
Abstract
The spike (S) protein of porcine transmissible gastroenteritis virus (TGEV) is located within the viral envelope and is the only structural protein that possesses epitopes capable of inducing virus-neutralizing antibodies. Among the four N-terminal antigenic sites A, B, C, and D, site A and to a lesser extent site D (S-AD) induce key neutralizing antibodies. Recently, we expressed S-AD (rS-AD) in recombinant form. In the current study, we used the rS-AD as an immobilized target to identify peptides from a phage-display library with application for diagnosis. Among the 9 phages selected that specifically bound to rS-AD, the phage bearing the peptide TLNMHLFPFHTG bound with the highest affinity and was subsequently used to develop a phage-based ELISA for TGEV. When compared with conventional antibody-based ELISA, phage-mediated ELISA was more sensitive; however, it did not perform better than semi-quantitative RT-PCR, though phage-mediated ELISA was quicker and easier to set up.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transmissible gastroenteritis virus (TGEV) is a member of the Coronaviridae family and is a major cause of enteric disease in pigs where it threatens swine production and triggers substantial economic losses in the industry [1–4]. Its genome is composed of positive-stranded RNA approximately 28.5-kb in length. The virus consists of four structural proteins: envelope (E), membrane (M), spike (S), and nucleocapsid (N) proteins [1, 3, 5]. Non-structural proteins, which comprise two-thirds of the 5′-proximal end, are encoded by open reading frames 1a and 1ab as well as the replicase. In contrast, the 3′ end of the genome encodes both non-structural and structural proteins (5′-S-3a-3b-E-M–N-7-3′) [6].
The S protein, which induces neutralizing antibodies, is important in the initiation of infection [7–9] and has been further delineated into four antigenic sites A, B, C, and D which are located within the N-terminal region of the S protein [8]. Among these, only site A and to a lesser extent site D (herein defined as S-AD) are involved in eliciting neutralizing antibodies. Recent work demonstrated that recombinant S-AD (rS-AD) was able to induce antibodies capable of neutralizing TGEV infection in vitro [10].
Attenuated or inactivated TGEV vaccines are less than optimal because they are capable of reverting back to virulent phenotypes and generally do not prevent viral shedding. Therefore, effective diagnostic tests have become important in virus management and control. Phage display is a proven technology for identifying small peptide ligands that can bind specific target proteins [11–14]. It has been utilized in antibody engineering [15], drug discovery [16], vaccine development [17], and molecular diagnosis. In virology, phage display has been used to identify peptides that interact with several viruses such as bovine rotavirus [18], adenovirus type 2 [19], Andes virus [20], Sin Nombre virus [21], coronavirus [22], and Avian H5N1 Virus [23]. Herein, we use similar technology and advance previous work by using the rS-AD as an immobilizing target to select phages from a peptide display library, with diagnostic potential for TGEV. Our results indicate that phages bearing peptide ligands that bind rS-AD can be used to develop a phage-mediated ELISA with high sensitivity and specificity to distinguish TGEV from other common swine viruses.
Materials and methods
Biopanning
Swine testis (ST) cells were purchased from ATCC and used to propagate TGEV strain PUR46-MAD [4]. The rS-AD was produced and purified as described elsewhere [10]. A 12-mer phage-display library was purchased from New England Biolabs for panning according to published protocols [11, 14, 24] using the rS-AD as a target at a concentration of 10 µg/well. The 96-well plates coated with rS-AD, were initially incubated with the phage library (1.5 × 1011 pfu/ml; 100 μl/well) suspended in TBST (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 0.05 % Tween-20) for 30 min. Subsequent pannings 2, 3, and 4 were performed using incrementally higher concentrations of Tween-20. The phage titers of the input, output (elution), and amplified phages were determined as defined by the manufacturer.
Binding of phage to TGEV or rS-AD protein
Indirect ELISA was used to assess the phages that remained after four rounds of biopanning. Either TGEV (0.61 mg/ml) or rS-AD (10 μg/well) in 0.1 M NaHCO3 pH 8.6 was used to coat 96-well plates at 4 °C for 12 h. The next day, the plates were blocked with 1 % bovine serum albumin (BSA) in TBS (TBSB) for 2 h, washed (3×) with TBST, and then incubated with phage (1.5 × 1012 pfu/ml in 0.1 M NaHCO3, pH 8.6; 100 μg/well) for 1 h at 37 °C. The plates were again washed with TBST, then incubated for 1 h at 37 °C with rabbit anti-M13 antibody (1:1000 in TBSB; Abcam), followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (GARP; 1:5000 in TBSB, Sigma). The OD490 nm was determined in triplicate as previously described [24].
Ten phages with the highest affinity for binding rS-AD as determined by ELISA were amplified, precipitated with polyethylene glycol-NaCl, and then used for DNA extraction according to the manufacturer’s instructions (New England Biolabs). Amplification of the genes encoding the exogenous peptides was performed using sense (5′-TCACCTCGAAAGCAAGCTGA) and anti-sense (5′-CCCTCATAGTTAGCGTAACG) M13 primers followed by DNA sequencing [14, 24]. The PCR conditions were as follows: 95 °C for 5 min, 30 cycles of 95 °C for 30 s, 57 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 7 min.
Comparison of antibody- and phage-mediated ELISA
To compare the sensitivities of phage-mediated ELISA to antibody-mediated ELISA, TGEV serially diluted in 0.1 M NaHCO3 (pH 8.6) was coated onto duplicate ELISA plates overnight at 4 °C followed by blocking with 5 % skim milk for 3 h at rt. The selected phages or unbound phage complexes (negative control) diluted in PBS (1.5 × 1012 pfu/ml) were added to one set of plates, followed by anti-M13 antibody (1:1000 in PBS + BSA). To the second set of duplicate plates, rabbit anti-TGEV polyclonal antiserum serially diluted in PBS + BSA, and normal rabbit serum were added as the primary and control antibodies, respectively. After incubating both sets of plates for 1 h at 37 °C followed by extensive washing, GARP (1:5000) was added as described above. The OD490 values were read on all plates; OD490 ratios where OD490 (sample−negative standard) (P)/OD490 (positive control−negative standard) (N) > 2 were judged as positive. All experiments were performed in triplicate.
RT-PCR for diagnosing TGEV
The TCID50 of TGEV was determined using the Reed–Muench method, and TGEV was adjusted to 0.61 mg/ml in PBS. Total RNA was extracted from 300 μl of virus (Fastgene, China) and dissolved in 20 μl of sterile water. Reverse transcription was performed in 20 µl using 2 μl of RNA (550 ng/μl), Oligo dT as primer, and M-MLV reverse transcriptase as recommended by the manufacturer (TaKaRa, China). The resulting cDNA (1 μl) was used as a template for PCR in 20 μl which included 0.2 μl of 10× Easy Taq polymerase (TaKaRa, China), 1 μl of dNTP (2.5 mM), 10× PCR Buffer (1 μl), and 0.2 μl each of sense (5′-CTTAGTAGTAATATTTTGCATAC) and anti-sense (5′-TATAGCAGATGATAGAATTAACA) primers. Amplification conditions were as follows: 94 °C for 5 min, then 30 cycles of 95 °C 30 s, 47.6 °C 30 s, and 72 °C 40 s followed by a final extension at 72 °C for 10 min. The amplified fragment was confirmed by DNA sequencing.
Phage expressing surface peptides for viral diagnosis
Phage specificity was evaluated against the following panel of porcine viruses: TGEV, strain HR/DN1 [25], porcine epidemic diarrhea virus (PEDV; strain HLJBY) [26], porcine reproductive and respiratory syndrome virus (PRRSV; strain JilinTN1) [27], porcine circovirus type II (PCV2; strain PCV2-LJR) [28], porcine parvovirus (PPV; strain PPV2010) [29], porcine pseudorabies virus (PrV; strain Kaplan) [23, 30], and porcine rotavirus (PRoV; isolate DN30209) [31]. All viruses were initially coated at 8 µg/ml then serially diluted in 0.1 M NaHCO3 (pH 8.6) and subjected to phage-ELISA as described above. Average OD490 values were obtained from three independent experiments.
Statistical analyses
Data were collated and the mean ± SD values were determined. Arithmetic means were compared between treatment groups using ANOVA (SPSS 15.0; SPSS Inc., Chicago, Illinois, USA) followed by Duncan’s multiple-range test. Values of p < 0.05 and p < 0.01 were defined as statistically significant (“*”) or highly significant (“**”), respectively.
Results and discussion
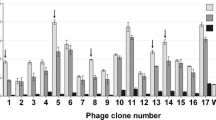
In this study, we used phage display to select 12-mer peptides that bind rS-AD [12] and that may function for diagnosing TGEV infections. After four rounds of panning, rS-AD-specific phages increased 11× from 4.7 × 104 in the first round to 5.3 × 105 in the fourth round (Table 1). Following the last screen, we selected 10 phage clones from the original 18 that bound both rS-AD and TGEV. This subset was characterized by ELISA with respect to their binding efficiencies (Fig. 1).

Phage binding to rS-AD and TGEV. [Eighteen selected phages designated phTGEV-SAD 1—phTGEV-SAD 18 were incubated with rS-AD or TGEV then screened with anti-M13 antibody and visualized with HRP-conjugated secondary antibody. The ELISA OD values represent the binding efficiencies of 18 different phages to rS-AD and TGEV. The experiment was performed in triplicate. Controls are (1) non-binding phage library, (2) secondary antibody-free, (3) phage-free, (4) porcine circovirus-cap protein, (5) M proteins of TGEV, (6) blocking buffer, and (7) virus dilution buffer.]
PCR amplification and sequencing indicated that nine distinct 12-mer peptides were identified among the 10 phages that were selected (Table 2). In contrast to previous reports [14, 22, 24], these peptides exhibited substantial sequence diversity in the number of peptides that bound to rS-AD. It is not known if this relates to the length of the target protein or to changes made in the panning process to enhance binding specificity.
As shown in Fig. 2, we selected four (phTGEV-SAD-15, phTGEV-SAD1/7, phTGEV-SAD11, phTGEV-SAD16) of the ten phages with the highest binding affinity to TGEV for further testing. The lowest detectable quantity of TGEV for the above defined phages was 0.1, 0.3, 0.2, and 0.4 mg, respectively, suggesting that phTGEV-SAD15 was the most sensitive when used in a phage-based ELISA. Binding directly to TGEV was uncharacteristically better than binding to the rS-AD used in the selection process (Figs. 1, 2). This is likely attributable to more complete folding of the native protein or to better accessibility of the binding epitope in the native form.

Detection limit of TGEV by phage-mediated ELISA. [TGEV serially diluted in PBS was used to coat ELISA plates. The plates were then incubated with phages (ph) phTGEV-SAD15, phTGEV-SAD1/7, phTGEV-SAD11, or phTGEV-SAD16, followed by anti-M13 antibody and GARP. The OD490 ratios [(P 490)/(N 490)] > 2 were designated as positive responses. The P/N values were derived from triplicate assays. Virus concentrations are indicated on the x axis.]
The minimum quantity of TGEV required for detection via antibody-based ELISA was 0.6 µg (P/N value > 2) (Fig. 3), whereas the minimum quantity of TGEV required for phTGEV-SAD15-based ELISA was 0.1 µg. This is consistent with the phage-mediated ELISA being more sensitive than conventional antibody ELISA. A number of ELISA-based assays have been developed over the years for detecting TGEV, many of which have been directed at differentiating TGEV from PRCV-infected animals. Among the earlier ones, Sestak et al. [32] targeted the S glycoprotein of TGEV in a competition ELISA where recombinant S protein was coated onto plates and used to capture host antibodies. Using a monoclonal Ab to epitope D and which is specific for TGEV, the investigators were able to differentiate the infectious agents. Liu et al. [33] cloned and expressed the nucleoprotein (N) to develop an ELISA. Compared to the Virus neutralization assay, they demonstrated 98 % sensitivity and specificity; however, they did not characterize or address the lower level of sensitivity in vitro or in vivo. In 2010, Elia et al. [34] used the recombinant S protein to develop an ELISA to assess swine-like TGEV coronaviruses in canine hosts. Given the novelty of the virus, they were unable to compare it to other assays currently in use. Zou et al. [35] use techniques similar to those developed here, i.e., peptide display, to target the M protein of TGEV in developing an ELISA-based diagnostic test. In this case, the sensitivity of the ELISA exceeded that developed when targeting the A–D regions of the S protein. This is likely the result of the location and overall accessibility of the M protein to the host immune response when compared to the S protein.

Detection limit of TGEV by antibody-based ELISA. [The TGEV serially diluted in PBS was used to coat ELISA plates. The bound virus was then screened with serially diluted rabbit anti-TGEV or with normal rabbit serum (negative control) followed by GARP. The OD490 ratios [(P 490)/(N 490)] > 2 were designated as positive responses. The P/N values were derived from triplicate assays. Virus concentrations are indicated on the x axis.]
When the phage-mediated ELISA and antibody ELISA were compared to RT-PCR which targeted a 208-base pair fragment of the S gene, the RT-PCR was most sensitive of all assays tested. This is not unexpected given the higher sensitivity of PCR assays in general. PCR amplification was positive using cDNA equivalents of 0.02 µg of TGEV (data not shown). Real-time PCR and/or nested PCR would clearly have generated even more sensitive results. In addition, phages expressing peptide that bind to TGEV S-AD did not bind to other selected viruses (Fig. 4).

Binding specificity of phTGEV-SAD15. [One phage, designated phTGEV-SAD15, was selected from the 10 phages analyzed in Fig. 2 and tested against other common swine viruses for potential cross-reactivity. Shown in this figure are ELISA values using phTGEV-SAD15 to screen the following porcine viruses: epidemic diarrhea virus (PEDV), respiratory syndrome virus (PRRSV), circovirus (PCV2), reproductive porcine parvovirus (PPV), pseudorabies virus (PrV), and rotavirus (PRoV) and visualized using rabbit anti-M13 antibody and GARP. The OD490 ratios between individual phages and a control phage are shown on the y axis; p < 0.01 is defined by ‘‘**’’ relative to other viruses. Bars show the standard deviation from three independent assays.]
In summary, we identified peptides that specifically bind to TGEV and can form the basis of new diagnostic tests where the sensitivity of phTGEV-SAD15 was 0.1 µg of TGEV. This sensitivity fared quite well when compared to the antibody-mediated ELISA which had a sensitivity of 0.6 µg but fell short of the sensitivity of RT-PCR; however, phTGEV-SAD-15 provides a quicker and less costly alternative to RT-PCR.
References
H. Laude, H.D. Rasschaert, B. Delmas, M. Godet, J. Gelfi, B. Charley, Vet. Microbiol. 23, 147–154 (1990)
L.J. Saif, R.D. Wesley, in Diseases of swine 7th Ed. ed. by A.D. Leman, B.E. Straw, W.L. Mengeling et al. (Iowa State University Press, Ames, IA 1992), pp. 362–386
W. Spaan, D. Cavanagh, M.C. Horzinek, J. Gen. Virol. 69, 2939–2952 (1988)
X. Ren, J. Glende, J. Yin, C. Schwegmann-Wessels, G. Herrler, Virus Res. 137, 220–224 (2008)
F. Almazán, J.M. González, Z. Pénzes, A. Izeta, E. Calvo, J. Plana-Durán, L. Enjuanes, Proc. Natl. Acad. Sci. U.S.A. 97, 5516–5521 (2000)
J.L. Moreno, S. Zúñiga, L. Enjuanes, I. Sola, J. Virol. 82, 3882–3893 (2008)
L. Enjuanes, C. Suñé, F. Gebauer, C. Smerdou, A. Camacho, I.M. Antón, S. González, A. Talamillo, A. Méndez, M.L. Ballesteros et al., Vet. Microbiol. 33, 249–262 (1992)
F. Gebauer, W.P. Posthumus, I. Correa, C. Suñé, C. Smerdou, C.M. Sánchez, J.A. Lenstra, R.H. Meloen, L. Enjuanes, Virology 183, 225–238 (1991)
C. Schwegmann-Wessels, G. Zimmer, B. Schröder, G. Breves, G. Herrler, J. Virol. 77, 11846–11848 (2003)
F. Meng, Z. Zhao, G. Li, S. Suo, N. Shi, J. Yin, D. Zarlenga, X. Ren, X. Virus Genes 43, 335–341 (2011)
M.M. Smith, L. Shi, M. Navre, J. Biol. Chem. 270, 6440–6449 (1995)
S.K. Kang, J.H. Woo, M.K. Kim, S.S. Woo, J.H. Choi, H.G. Lee, N.K. Lee, Y.J. Choi, Biotechnology 135, 210–216 (2008)
J. Latzka, S. Gaier, G. Hofstetter, N. Balazs, U. Smole, S. Ferrone, O. Scheiner, H. Breiteneder, H. Pehamberger, S. Wagner, PLoS ONE 6, e19383 (2011)
X. Ren, M. Wang, J. Yin, G. Li, J. Clin. Microbiol. 48, 1875–1881 (2010)
M.S. Hayden, L.K. Gilliland, J.A. Ledbetter, Curr. Opin. Immunol. 9, 201–212 (1997)
B.K. Kay, A. Kurakin, R. Hyde-DeRuyscher, Drug Discov. Today 3, 370–378 (1998)
G.B. Lesinski, M.A. Westerink, J. Microbiol. Methods 47, 135–149 (2001)
X. Wang, G. Li, Y. Ren, X. Ren, PLoS ONE 6, e28667 (2011)
S.S. Hong, P. Boulanger, EMBO J. 14, 4714–4727 (1995)
P.R. Hall, B. Hjelle, H. Njus, C. Ye, V. Bondu-Hawkins, D.C. Brown, K.A. Kilpatrick, R.A. Larson, J. Virol. 83, 8965–8969 (2009)
R.S. Larson, D.C. Brown, C. Ye, B. Hjelle, J. Virol. 79, 7319–7326 (2005)
X. Ren, B. Liu, J. Yin, H. Zhang, G. Li, Virology 410, 299–306 (2011)
X. Ren, J. Yin, G. Li, G. Herrler, Curr. Microbiol. 62, 261–266 (2011)
D. Wu, G. Li, C. Qin, X. Ren, PLoS ONE 6, e23058 (2011)
X. Sun, Y. Ren, Y. Li, J. Zhu, W. Zhu, F. Ding, G. Li, C. Wang, M. Gao, Y. Gao, L. Cao, X. Ren, Monoclon. Antib. Immunodiagn. Immunother. 32, 36–40 (2013)
W. Yang, G. Li, Y. Ren, S. Suo, X. Ren, Acta Vet. Hung. 61, 257–269 (2013)
M. Gao, J. Cui, Y. Ren, S. Suo, G. Li, X. Sun, D. Su, T. Opriessnig, X. Ren, J. Virol. Methods 185, 18–23 (2012)
W. Zhu, X. Ren, Pak. Vet. J. 32, 165–170 (2012)
J. Cui, X. Wang, Y. Ren, S. Cui, G. Li, X. Ren, J. Virol. 86, 2379 (2012)
T.C. Mettenleiter, L. Zsak, A.S. Kaplan, T. Ben-Porat, B. Lomniczi, J. Virol. 61, 4030–4032 (1987)
J. Zhu, Q. Yang, L. Cao, X. Dou, J. Zhao, W. Zhu, F. Ding, R.E. Bu, S. Suo, Y. Ren, G. Li, X. Ren, Virol. J. 10, 91 (2013)
K. Sestak, Z. Zhou, D.I. Shoup, L.J. Saif, J. Vet. Diagn. Invest. 11, 205–214 (1999)
C. Liu, T. Kokuho, T. Kubota, S. Watanabe, S. Inumaru, Y. Yokomizo, T. Onodera, J. Vet. Med. Sci. 63, 1236–1253 (2001)
G. Elia, N. Decaro, V. Martella, E. Lorusso, V. Mari, S.L. Maria, P. Cordioli, C. Buonavoglia, J. Virol. Methods 163, 309–312 (2010)
H. Zou, D.S. Zarlenga, K. Sestak, S. Suo, X. Ren, X. Antiviral Res. 99, 383–390 (2013)
Acknowledgments
This work was supported by Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2013BAD12B04), Research Team Program on Scientific and Technological Innovation in Heilongjiang Provincial University (2011TD001), the Harbin Science and Technology Bureau (RC2012XK002003), and sponsored by Chang Jiang Scholar Candidates Program for Provincial Universities in Heilongjiang (2013CJHB002).
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Keizo Tomonaga.
Rights and permissions
About this article

Cite this article
Suo, S., Wang, X., Zarlenga, D. et al. Phage display for identifying peptides that bind the spike protein of transmissible gastroenteritis virus and possess diagnostic potential. Virus Genes 51, 51–56 (2015). https://doi.org/10.1007/s11262-015-1208-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1208-7