene as described previously by Hoffman et al. [14], HAF1:5′-AGCAAAAGGAGGGG-3′, HAR1:-5′-GTGYCCATACCATGGRGC-3′, NA2-1:5′-TCCGTTTCATTTGGGAACC-3′, and NA2-2:-5′-CTGACAATGGRCTAATGTG-3′. The amplification was implemented in a final volume of 25 μL, containing 5 μL of cDNA, 12.5 μL of SinaClon 2× PCR master mix (SinaClon, Iran), 1 μL (10 μlM) of each primer, and 5.5 μL of distilled water. The amplification program of HA gene was as follows: 95 °C for 4 min, 35 cycles of 94 °C for 1 min, 48 °C for 1 min, and 72 °C for 1 min followed by 72 °C for 10 min. In addition, the amplification program of the NA gene was 94 °C for 3 min, 35 cycles of 94 °C for 30 s, 47 °C for 30 s, and 72 °C for 30 s followed by 72 °C for 10 min. The PCR products were assessed via gel electrophoresis on 1.5% agarose gel, followed by staining with GelRed™ (Biotium, USA) and visualized under UV light.
Phylogenetic analysis
The PCR products were purified by the PCR AccuPrep® PCR purification kit (Bioneer Co., Korea). Sequencing was carried out with the same primers (both directions) that were employed for amplification of HA and NA genes (Bioneer Co., Korea). Chromatograms were evaluated by CromasPro (CromasPro version 1.5). To determine the genetic diversity of these viruses, all data related to the nucleotide sequence of HA and NA genes of the present study were edited via the CLC Sequence Viewer software (version 7.6.1). The phylogenetic analysis was performed by the MEGA5 software. Furthermore, distance-based neighbor-joining trees were constructed, using the Tamura–Neimodel available in MEGA5 program [15]. The robustness of the phylogenetic trees was assessed by 1000 bootstrap replicates. Bootstrap values lower than 60 were omitted. The MEGA5 was deployed for determining and comparing the percentage of sequence similarity/difference of the six Iraqi H9N2 viruses in the present study with those of other sequences of AIV belonging to the main H9N2 lineages, i.e., Asian and European countries, which were extracted from Gene Bank database as shown in Table 1. The sequences of HA and NA of H9N2 subtype in this study were submitted to the Gene Bank database, and the accession numbers were (KX944335–KX944340) and (KX944341–KX944346), respectively.
Results
Virus detection
During the period of 2014–2015, six H9N2 AIVs (QMG-1 to QMG-6) were detected in five Iraqi provinces (Table 2), one virus from every province except for An-Najaf province which has two representative viruses from different farms. Subsequently, these viruses were analysis for genetic studies in the present study.
Phylogenetic analysis
HA gene
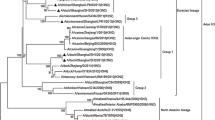
Genetic comparison of the partial sequences of HA gene of the Iraqi H9N2 viruses demonstrated that all the viruses were related to each other and could be divided into two subgroups (Table 2) based on the degree of similarity; moreover, the viruses in the subgroups exhibited high percentages of identity, i.e., 99% and 98.7–100%, in the first and the second subgroups, respectively. Our study revealed that the HA gene of all Iraqi viruses shared a close relationship with gene sequences of other H9N2 isolates from the Middle Eastern strains, in particular, those of Iraq’s neighbors (Iran, Kuwait, Saudi Arabia, and Jordan). In addition to the strains from non-neighboring countries, including two UAE strains (UAE-06 and UAE-05) and Israel (IS-2007) strain, the viruses of the second subgroup showed high identity of 96.7–97.2% and 95–95.5% among Iranian strains (Iran-121 and Iran-B326), respectively. Interestingly, all strains from Pakistan shared high nucleotide sequence identity (Pak-294, P.UDL-01, P.UDL-02, and Karachi-03) among viruses of both subgroups, especially those of the first subgroup which shared the highest identity of 97–97.6% and 95.2–95.8% with Pak-294 and P.UDL-02, respectively. The results indicated that sequences identity among these six Iraqi viruses showed different percentages of similarity with a previous Iraqi isolate (Iraq EKI14); additionally, the viruses belonging to the first subgroup exhibited a range of minimum identity (89.9%), while those belonging to the second subgroup displayed a range of maximum identity (96.3%). The scores of similarity among nucleotide sequences were calculated by pairwise alignments of similarity which supported the phylogenetic results. Tree analysis of the HA gene demonstrated that all viruses in the present study belonged to the G1 lineage (Fig. 2) and shared 86.7–88.6% identity with the A/Hong Kong/1073/99 G1 ancestor (Table 3).

Phylogenetic relationships of HA genes from H9N2 viruses diagnosed in Iraq during 2014–2015, Middle Eastern, and Eurasian countries. The nucleotide ranges of segments that were used for drawing phylogenic trees are as follows: HA (1–776). Trees were generated by the neighbor-joining method with the MEGA 5 software. The reliability of the trees was assessed by bootstrap analysis with 1000 replications. Numbers above branches indicate neighbor-joining bootstrap values. The length of the horizontal lines is proportional to the minimum number of nucleotide differences required to join nodes. The vertical lines are for spacing branches and labels. The viruses characterized in this study are indicated in a black triangle
NA gene
In this study, 314 base pairs of NA gene were sequenced. The nucleotide sequences of the NA protein of the all studied Iraqi viruses were very similar (95.2–100% identity), and shared the highest identities of 94.3–100%, 93.9–98%, and 93.1–98% with the Iranian (Ir-121), Pakistani (P.UDL-1), and Lebanese (Leb-2010) strains, respectively. The phylogenetic tree (Fig. 3) revealed that all viruses in the present study belonged to a single lineage (G1) and shared 91.8–96.8% identity with the A/Quail/Hong Kong/G1/97 ancestor (Table 4).

Phylogenetic relationships of NA genes from H9N2 viruses isolated in Iraq during 2014–2015, Middle Eastern, and Eurasian countries. The nucleotide ranges of segments that were used for drawing phylogenic trees are as follows: NA (1–314). Trees were generated by the neighbor-joining method with the MEGA 5 software. The reliability of the trees was assessed by bootstrap analysis with 1000 replications. Numbers above branches indicate neighbor-joining bootstrap values. The length of the horizontal lines is proportional to the minimum number of nucleotide differences required to join nodes. The vertical lines are for spacing branches and labels. The viruses characterized in this study are indicated in a black triangle
HA receptor-binding sites
All the six recent Iraqi viruses from this study possessed N98, W153, T155, H183, A190, L194, Y195, and L226, respectively, which are the key residues in receptor-binding sites (numbered according to H3). The left edge (amino acid residues at position 234–239) of the binding pocket motif was NGLIGR. Two kinds of right edge (amino acid residues at position 148–152) of the binding pocket motif (GTSKA and GTSKS) in Iraqi viruses have been detected in this study (Table 5).
Discussion
During the past 12 years, AIVs of H9N2 subtype have been the leading cause of high economic losses in poultry in various provinces of Iraq [16]. As a response to these outbreaks, several investigations have been carried out on AIVs in Iraqi broiler flocks, and the presence of H9 subtype has been proved, being responsible for high mortality rates of 20–90% and 30–70% [17, 18]. However, little is known about the genetic information of the Iraqi H9N2 viruses; therefore, this study provides the genetic data for the H9N2 viruses that are currently circulating in the poultry population in Iraq. In the present study, we described the detection of six H9N2 viruses from different chicken flocks in Al-Basra, Wasit, An-Najaf, Al-Muthanna, and Al-Qadisyia province, and this result indicated the endemic situation of H9N2 AIVs with broad geographical distribution in central and southern parts of Iraq which represents another risk factor to the poultry industry in Iraq, especially with the presence of other respiratory pathogens like IBV, NDV, and MG, as well as the low biosecurity level in some commercial sectors. The main aspect of this study is the sequencing of the HA and NA genes from six Iraqi H9N2 strains. It is interesting to know that these six strains, in addition to the previous isolate (Iraq EKI14), clustered within a broad grouping that included G1-like lineage in the H9N2 subtype, and fell together into a distinct cluster along with Iran, Pakistan, UAE, Israel, India, and Saudi Arabia’s isolates, indicating a close relationship. These results are basically in agreement with those of the previous studies which suggested that the viruses from Pakistan, Iran, Germany, and Saudi Arabia all showed very close relationships and that they might have a common origin [5]. Despite the disappearance of A/quail/Hong Kong/G1/97 H9N2 prototype virus from Hong Kong due to withdrawal of quail from live poultry markets in this part of china being followed by the first isolation of this prototype, G1 viruses, however, continue to be detected in South Asia and Middle-East countries [5, 19]. The presence of the Middle Eastern isolates on this lineage may be owing to a close geographical relationship among these countries that had given the impression that the outbreaks seen in these countries resulted from a major epizootic caused by the spread of a single virus. Genetic comparison of the nucleotide sequences of the HA gene of Iraqi viruses confirmed that these Iraqi viruses are genetically related to and divided into two subgroups based on the degree of similarity. This finding was in accordance with that of Langeroudi et al. who reported different ranges of identity among some Iranian H9N2 isolates extended from the minimum (91.1%) to the maximum identity (99.11%) [20]. Previous studies suggested that H9N2 group was not homogenous so that the viruses may be distributed into some subgroups, associated with the time of their isolation [21, 22]. A recent study has shown that the HA sequences of the Egyptian H9N2 viruses segregate into three subgroups, and this segregation is not correlated with the time of occurrence, region, or host species and seems to reflect a star-like diversification of these sequences [23]. Genetic diversity was demonstrated in all gene segments of AIVs, but most notably in the HA gene [24]. Molecular analysis of NA nucleotide sequences proved that current Iraq H9N2 viruses are closely connected to each other with a high identity (94.3–100%). These findings are in agreement with those of Naguib et al. [23] who stated that the NA gene sequences of the Egyptian H9N2 viruses similarly reveal a shallow sub-clustering. In contrast, the antigenic sites of NA in the Iranian H9N2 isolates have varied in a yearly manner and can be divided into two main subgroups [25]. Nevertheless, some exceptions are seen in the Iranian H9N2 isolates (A/chicken/Iran/RZ53/2008/H9N2) which were placed in the first subgroup, including viruses isolated from 1998 to 2004 [21]. These variations in the range of similarity may be attributed to the accumulation of mutation among the viruses circulating within the poultry reservoirs [26]. Genetic comparisons of the nucleotide sequences of the HA and NA genes in the Iraqi viruses with the corresponding gene of the H9N2 viruses isolated in Iran, Pakistan, Lebanon, and Israel showed that these viruses are genetically related to each other. Recently, Ye et al. [27] have demonstrated that the Israeli (Is- 2007) H9N2 isolate, which has shown more than 90% nucleotide identity with HA and NA gene sequences of the Iraqi H9N2 viruses in the present study, clustered together with H9N2 viruses isolated in Iran and Pakistan during the period of 2005–2008. It has been observed that HA gene sequence of the Iranian isolate (Ck/IR/ZMT-101/98) shared a common ancestor along with the Pakistani isolates; in addition, all the Pakistani isolates were closely related to H9N2 viruses isolated from Iran between 2004 and 2007 [19, 28]. In other study, Norouzian et al. [25] suggested that the NA gene sequences of the Iranian and the Pakistani H9N2 isolates are not different in regard to the progenitor. They concluded that the origin of the isolates circulating in poultry farms of Iran may be traced back to Pakistan. The phylogeny-based analysis of geographical association identifies avian populations from Pakistan and Iran as two possible sources of the H9N2 viruses in Central Asia and the Middle East [29]. Interestingly, the phylogenetic analysis of NA genes showed a 93.1–95.6% identity between all the Iraqi viruses and Ahwaz strain. These results may be attributed to the geographical location of Ahwaz province which is situated close to the Iraqi border. In addition to the presence of marshes in this Iranian province, which are closely related to the marsh of southern Iraqi provinces, the presence of migratory birds coming from Russia and China staying for several months in these marshes contributes to the transmission of the disease to poultry farms of Iraq and Iran. It is well documented that the migratory water birds are responsible for the distribution of different AIV subtypes to humans and animals all over the world [30, 31]. The receptor-binding site motif of HA is critical for cellular receptor specificity and determining virus host range [32]. In this study, the key residues in the receptor-binding sites of HA protein were studied, with a comparative approach to the previous AIVs. All HA proteins of Iraqi H9N2 viruses possessed H, A, and L at positions 183, 190, and 226 in the receptor-binding sites, respectively. A similar result was reported by Iqbal et al. [19] that all UDL viruses from Pakistan in 2005–2008 possessed H, A, and L at positions 183, 190, and 226. The current study revealed that the six recent H9N2 viruses with previous Iraqi strain (A/chicken/IraqEKI14/2008) possessed amino acid L226 at the left edge of binding pocket (NGLIGR), indicating its potential to infect humans. The substitution of Gln to Leu at residue 226 in HA allows H9N2 viruses to replicate more efficiently in human airway epithelial cells cultured in vitro [33]. Two kinds of motif GTSKA and GTSKS were detected in this study and this result in accordance with that of Langeroudi et al. [20] who reported new motif in this region in viruses that were isolated after 2006 (TH85, TH186, TH286, TH386). Remarkably, the residue at position 190 within the receptor-binding sites has been recorded to influence the affinity to the sialyl-α2, 6-galactose linkage. The binding affinity is at the weakest level with A at position 190. Also, it is at the intermediate level with T, and at the highest level with V at the same position [34]. It can be predicted that the Iraqi viruses have a weaker affinity binding to a human-like receptor, but it seems that the potential of Iraqi AIVs to infect human should be considered. Our analysis indicated that the presence of H9N2 in Iraq is an expected event because of its endemic circulation in the neighboring countries. Although vaccination campaigns were implemented in several Asian countries, to date H9N2 is still evolving, spreading, and causing severe problems to the poultry industry. It is, however, difficult to determine the time and the mechanism of introduction of this subtype in Iraq. The remarkable similarity between the Iraqi viruses and those from Iran and Pakistan indicates possible epidemiological links and the fact that these viruses could stem from a common origin. Especially in recent years, the Iraqi poultry and poultry products have not met the standards of the Iraqi markets; consequently, Iraq has been forced to import these products from the neighboring countries such as Iran, Turkey, and Saudi Arabia, which may have facilitated virus transmission to and from Iraq to these countries. The trade in poultry and poultry products seems to play the major role in the spread of H9N2 viruses among Central Asian and Middle Eastern countries [29]. Finally, H9N2 viruses from Iraq, Iran, and Pakistan share a common ancestor. Indeed, it is not easy to predict the time of virus existence due to a gap of sequence data for Iraq. From 2008 up to now, there have been single available HA gene data of an H9N2 virus detected in a chicken in Iraq. The present study highlights the need to continue monitoring AIV in Iraq poultry farms and to confirm the role of migratory birds to disseminate AIV in Iraq marshes throughout the years. Obtaining further information on genetic changes could lead to the prediction and control the rapid spread of H9N2 virus in the field. In conclusion, quarantine and control measures seem to be very critical for controlling the H9N2 circulation in the Middle Eastern countries, including Iraq.
References
D.J. Alexander, Vet. Microbiol. 74, 3–13 (2000)
K. Butt, G.J. Smith, H. Chen, L. Zhang, Y.C. Leung, K. Xu, W. Lim, R.G. Webster, K. Yuen, J.M. Peiris, J. Clin. Microbiol. 43, 5760–5767 (2005)
W. Chen, P.A. Calvo, D. Malide, J. Gibbs, U. Schubert, I. Bacik, S. Basta, R. O’Neill, J. Schickli, P. Palese, Nat. Med. 7, 1306–1312 (2001)
R.A. Fouchier, V. Munster, A. Wallensten, T.M. Bestebroer, S. Herfst, D. Smith, G.F. Rimmelzwaan, B. Olsen, A.D. Osterhaus, J. Virol. 79, 2814–2822 (2005)
J. Banks, E. Speidel, P. Harris, D. Alexander, Avian Pathol. 29, 353–359 (2000)
K. Urbaniak, A. Kowalczyk, I. Markowska-Daniel, Acta Biochim. Pol. 61, 433–439 (2014)
B. Mänz, M. Schwemmle, L. Brunotte, J. Virol. 87, 7200–7209 (2013)
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56, 152–179 (1992)
L.V. Gubareva, L. Kaiser, F.G. Hayden, The Lancet 355, 827–835 (2000)
Y. Guan, K. Shortridge, S. Krauss, P. Chin, K. Dyrting, T. Ellis, R. Webster, M. Peiris, J. Virol. 74, 9372–9380 (2000)
X.J. Xu, G.Y. Xu, H.B. Zhou, Z.J. Yu, A.D. Zhang, Y.F. Song, M.L. Jin, H.C. Chen, Virus Genes 36, 79–83 (2008)
K.M. Abdul-Sada, Int. J. Adv. Res. 3, 170–176 (2015)
I. Monne, S. Ormelli, A. Salviato, C. De Battisti, F. Bettini, A. Salomoni, A. Drago, B. Zecchin, I. Capua, G. Cattoli, J. Clin. Microb. 46, 1769–1773 (2008)
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Petez, Arch. Virol. 153, 651–655 (2001)
K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol. Biol. Evol. 28, 2731–2739 (2011)
E.J. Khamas, Iraqi Vet. Med. J. 32, 223–230 (2008)
A. Al-Dabhawe, H. Kadhim, H. Samaka, Iraqi J. Vet. Sci. 27, 97–101 (2013)
A.M. Al-Mohana, H.M. Kadhimv, A.H. Al-Charrakh, Z. Al-Habubi, F.H. Nasir, S.A. Al-Hilali, Z.J. Hadi, Iraq Sci. Res. Essays 8, 1191–1195 (2013)
M. Iqbal, T. Yaqub, K. Reddy, J.W. McCauley, PLoS ONE 4, 1–18 (2009)
A.G. Langeroudi, V. Karimi, M.T. Kheiri, A. Barin, Comp. Clin. Pathol. 22, 321–330 (2013)
M. Soltanialvar, H. Shoushtari, M. Bozorgmehrifard, S. Charkhkar, F. Akbarnejad, Trop. Anim. Health Pro. 44, 419–425 (2012)
M. Soltanialvar, H. Shoushtari, M. Bozorgmehrifard, S. Charkhkar, F. Eshratabadi, J. Biol. Sci. 10, 145–150 (2010)
M.M. Naguib, A.S.A. Arafa, M.F. El-Kady, A.A. Selim, V. Gunalan, S. Maurer-Stroh, K.V. Goller, M.K. Hassan, M. Beer, E. Abdelwhab, Infect. Genet. Evol. 34, 278–291 (2015)
A. Lorusso, A.L. Vincent, M.L. Harland, D. Alt, D.O. Bayles, S.L. Swenson, M.R. Gramer, C.A. Russell, D.J. Smith, K.M. Lager, N.S. Lewis, J. Gen. Virol. 92, 919–930 (2011)
H. Norouzian, M. Bashashati, M. Vasfimarandi, Iran J. Microbiol. 6, 91–97 (2014)
A. Bozorgi, H. Keyvanfar, H. Shushtari, M. Bahmaninejad, F. Eshratabadi, Afr. J Microbiol. Res. 6, 4550–4556 (2012)
T. Ye, N. Golender, I. Shkoda, M. Drabkin, K. Lapin, A. Panshin, Actual Probl. Transp. Med. 1, 112–136 (2015)
M. Bashashati, M.V. Marandi, F. Sabouri, Arch. Virol. 158, 2089–2100 (2013)
A. Fusaro, I. Monne, A. Salviato, V. Valastro, A. Schivo, N.M. Amarin, C. Gonzalez, M.M. Ismail, A.R. Al-Ankari, M.H. Al-Blowi, O.A. Khan, A.S. Maken Ali, A. Hedayati, J. Garcia Garcia, G.M. Ziay, A. Shoushtari, K.N. Al Qahtani, I. Capua, E.C. Holmes, G. Cattoli, J. Virol. 85, 8413–8421 (2011)
S.N. Bevins, K. Pedersen, M.W. Lutman, J.A. Baroch, B.S. Schmit, D. Kohler, T. Gidlewski, D.L. Nolte, S.R. Swafford, T.J. DeLiberto, PLoS ONE 9, 1371–1379 (2014)
D.J. Prosser, L.L. Hungerford, R.M. Erwin, M.A. Ottinger, J.Y. Takekawa, E.C. Ellis, Front. Public Health 1, 1–11 (2013)
A. Gambaryan, R. Webster, M. Matrosovich, Arch. Virol. 147, 1197–1208 (2002)
H. Wan, D.R. Perez, J. Virol. 81, 5181 (2007)
M.N. Matrosovich, S. Krauss, R.G. Webster, Virology 281, 156–162 (2001)
Acknowledgements
We are thankful to the staff members of the Iraqi Veterinary Hospital, specially Dr. Haider Kadhim and Dr. Atheer Qasim for their excellent supports. This project was financially supported by a Grant (No. 28088/6/12) from the research council of the University of Tehran and a Grant (No. 3.11.2420) from ministry of higher education and scientific research of Iraq, University of Basra. Finally, the authors gratefully acknowledge Dr. Iraj Ashrafi, Mr. Ahmad Vahedi and Mr. Behrooz Asadi for their extensive technical supports.
Author’s contribution
Qayssar Ali Kraidi, Omid Madadgar, Vahid Karimi, and Arash Ghalyanchi Langeroudi made substantial contributions to the conception and design, acquisition of data, analysis and interpretation of data. Qayssar Ali Kraidi, Omid Madadgar, and Arash Ghalyanchi Langeroudi participated in the sampling, diagnosis, molecular detection, technical supports, and preparation of manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Edited by Keizo Tomonaga.
Rights and permissions
About this article

Cite this article
Kraidi, Q.A., Madadgar, O., Ghalyanchi Langeroudi, A. et al. Genetic analysis of H9N2 avian influenza viruses circulated in broiler flocks: a case study in Iraq in 2014–2015. Virus Genes 53, 205–214 (2017). https://doi.org/10.1007/s11262-016-1407-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-016-1407-x