
Abstract
The nucleocapsid protein (NP) of rice stripe virus (RSV) encapsidates viral genomic RNAs to form virion. The binding of NP with RNA is essential for the formation of virus particle. In this study, the binding specificity of RSV NP to RNA and the domains within the NP that mediate this interaction were investigated by gel electrophoretic mobility shift assays and Northwestern blot analysis. The results demonstrated that RSV NP was able to bind to all synthetic RNAs and DNAs without sequence specificity. Using a series of truncated NPs expressed in E. coli and synthetic peptides, we mapped the RNA-binding domain of NP to the central region from amino acid residues 201–232. Further alanine substitution analysis revealed that Lys206, Lys207, Lys220, and Tyr221 in the RNA-binding domain were essential for NP to bind with RNA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice stripe disease is one of most serious viral diseases of rice and causes severe damage to rice crops in many East Asian countries. This disease is caused by rice stripe virus (RSV) and mainly transmitted by the small brown planthopper (Laodelphax striatellus) in a persistent-propagative manner [1].
RSV is the type species of genus Tenuivirus and has filamentous particle that contains four negative-sense single-stranded RNA segments encapsidated by nucleocapsid proteins (NP) and several copies of RNA-dependent RNA polymerase (RdRp) [2]. RNA1 is negative-sense and encodes viral RdRp which is responsible for both transcription and replication of genomic RNAs [3]. RNA2, RNA3, and RNA4 are all ambisense and each encodes two proteins by viral sense RNA (vRNA) and viral complementary-sense RNA (vcRNA), respectively. vRNA2 and vcRNA2 encode a RNA-silencing suppressor P2 [4] and a membrane protein Pc2 [5, 6], respectively. vRNA3 and vcRNA3 encode a second viral suppressor P3 [7] and NP [8], respectively. vRNA4 and vcRNA4 encode a disease-specific protein P4 [9] and a movement protein Pc4 [10], respectively.
Tenuiviruses have a close phylogenetic relationship with the viruses in the family Bunyaviridae because they have similar genome organization and gene expression strategies [2]. Furthermore, some proteins of tenuiviruses share sequence homologies to those of bunyaviruses [3, 5]. For the segmented, negative-sense RNA viruses, encapsidation of viral genomic RNA by NP plays multiple important roles in the viral replication cycles including transcription, replication and the assembly of viral particles. Viruses in the family Bunyaviridae only use the ribonucleoprotein complexes (RNPs), but not the naked RNAs as the template for viral genome replication and gene transcription [11]. The genomic and antigenomic RNAs of these viruses contain highly conserved, complementary terminus sequence which might serve as the signal for encapsidation. The location and specificity of RNA responsible for binding with NP have been reported for several bunyaviruses but the data were conflicting. The N protein of Jamestown Canyon virus (JCV) has been reported to prefer to bind the 5′-end of genomic RNAs [12]. The trimeric Hantavirus N protein specifically recognized the panhandle structure formed by the base pairing of the 5′ and 3′ complementary ends [13]. However, Tomato spotted wilt virus (TSWV) N protein has been shown to bind ssRNA irrespective of sequence [11].
RSV NP is the major component of the viral particle. The terminal ten bases of all four genomic RNA segments are highly conserved and the 5′ and 3′ termini of each segment are complementary to each other to support the formation of base-paired panhandle structure [2]. The 3′-terminal conserved sequence has been proposed to serve as promoter for the viral RdRp to drive transcription [14]. It has been reported that the C-terminal part of RSV NP could bind with nucleic acids in vitro [15]. However, it is unclear that the precise RNA-binding domain of RSV NP and whether NP specifically recognize the specific sequence of genomic RNA. In order to understand the process of RSV NP encapsidating genome RNA, we have investigated the specificity of RNA-NP interaction and the RNA-binding domains of RSV NP by electrophoretic mobility shift assay (EMSA) and Northwestern blot analysis in this study. The NP was found to bind RNA in a sequence-independent manner and the amino acid residues Lys206, Lys207, Lys220, and Tyr221 on NP were essential for RNA binding.
Materials and methods
Virus and RNA extraction
Rice leaves infected with RSV were collected from a rice field in Jiangsu Province, China. Total RNA was extracted from the infected leaves using RNAsimple Total RNA Kit (Tiangen) according to the manufacturer’s instruction.
Construction of recombinant plasmids used for expression of truncated RSV NP
The full length of RSV NP coding region was amplified by RT-PCR using primer pair np-F1/np-R1 (Table 1). The N-terminus deletion mutants (ΔN70, ΔN130, ΔN170, ΔN230, and ΔN280) were generated using the same reverse primer (np-R1) and different forward primers (Table 1). The C-terminus deletion mutants (ΔC40, ΔC90, ΔC150, ΔC190, and ΔC250) were created using the same forward primer (np-F1) and different reverse primers (Table 1). The mutants 171–282 and 171–232 were created using the primer pairs np-F4/np-R2 and np-F4/np-R3 (Table 1), respectively. Each of the forward and reverse primers contained a BamHI and a HindIII restriction enzyme site, respectively. The PCR products were purified, digested by BamHI and HindIII, ligated with pMal-c2x (New England Biolabs), and transformed into competent E. coli DH5α cells. The recombinant expression plasmids were extracted and identified by BamHI/HindIII.
Expression and purification of wile-type NP and its deletion mutants
The recombinant expression plasmids (pMal-NP, pMal-ΔN70, pMal-ΔN130, pMal-ΔN170, pMal-ΔN230, pMal-ΔN280, pMal-ΔC40, pMal-ΔC90, pMal-ΔC150, pMal-ΔC190, pMal-ΔC250, pMal-171–282, and pMal-171–232) were, respectively, transformed into BL21 (DE3) competent cells. Protein expression was induced with 0.4 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 3 h and purified as MBP-tagged fusion proteins by amylose affinity chromatography according to the protocol of pMal protein fusion and purification system (New England Biolabs). Purified proteins were concentrated by centrifugation using Amicon® Ultra-15 centrifugal filters (Millipore) and protein concentrations were measured by the BCA Protein Assay Kit (Tiangen). Then, purified proteins were separated on 10 % SDS-PAGE followed by staining with Coomassie Brilliant Blue-R250.
Preparation of RNA and DNA
The full length of RSV S4 segment was RT-PCR amplified from the total RNA extracted from RSV-infected rice leaves using the primer S4-F1 (5′-TAATACGACTCACTATAGACACAAAGTCCAGGGCATTTG-3′) and S4-R1 (5′-ACACAAAGTCAGGGCATATC-3′). The 5′-terminus 271 bp region of S4 was amplified using the primer S4-F1 and S4-R2 (5′-AAAGGGTGTCAGTCTCCA-3′). The 3′-terminus 173 bp of S4 was amplified using the primer S4-F2 (5′-TAATACGACTCACTATAGAGGCCTCTTGGAGAGAGC-3′) and S4-R1. The primers p4-F (5′-TAATACGACTCACTATAGATGCAAGACGTACAAAGG-3′) and p4-R (5′-CTATGTTTTATGAAGAAG-3′) were used to amplify the open reading frame (ORF) of P4. T7 promoter was added to the 5′-end of each forward primer. The PCR products were purified and ligated with pMD19-T vector (TaKaRa) to generate pMD-S4, pMD-S4-5′, pMD-S4-3′, and pMD-P4, respectively. The cDNA sequence of S4 contained two StuI sites, so the recombinant plasmid pMD-S4-5′-3′ containing both 5′-terminus 102 bp and 3′-terminus 173 bp of S4 was constructed by digestion of pMD-S4 with StuI. After being separated by gel electrophoresis, the larger linearized fragment containing both 5′-terminus 102 bp and 3′-terminus 173 bp of S4 was purified and self-ligated.
For generation of ssRNA, the above constructed plasmids pMD-S4, pMD-S4-5′, pMD-S4-3′, pMD-P4, and pMD-S4-5′-3′ were linearized by SalI and then used for in vitro transcription reactions. pGEM-7Zf (+) was digested with SmaI and used for in vitro transcription of a nonviral RNA. RNA transcripts were produced from the linear DNAs with a MEGAscript T7 Transcription Kit (Invitrogen) as instructed in the protocols.
For generation of dsRNA, S4 segment was amplified with the primer S4-F1 and S4-R3 (5′-CATTTAGGTGACACTATAACACAAAGTCAGGGCATATC-3′). T7 and SP6 promoters were added to the 5′-ends of forward and reverse primers, respectively. PCR product was purified and used as the template for in vitro transcription. Plus- and minus-sense ssRNA were produced by in vitro transcription with T7 and SP6 RNA polymerase, respectively. The dsRNA was generated by mixing the equimolar amounts of plus- and minus-sense ssRNA, heating at 95 °C for 10 min and then annealing at room temperature. The dsDNA was PCR product of RSV S4. The ssDNA was generated by heating the dsRNA 95 °C for 10 min and then placing on ice immediately.
Electrophoretic mobility shift assay
For the electrophoretic mobility shift assay (EMSA), RNA or DNA was mixed with purified protein or synthetic peptides in binding buffer (10 mM Tris–HCl [pH 7.4], 100 mM NaCl, 0.2 mM DTT, 5 % glycerol) and incubated for 30 min at room temperature in a total volume of 10 μl. Subsequently, the mixtures were loaded onto 0.8 % agarose gels and separated by electrophoresis. After electrophoresis, RNA or DNA was stained with ethidium bromide (EB) and visualized by UV light.
Northwestern blot
After separated by 10 % SDS-PAGE, recombinant proteins were transferred to nitrocellulose membrane. The membrane was blocked for 2 h at room temperature in binding buffer (10 mM Tris–HCl [pH6.8], 1 mM EDTA, 25 mM NaCl, 0.05 % Ficoll400, 0.05 % polyvinyl pyrrolidone, 0.05 % BSA) containing 10 μg/ml tRNA. The biotin-labeled RNA probe was synthesized from the linearized pMD-S4 described above using biotin RNA labeling mix (Roche). Subsequently, the biotin-labeled RNA probe was added and incubated for 2 h at room temperature, followed by the addition of streptavidin–alkaline phosphatase conjugate (Pierce). Finally, the membrane was developed with BCIP/NBT alkaline phosphatase substrate (Beyotime).
Results
Expression and purification of NP and its truncated mutants
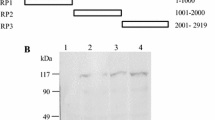
To map the RNA-binding domain of RSV NP, 12 truncated mutants were generated to remove the N-terminal or/and C-terminal regions. The N-terminally truncated mutants included ΔN70, ΔN130, ΔN170, ΔN230, and ΔN280, which removed 70, 130, 170, 230, and 280 amino acids (aa) from the N-terminus. The C-terminally truncated mutants included ΔC40, ΔC90, ΔC150, ΔC190, and ΔC250, which removed 40, 90, 150, 190, and 250aa from the C-terminus. The mutants 171–282 and 171–232 had both N-terminal and C-terminal deletions. A schematic diagram of all of these truncated NPs is shown in Fig. 1a. Each of wild-type and truncated NPs was expressed as an N-terminally MBP-tagged protein in E. coli, purified by amylose affinity chromatography and examined for its molecular weight and level of purity by 10 % SDS-PAGE. SDS-PAGE showed that only a single band at the expected size for NP, ΔN70, ΔN130, ΔN170, ΔN280, ΔC40, ΔC90, ΔC150, ΔC190, ΔC250, and 171–282, which suggest a high level of purity. ΔN230 and 171–232 showed less homogenous and two bands were found for them. One band was at the expected size for MBP-fused protein, and another band was at a lower molecular mass of about 43 kDa (Fig. 1b). Further analysis by Western blotting using rabbit polyclonal antiserum raised against NP confirmed these purified proteins were NPs (Fig. 1c). Only a single band was detected for all purified proteins (Fig. 1c), suggesting the band with a lower molecular mass found in ΔN230 and 171–232 was not the NP and it might represent cleaved MBP.

The full-length and truncated RSV NPs used for EMSA. a Schematic diagram of recombinant RSV NP and its deletions. The names of the deletions were shown on the left and the amino acids and predicted molecular weight were shown on the right. The symbols ΔN and ΔC along with the number referred to the amino acids truncated in the N or C-terminus, respectively. b SDS-PAGE analysis of wild and truncated RSV NPs purified by affinity chromatography. The sizes of the molecular weight standards (lane M) were indicated on the left. c Western blot analysis of purified proteins using anti-NP polyclonal antibody
EMSA analysis of the binding affinity of NP to RSV genomic RNA
The interaction between NP and genomic RNA of RSV was investigated with EMSA experiment. The increasing amount of purified MBP-NP was incubated with the in vitro transcribed RSV S4 ssRNA and then separated by agarose gel electrophoresis. In the absence of NP, RNA was found totally in unbound form. In the presence of small amount of NP, RNA was found mostly in unbound form. With increasing the amount of NP in the reaction mixtures, more and more RNA was transited from unbound form to bound form (data not shown), which indicated that NP was able to bind to RSV S4 ssRNA.
Specificity of RNA–NP interaction
It has been reported with some segmented RNA viruses that nucleocapsid proteins preferred to bind with 5′-terminus [12] or the panhandle structure formed by 5′- and 3′-termini of genomic RNA [13]. To examine whether the termini of RSV genomic RNA were also responsible for the interaction with NP, the 5′-end, 3′-end, the region including both 5′ and 3′-ends, P4 coding region containing neither 5′-end nor 3′-end of RSV S4, and nonviral RNA were generated by in vitro transcription and used for EMSA. EMSA showed that all of these RNAs could from protein-RNA complexes with RSV NP (Fig. 2a), suggesting the interaction between RSV NP and RNA did not depend on the sequence of RNA.

EMSA of RSV NP binding to various synthetic RNA and DNA. a The binding of RSV NP to ssRNA with different sequence. 300 ng of each ssRNA was incubated with (plus) or without (minus) 1.0 μg RSV NP in EMSA buffer. Then, the reaction mixtures were loaded on 0.8 % agarose gel, separated by electrophoresis, stained with EB and detected by a UV transilluminator. S4, S4-5′, S4-3′, S4-5′3′, P4 ORF, and pGEMZf represented the full length of RSV S4, 5′-terminus of S4, 3′-terminus of S4, both 5′- and 3′-ends of S4, the ORF of P4 gene and nonviral RNA transcribed from the plasmid pGEMZf, respectively. b The binding of RSV NP to ssRNA, dsRNA, ssDNA, and dsDNA
To investigate whether recombinant NP was able to bind to other nucleic acids as well as ssRNA, we also tested the interactions of NP with dsRNA, ssDNA, and dsDNA via EMSA. EMSA revealed that migration of dsRNA, ssDNA, and dsDNA was significantly retarded as well as ssRNA in the presence of NP (Fig. 2b), indicating NP could also bind to dsRNA and DNA.
RNA-binding domain of NP
Five N-terminal and five C-terminal deletions of NP were firstly produced for analysis of their interactions with vitro transcribed RSV S4 RNA by EMSA. MBP protein was used as a negative control. In EMSA, Protein-RNA complexes were observed in the reaction mixtures containing wild-type NP, ΔN70, ΔN130, ΔN170, ΔC40, and ΔC90, while no Protein-RNA complexes was observed in reactions containing ΔN230, ΔN280, ΔC150, ΔC190, ΔC250 as well as negative control MBP (Fig. 3a). These results indicated that the region 171–232aa was essential for the RNA-binding activity of NP. To confirm this conclusion, we also expressed and purified 171–282 and 171–232, and examined for their binding affinities with RNA. Protein-RNA complex was observed in reaction mixture containing either 171–282 or 171–232, which demonstrated that these two proteins were able to bind with RNA (Fig. 3a). To validate the EMSA results, we next performed Northwestern blot experiment to test the interactions between these truncated NPs and RNA. Those truncated NPs that maintained 171–232aa (NP, ΔN70, ΔN130, ΔN170, ΔC40, ΔC90, 171–282, and 171–232) retained the RNA-binding activity, while other truncated NPs that did not contain this region (ΔN230, ΔN280, ΔC150, ΔC190, ΔC250) could not bind to RNA (Fig. 3b). Therefore, the results obtained from Northwestern blot assays were in agreement with those obtained with EMSA experiments: the region from 171 to 232aa contained the RNA-binding domain.

Mapping the RNA-binding domain in RSV NP. a Analysis of the interaction between truncated NPs and RNA by EMSA. Wild and truncated NPs were incubated with RSV S4 ssRNA followed by separating by gel electrophoresis. Each protein used in EMSA was shown on the top of each lane. “0” indicated that no protein was added in the reaction mixtures. b Northwestern blot analysis of the interaction between truncated NPs and RNA. The purified recombinant proteins were transferred to nitrocellulose membrane and then probed with biotin-labeled ssRNA probe
To further localize the RNA-binding domain in 171–232aa, two peptides encompassing 171–200aa and 201–232aa were synthesized and analyzed their RNA-binding activities. One peptide 201–232aa had good binding activity, while another peptide 171–200aa had little RNA-binding activity as nearly no protein-RNA complex was observed (Fig. 3a). These data suggested a region from amino acid residues 201–232 of NP mainly responsible for NP–RNA interaction.
RNA-binding sites on NP
It was reported that the charged and polar amino acids (Lys, Arg, Asp, Glu, Thr, Ser and Tyr) usually interacted with nucleic acids. To map the critical amino acids involved in NP–RNA interaction, the charged and polar amino acids within the RNA-binding domain were selected for point mutagenesis. We then synthesized seven peptides with one or more amino acids substitution (K206A/K207A, R210A, K214A, D215A/217A, K220A/Y221A, T222A/T223A, and K231A) and evaluated the RNA-binding activity of these mutants. EMSA showed that the mutants of R210A, K214A, D215A/217A, T222A/T223A, and K231A could interact with RNA to form protein-RNA complexes (Fig. 4). In contrast, nearly no protein-RNA complex was detected in K206A/K207A or K220A/Y221A (Fig. 4). These data demonstrated that amino acids Lys206, Lys207, Lys220, and Tyr221 on NP were essential for RNA binding.

EMSA of alanine substitution mutants of NP. Each NP mutant with one or more amino acids substitution was incubated with RSV S4 vRNA. The reaction mixtures were loaded onto a 0.8 % agarose gel and the protein-RNA complexes were separated from free RNA by gel electrophoresis as described in “Materials and methods” section
Discussion
RSV NP is the main structural protein that plays an important role in the assembly of the virion [2]. During the assembly of viral particles, the NP need to recognize and selectively bind to viral RNAs and then package them into the virion. Thus, here we examined the domains of NP and specific nucleotide sequence of RNA responsible for the interaction between NP and genomic RNA.
The data here showed that no specific sequence of RNA was required for binding with RSV NP. This finding was consistent with recent data from TWSV and Bunyamwera virus (BUNV) studies [11, 16]. Study on TWSV N protein showed that it could bind nonspecifically to E. coli cellular RNA [11]. Study on BUNV suggested that N protein did not obligatorily require a sequence or structure for RNA encapsidation because neither common linear sequence nor structure was present in the extracted RNAs bound to RNPs [16]. Normally there must exist a mechanism to distinguish viral genomic RNAs and other RNAs so as to ensure that the genomic RNAs are correctly encapsidated into viral particles. In this study, the interaction between RSV NP and RNA was examined in vitro. In the virus-infected cells, replication of genomic RNAs or assembly of viral particles happened in the particular regions of cells. In these regions, the amount of genomic RNAs was probably much higher than that of other cellular RNAs so that NP preferred to bind with genomic RNA. It was also probable that other factors in virus-infected cells could help NP to selectively recognize genomic RNA. Additional studies will require establishment of reverse genetic system to analyze which mutant genomic RNAs could be packaged into virions in vivo.
Both EMSA and Northwestern blot analysis using a series of truncated NPs identified a RNA-binding region from the amino acid 201–232. No sequence similarity between RSV NP binding domain and other characterized nucleic acid binding proteins was found. However, like other identified nucleic acid binding domains, a vast amount of charged and polar amino acids were found in the RNA-binding domain of RSV NP. The charged or polar amino acids on NPs of some bunyaviruses were previously reported to be responsible for NP–RNA interaction [11, 17, 18]. For example, the residues of BUNV NP that interact with the RNA were primarily charged residues, including S14, S15, D18, R47, K50, T75, R94, Y176, R182, Q183, R184, and N217 [17]. The residues of E192, Y206, and S217 of Hantaan virus (HTNV) were critical for RNA recognition [18]. Four amino acid residues R94, R95, K183, and Y184 on the surface cleft of TWSV N protein were important for NP binding to RNA [11]. Here, K206, K207, K220, and Y221 on RSV NP were identified as being essential for RNA binding, which also support that the charged or polar amino acids were involved in nucleic acid binding.
In a recent study, the N-terminal 1–47aa was reported to be important for RSV NP self-interaction [8]. Here, we showed that the deletion of N-terminal 1–70aa did not affect the RNA-binding activity of RSV NP. Therefore, these results suggested that NP multimerization and NP–RNA binding might be independent. Similar findings have been reported with other viral nucleocapsid proteins. For example, the RNA-binding domain of HTNV N protein has been mapped to a central region from amino acids 175–217 [19], while both the N-terminal 43 amino acid residues and the C-terminal region between 383 and 398 amino acids contributed the homotypic interaction of N protein [20]. Another example is TWSV, a plant virus in the family Bunyaviridae. Studies on TWSV showed that neither the higher oligomeric formation of N protein depended on the binding of RNA nor the ability of binding RNA required the oligomerization of N protein [11].
In conclusion, we found that RSV NP bound to either RNA or DNA with sequence-independence in vitro and mapped the region of NP mainly responsible for this interaction to amino acid residues 201–232. Lys206, Lys207, Lys220, and Tyr221 on NP were identified by further alanine substitution analysis as the important amino acids for RNA binding. Further studies will be required to detect the specificity of NP–RNA interaction in vivo and explore the structure basis for this interaction.
References
Y. Huo, W. Liu, F. Zhang, X. Chen, L. Li, Q. Liu, Y. Zhou, T. Wei, R. Fang, X. Wang, PLoS Pathog. 10, e1003949 (2014)
B. Falk, J. Tsai, Annu. Rev. Phytopathol. 36, 139–163 (1998)
S. Toriyama, M. Takahashi, Y. Sano, T. Shimizu, A. Ishihama, J. Gen. Virol. 75, 3569–3579 (1994)
Z. Du, D. Xiao, J. Wu, D. Jia, Z. Yuan, Y. Liu, L. Hu, Z. Han, T. Wei, Q. Lin, Z. Wu, L. Xie, Mol. Plant Pathol. 12, 808–814 (2011)
S. Zhao, G. Zhang, X. Dai, Y. Hou, M. Li, J. Liang, C. Liang, Virology 429, 148–154 (2012)
M. Yao, X. Lu, S. Li, Y. Xu, Y. Zhou, X. Zhou, X. Tao, J. Virol. 88, 3223–3234 (2014)
R. Xiong, J. Wu, Y. Zhou, X. Zhou, Virology 387, 29–40 (2009)
S. Lian, W. Cho, Y. Jo, S. Kim, K. Kim, Virus Res. 183, 6–14 (2014)
W. Wu, L. Zheng, H. Chen, D. Jia, F. Li, T. Wei, PLoS One 9, e88636 (2014)
C. Zhang, X. Pei, Z. Wang, S. Jia, S. Guo, Y. Zhang, W. Li, Virology 425, 113–121 (2012)
J. Li, Z. Feng, J. Wu, Y. Huang, G. Lu, M. Zhu, B. Wang, X. Mao, X. Tao, J. Biol. Chem. 290, 3950–3961 (2015)
M. Ogg, J. Patterson, J. Virol. 81, 13754–13760 (2007)
M. Mir, B. Brown, B. Hjelle, W. Duran, A. Panganiban, J. Virol. 80, 11283–11292 (2006)
P. Barbier, M. Takahashi, I. Nakamura, S. Toriyama, A. Ishihama, J. Virol. 66, 6171–6174 (1992)
D. Liang, X. Ma, Z. Qu, R. Hull, Virus Genes 31, 203–209 (2005)
B. Mohl, J. Barr, RNA 15, 391–399 (2009)
B. Li, Q. Wang, X. Pan, C. de Fernandez, Y. Sun, Y. Guo, X. Tao, C. Risco, S. Sui, Z. Lou, Proc. Natl. Acad. Sci. USA. 110, 9048–9053 (2013)
W. Severson, X. Xu, M. Kuhn, N. Senutovitch, M. Thokala, F. Ferron, S. Longhi, B. Canard, C. Jonsson, J. Virol. 79, 10032–10039 (2005)
X. Xu, W. Severson, N. Villegas, C. Schmaljohn, C. Jonsson, J. Virol. 76, 3301–3308 (2002)
P. Kaukinen, A. Vaheri, P. Alexander, J. Virol. 77, 10910–10916 (2003)
Acknowledgments
This research was supported by Natural Science Foundation of Jiangsu Province (BK20130441).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Thomas Hohn.
Rights and permissions
About this article

Cite this article
Zhao, S., Xue, Y., Hao, J. et al. The RNA-binding properties and domain of Rice stripe virus nucleocapsid protein. Virus Genes 51, 276–282 (2015). https://doi.org/10.1007/s11262-015-1235-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1235-4