
Abstract
Introduction
Tick-borne diseases (TBDs) pose a major hindrance to livestock production in countries with limited resources. Effective prevention and management of TBDs require a thorough understanding of disease vectors and pathogens. However, there is limited information on studies of bovine tick-borne pathogens (TBPs) using molecular methods in Malawi. This study aimed to detect TBPs of cattle populations in southern Malawi, which has the largest cattle population in the country.
Methodology
A total of 220 blood samples from apparently healthy cattle were collected in six districts, and were screened for selected TBPs using polymerase chain reaction (PCR).
Results
The overall detection rate of TBPs was 72.3%. Among the detected pathogens, Babesia bigemina had the highest detection rate (34.5%), followed by Anaplasma marginale (23.2%), Anaplasma phagocytophilum (22.3%), Theileria taurotragi (22.3%), Theileria parva (15.5%), Anaplasma bovis (9.6%), Babesia bovis (7.3%), Theileria mutans (4.1%), and Babesia naoakii (2.7%). Among the positive samples, 64.2% were found to be co-infected with two or more TBPs, with the highest number of seven pathogens detected in a single sample. The study documents the existence of A. phagocytophilum, B. bovis, and B. naoakii in Malawian cattle for the first time.
Conclusion
The findings herein demonstrate a significant burden of TBPs on cattle in Malawi, which gives a challenge in combating TBDs. The high TBP burden, along with the high co-infection frequencies in Malawian cattle necessitates the urgency to implement effective control strategies to enhance cattle production in the country.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tick-borne pathogens (TBPs), transmitted by vectors, account for a substantial number of infections in humans and animals (Parola and Raoult 2001). Globally, 80% of the bovine population is greatly afflicted by TBDs, resulting in decreased livestock production and loss of resources in fighting the diseases (Mangold et al. 1998). It is a key burden on livestock farming, and has a substantial economic impact on rural people, disturbing their food accessibility and agricultural undertakings (Minjauw and McLeod 2003). Farmers are subjected to economic losses due to huge cattle deaths, poor body condition of the animals, reduced milk production, and huge amounts of costs used in dealing with the tick burden and diseases they transmit (Uilenberg 1992). In Africa, anaplasmosis, babesiosis, ehrlichiosis, and theileriosis are the main health and management drawbacks for cattle and small ruminant production (Ocaido et al. 2009).
Bovine babesiosis is transmitted by Rhipicephalus microplus ticks and is caused by Babesia species that include B. bigemina, B. bovis and B. naoakii. However, B. bovis is more virulent among the pathogens, with catastrophic consequences in cattle (Woodford et al. 1990). It is clinically manifested as pyrexia, hemoglobinuria, anemia, and jaundice (Bock et al. 2004). Similarly, Babesia naoakii, which can cause bovine clinical babesiosis, is a recent challenge in Asian countries, causing mortalities in the cattle population (Sivakumar et al. 2020). Meanwhile, Anaplasma marginale, which causes bovine anaplasmosis, is marked by anemia, jaundice, pyrexia, cachexia, abortions, and stupor. In the worst cases, it can lead to death (Mohanta et al. 2023a). On the contrary, A. phagocytophilum causes bovine granulocytic anaplasmosis, also known as tick-borne fever. It can cause clinical disease in humans and animals with depression, loss of appetite, and fever as clinical symptoms (Woldehiwet 2009), although most of the human cases have been reported in Europe (Dugat et al. 2015). Despite Ixodes ticks being responsible for the transmission of A. phagocytophilum, mechanical transfer of infected blood via hematophagous arthropods, fomites and transplacental transmission during pregnancy have also been reported (Jurković et al. 2020).
East Coast Fever (ECF), also known as theileriosis, is a disease caused by Theileria parva, which is transmitted by Rhipicephalus appendiculatus ticks (Walker et al. 2003). It leads to acute lymphoproliferative diseases, usually leading to death in most affected cattle populations (Olds et al. 2018). Moreover, T. mutans might cause severe illness in cattle, with clinical symptoms that may be confused with a mild form of T. parva in cattle populations (Chaisi et al. 2013). However, T. mutans is transmitted by ticks belonging to the genus Amblyomma (Walker et al. 2003). Furthermore, T. taurotragi, causes mild and cerebral theileriosis in cattle and elands. It is associated with bovine cerebral disease known as turning disease, thereby making it an economic disease of veterinary importance (De Vos et al. 1981).
The livestock subsector makes a significant contribution to Malawi’s economy and food security. It contributes approximately 10.5% to the national income and roughly 37.4% to the agricultural gross domestic product (MoAIWD 2022). About 60% of Malawians, mostly smallholder farmers, own various livestock species. Malawi has a cattle population of about 1,959,101, of which 92% is composed of the indigenous Malawi zebu, and 8% dairy crossbreds and exotic breeds (MoAIWD 2022). Ticks belonging to the genera Amblyomma, Rhipicephalus, and Hyalomma have been reported in Malawi (Chikufenji et al. 2024; Berggren 1978). On the contrary, TBPs of genera Theileria, Anaplasma, and Babesia have been reported mostly in central and northern Malawi where ECF had been declared endemic (DAHLD 2006; Chatanga et al. 2022). Less sensitive and conservative diagnostic methods, such as the use of blood smears, have been used in Malawi. Nevertheless, information on the detection of TBPs in cattle in Malawi is scarce. This study, therefore, aimed to investigate the prevalence of TBPs in cattle in southern Malawi using molecular methods.
Materials and methods
Sampling locations, sample collection, and DNA extraction
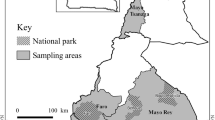
Two hundred and twenty blood samples were collected from randomly selected, and communally grazed local heads of cattle in six districts of southern Malawi (SI Fig. 1) (Blantyre: n=47; Chikwawa: n=43; Chiradzulu: n=37; Mulanje: n=28; Thyolo: n=29 and Zomba: n=36), between October and December 2021. Approximately 2–5 mL of blood was obtained from each animal into an ethylenediaminetetraacetic acid (EDTA) containing tube (BD Bioscience, Bergen County, NJ, USA). After collection, blood samples were briefly kept on ice in the field, and at -20°C in the laboratory. DNA from the collected samples was extracted as described by Ringo et al. (2022).
Molecular detection of tick-borne pathogens and sequencing
Selected tick-borne pathogens were screened from the DNA samples using the primers listed in SI Table 1. The PCR assays were performed as described by Chikufenji et al. (2024). The PCR bands were cut from the gel and purified using Nucleospin® Gel and PCR Clean-up kit (Macherey–Nagel, Düren, Germany), following the manufacturer’s instructions. The purified PCR products were directly sequenced using a BigDye™ Terminator Cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) and ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Codon Code Aligner version 9 software (Codon Code Corporation, Centerville, MA, USA) was used for trimming, assembling, and generating the consensus sequences for the reads. The BLASTn analysis was performed to confirm the identity of the sequences.
Phylogenetic analysis and accession numbers assigned
The nucleotide sequences obtained from the TBPs in this study were assembled by using Clustal W multiple alignments in MEGA XI software (Tamura et al. 2021), and the maximum likelihood (ML) method was used to construct phylogenetic trees for the sequences generated, with those previously deposited in the GenBank. The sequences obtained were submitted to the GenBank of the National Center for Biotechnology Information (NCBI) through BankIt and GenBank for DNA sequences and ribosomal RNA sequences, respectively, and the following accession numbers were assigned: OP839189–OP839190 for B. bovis; OP854628–OP854629 for B. naoakii; OP866967–OP866971 for B. bigemina; OP868837–OP868840 for A. marginale; OP824619–OP824620 for A. phagocytophilum; and OP824766 for A. bovis; OP866888–OP866892 for T. parva; OP821414–OP821415 for T. mutans; OP824492–OP824498 for T. taurotragi.
Results
Tick-borne pathogen detection rates
A total of 159 (72.3%) samples were positive for at least one of the screened TBPs. The pathogens detected were B. bigemina (34.5%), A. marginale (23.2%), A. phagocytophilum (22.3%), T. taurotragi (22.3%), T. parva (15.5%), A. bovis (9. 5%), B. bovis (7.3%), T. mutans (4.1%), and B. naoakii (2.7%) (Table 1). A significant difference in the detection rate of B. bigemina was observed among the study locations, while a significantly higher detection rate of T. parva was observed in Chiradzulu (32.4%), Mulanje (17.9%) and Chikwawa (20.9%). In terms of infection rate by sex, 41 males (47.7%) and 118 females (88.1%) were infected with at least one of the examined pathogens. Significantly higher detection rates for T. taurotragi (26.9%; p < 0.05) and B. bovis (10.4%; p < 0.05) were observed in females than males (Table 2). Coxiella burnetii, E. ruminantium, A. platys, T. orientalis, and Rickettsia spp., were not detected in any of the screened samples.
Co-infections were observed in 64.2% of the positive samples, with up to septuple different pathogens simultaneously detected in one sample. Co-infections with double, triple, quadruple, quintuple, hextuple, and septuple pathogens were observed in 44.0% (70/159), 15.1% (24/159), 1.9% (3/159), 1.3% (2/159), 1.3% (2/159), and 0.6% (1/159) of the TBP-positive samples, respectively (SI Table 2).
Gene sequence analysis
The groEL gene sequences of A. marginale (OP868837–OP868840) were well conserved among themselves, with high identity values of 97.2–99.6%. The identity values of these sequences ranged from 99.1–100% when compared with A. marginale sequences from Cattle in Benin (KX685364) and Tanzania (OP414689).
Additionally, the percent identity values of B. bigemina (OP866967–OP866971) in this study ranged from 91.2–99.8% among themselves. The sequences had similarity scores ranging from 98.4–100% with the bovine B. bigemina sequences from Kenya (KP347559) and South Africa (MK481015). On the other hand, the sequences of B. bovis sbp-4 gene (OP839189–OP839190) shared identity values ranging from 94.4–99.2% and showed higher identities of up to 99.8–100% with sequences OQ144958 (Bangladesh) and KF626632 (South Africa). Furthermore, two B. naoakii ama-1 sequences (OP854628 and OP854629) showed identity values of 99.5–100%. The sequences were identical to the sequences LC385804 (Sri Lanka) and OQ148404 (Bangladesh). In contrast, p104 sequences of T. parva (OP866888–OP866892) shared percent identity between 93.4% and 99.6%. The sequences showed 96.3% and 100% identity with T. parva sequences from cattle in Cameroon (MK568804) and Tanzania (MZ798151 and OP390278), respectively. In addition, the percent identity of T. taurotragi (OP824492–OP824498) and T. mutans (OP821414–OP821415) in this study ranged from 97.5–99.6% and 99.2–100%, respectively.
Phylogenetic analyses
Babesia bovis SBP-4 sequences (OP839189 and OP839190) clustered in the clade with those from Bangladesh (OQ144958), Sudan (LC611418), Mongolia (AB569302), Syria (AB617641), Egypt (KF192805 and MZ197894), Japan (AB594481), Benin (KX685399), and Malawi (OR818703) (Fig. 1). In addition, the B. naoakii ama-1 sequences (OP854628 and OP854629) clustered with LC506533 from Mongolia, LC486029 from Argentina, LC486011 from the Philippines, LC486017 from Vietnam, OQ148404 from Bangladesh, and OR601001 from Egypt (Fig. 2). Moreover, Babesia bigemina Rap-1a gene sequences (OP866967-OP866971) clustered in the same clade with isolates from Benin (KU042084), Tanzania (OP390284), Burkina Faso (OK323209), Kenya (KP347559), Bangladesh (OQ162126), Uganda (MG426198), South Africa (MK481015), Nigeria (OM406333), and Turkey (KT220512) (SI Fig. 2).

Phylogenetic analysis of Babesia bovis identified in this study is based on the SBP-4 gene. The tree was constructed by MEGA XI using the maximum likelihood method based on Kimura 2-parameter model. Numbers on the nodes indicate the percentage of 1000 bootstrap replicates. The sequences of this study are shown in red. Babesia caballi (MT032180) was used as an outgroup

Phylogenetic analysis of Babesia naoakii identified in this study is based on the AMA-1 gene. The tree was constructed by MEGA XI using the maximum likelihood method based on Kimura 2-parameter model. Numbers on the nodes indicate the percentage of 1000 bootstrap replicates. The sequences of this study are shown in red. Babesia bigemina (OR640338) was used as an outgroup
The A. marginale sequences (OP868837—OP868840) obtained herein, clustered in a clade with the sequences OQ148410 (Bangladesh), FJ226455 (Japan), OQ185223 (China), KC113455 (Philippines), OP414689 (Tanzania), OR767905 and LC664079 (Malawi), KY522983 (Uganda), MN870643 (Egypt), and KX685364 (Benin) (SI Fig. 3). The phylogeny inferred from 16S rRNA of A. phagocytophilum and for A. bovis resulted in clustering in the respective clades (Fig. 3). The A. bovis isolate had a close similarity to those deposited from Bangladesh (OQ135122), Malawi (OR823813), and Iran (KU242422). However, A. phagocytophilum isolates generated herein had similarities to that reported from South Korea (MF787269).

The phylogenetic analysis of A. phagocytophilum and A. bovis identified in this study is based on the 16S rRNA gene. The tree was constructed by MEGA XI using the maximum likelihood method based on Hasegawa-Kishino Yano model. Numbers on the nodes indicate the percentage of 1000 bootstrap replicates. The sequences of this study are shown in red. Anaplasma platys (MK121782) was used as an outgroup
The T. parva p104 gene sequences OP866888–OP866892 clustered together with MZ798149 from South Africa, MN810052 from Uganda, MK568804 from Cameroon, MZ798151 from Tanzania, KP347566 from Kenya, ON376062 from Mozambique, and EF469604 from Sudan (SI Fig. 4). The sequences of T. mutans (OP821414 and OP821415) were conserved and clustered in the same clade with those from South Africa (MH751463), Angola (MT898574), and Mozambique (FJ869899), whereas T. taurotragi sequences (OP824492-OP824498) formed a monophyletic clade with those previously obtained from Malawi (LC664058), South Africa (L19082), Tanzania (MN726635), Kenya (MT459438) and Zambia (MT814757) (SI Fig. 5).
Discussion
In this study, PCR was used to detect cattle TBPs from southern Malawi, and relatively high detection rates of TBP infections among cattle populations were recorded. The molecular techniques used in this study offer a more sensitive and dependable diagnostic tool than the conservative methods previously used in Malawi.
This study reports B. bovis and B. naoakii for the first time in Malawi using molecular techniques. We report the detection rate of 7.3% for B. bovis in this study which was higher than 4.5% from Tanzania (Ringo et al. 2018). However, this was lower than 7.7% from Zambia (Tembo et al. 2018), 82% from Mozambique (Martins et al. 2008), and 12.3% from Kenya (Adjou Moumouni et al. 2015). The existence of B. bovis in Malawi was not surprising because the pathogen has already been reported from Zambia, Tanzania and Mozambique, the neighboring countries of Malawi. The detection of B. bovis was observed in all study locations except Zomba district. This observation could be due to differences in cattle-rearing systems among the study sites. Although the semi-intensive system is practiced in Zomba, the extensive system is used in other study areas which possibly exposed the animals more to tick vectors. The phylogenetic analysis of sbp-4 gene sequences showed that the two sequences obtained in this study clustered in one clade along with those from Mongolia (AB569302), Egypt (MZ197894), Sudan (LC611418), Indonesia (KY484532), and Japan (AB594813). This result suggests that there might be one strain of B. bovis infecting cattle in southern Malawi.
We detected B. naoakii in 2.7% of the screened samples. This was similar to 1.09% reported from Bangladesh (Mohanta et al. 2023a). However, the detection rate in this study was lower than 9.6% from Vietnam (Sivakumar et al. 2020), 27.9% from Mongolia (Otgonsuren et al. 2020), and 11.3% from the Philippines (Sivakumar et al. 2020). The detection of this pathogen in Malawi was surprising because it has never been reported in any of the sub-Saharan African countries. However, it has been reported recently in Egypt (Sivakumar et al. 2020). The phylogenetic analysis of B. naoakii ama-1 sequences clustered in one clade with previously reported isolates from Mongolia, Argentina, the Philippines, Vietnam, Bangladesh, and Egypt (Fig. 2). This finding indicates that B. naoakii isolates reported in Asian and other countries in the world is the same strain affecting cattle in Malawi. However, more studies on tick vectors of this pathogenic Babesia spp. and its impact on livestock in Malawi is urgently warranted.
We report a detection rate of 23.1% for A. marginale in Malawian cattle, which is consistent with a previous report (24.0%; Chatanga et al. 2022). However, a lower detection rate was recorded in other countries, 16% in Tanzania (Ringo et al. 2018), 7.9% in Kenya (Adjou Moumouni et al. 2015), and 10.51% in Bangladesh (Mohanta et al. 2023b). Extensive type of livestock rearing practice in the study locations herein might have contributed to the higher detection rates. We also report a detection rate of 22.3% for A. phagocytophilum, a zoonotic parasite causing acute and subclinical disease in the animal host (Inokuma et al. 2005). Compared to the present study, the lower detection rates of this zoonotic pathogen were reported in Ethiopia, 2.7% (Teshale et al. 2018), South Africa, 7.0% (Mtshali et al. 2016) and Bangladesh, 0.72% (Mohanta et al. 2023b). This variation might be a result of different sample sizes in these study locations.
Zoonotic cases of A. phagocytophilum have been reported in South Africa (Inokuma et al. 2005), and China (Cao et al. 2000) in acute and subclinical forms with fever, and central nervous system dysfunction (Inokuma et al. 2005), while anorexia and lameness in the animal hosts (Mtshali et al. (2016). This study reports the existence of A. phagocytophilum for the first time in Malawi.
The detection rate of 15.5% for T. parva, the causative agent for ECF, was found herein. The detection of this pathogen in southern Malawi suggests that the pathogen has geographically spread out in Malawi, and this may be attributed to uncontrolled animal movements from the endemic central and northern Malawi. The detection rate herein is lower than that previously reported from Malawi (33.0%, Chatanga et al. 2022), which could be a result of differences in sampling locations. In this study, sampling was done from the non-endemic region, while the previous study was done in the endemic central region. The significantly higher detection rates observed in Chiradzulu (32.4%), Mulanje (17.9%) and Chikwawa (20.9%) were due to the introduction of dairy animals from endemic regions of central and northern Malawi, and uncontrolled cattle movements from the neighboring Mozambique as well (MoAIWD 2022).
The detection rate of 4.1% for T. mutans found herein was notably lower than that previously reported from Malawi (73.8%, Chatanga et al. 2022), Tanzania (34.4%, Ringo et al. 2018) and Uganda (88.3%, Byaruhanga et al. 2016). The detection rates in different geographies might vary with tick activity in dry and wet or rainy seasons. In the present study, samples were collected during the dry season (October to December), while in the other studies, the sampling was done during or immediately after the rainy season.
The higher detection rates of co-infections (64.2%) with up to seven pathogens in this study and that reported previously in Malawi (79.6%, Chatanga et al. 2022) compared to those reported from neighboring Tanzania (44.8%, Ringo et al. 2022), and Mozambique (52%, Martins et al. 2008), indicate simultaneously multiple parasites in the same host, which further imply that the animals in the area are prone to multiple TBD outbreaks.
In conclusion, this study has revealed high frequencies of TBPs in cattle in southern Malawi. In addition, the detection of A. phagocytophilum, B. bovis, and B. naoakii for the first time in Malawi reveals the need for extra effort in dealing with TBDs, including zoonoses in Malawi. The findings herein provide critical information for the control clues against ticks and TBDs in Malawi.
Data availability
The data will be available on request from the corresponding authors.
References
Adjou Moumouni PF, Aboge GO, Terkawi MA, Masatani T, Cao S, Kamyingkird K, Jirapattharasate C, Zhou M, Wang G, Liu M, Iguchi A, Vudriko P, Ybanez AP, Inokuma H, Shirafuji-Umemiya R, Suzuki H, Xuan X (2015) Molecular detection and characterization of Babesia bovis, Babesia bigemina, Theileria species and Anaplasma marginale isolated from cattle in Kenya. Parasit Vectors 8:496. https://doi.org/10.1186/s13071-015-1106-9
Berggren SA (1978) Cattle ticks in Malawi. Vet Parasitol 4:289–297. https://doi.org/10.1016/0304-4017(78)90055-9
Bock R, Jackson L, de Vos A, Jorgensen W (2004) Babesiosis of cattle. Parasitology 129:247–269. https://doi.org/10.1017/S0031182004005190
Byaruhanga C, Collins NE, Knobel D, Chaisi ME, Vorster I, Steyn HC, Oosthuizen MC (2016) Molecular investigation of tick-borne haemoparasite infections among transhumant zebu cattle in Karamoja Region, Uganda. Vet Parasitol Reg Stud Rep 3–4:27–35. https://doi.org/10.1016/j.vprsr.2016.06.004
Cao WC, Zhao QM, Zhang PH, Dumler JS, Zhang XT, Fang LQ, Yang H (2000) Granulocytic Ehrlichiae in Ixodes persulcatus ticks from an area in China where Lyme disease is endemic. J Clin Microbiol 38:4208–4210. https://doi.org/10.1128/jcm.38.11.4208-4210.2000
Chaisi ME, Collins NE, Potgieter FT, Oosthuizen MC (2013) Sequence variation identified in the 18S rRNA gene of Theileria mutans and Theileria velifera from the African buffalo (syncerus caffer). Vet Parasitol 191:132–137. https://doi.org/10.1016/j.vetpar.2012.08.005
Chatanga E, Maganga E, Mohamed WMA, Ogata S, Pandey GS, Abdelbaset AE, Hayashida K, Sugimoto C, Katakura K, Nonaka N, Nakao R (2022) High infection rate of tick-borne protozoan and rickettsial pathogens of cattle in Malawi and the development of a multiplex PCR for Babesia and Theileria species identification. Acta Trop 231:106413. https://doi.org/10.1016/j.actatropica.2022.106413
Chikufenji B, Chatanga E, Galon EM, Mohanta UK, Mdzukulu G, Nkhata M, Ma Y, Shirafuji-Umemiya R, Xuan X (2024) First report of dog ticks and tick-borne pathogens they are carrying in Malawi. J Vet Med Sci 86:150–159. https://doi.org/10.1292/jvms.23-0397
DAHLD, 2006. Policy document on livestock in Malawi. Ministry of Agriculture, Irrigation and Water Development (MoAIWD); Department of Animal Health and Livestock Development (DAHLD): Lilongwe, Malawi
De Vos AJ, Bessenger R, Banting LF (1981) Research communication Theileria taurotragi: A probable agent of bovine cerebral theileriosis Onderstepoort. J Vet Res 48:177–178
Dugat T, Lagrée AC, Maillard R, Boulouis HJ, Haddad N (2015) Opening the black box of Anaplasma phagocytophilum diversity: Current situation and future perspectives. Front Cell Infect Microbiol 5:00061. https://doi.org/10.3389/fcimb.2015.00061
Inokuma H, Oyamada M, Kelly PJ, Jacobson LA, Fournier PE, Itamoto K, Okuda M, Brouqui P (2005) Molecular detection of a new Anaplasma species closely related to Anaplasma phagocytophilum in canine blood from South Africa. J Clin Microbiol 43:2934–2937. https://doi.org/10.1128/jcm.43.6.2934-2937.2005
Jurković D, Mihaljević Ž, Duvnjak S, Silaghi C, Beck R (2020) First reports of indigenous lethal infection with Anaplasma marginale, Anaplasma bovis and Theileria orientalis in Croatian cattle. Ticks Tick-Borne Dis 11:101469. https://doi.org/10.1016/j.ttbdis.2020.101469
Mangold AJ, Bargues MD, Mas-Coma S (1998) Mitochondrial 16S rDNA sequences and phylogenetic relationships of species of Rhipicephalus and other tick genera among Metastriata (Acari: Ixodidae). Parasitol Res 84:478–484. https://doi.org/10.1007/s004360050433
Martins TM, Pedro OC, Caldeira RA, do Rosário VE, Neves L, Domingos A, (2008) Detection of bovine babesiosis in Mozambique by a novel semi-nested hot-start PCR method. Vet Parasitol 153:225–230. https://doi.org/10.1016/j.vetpar.2008.01.037
Minjauw B, McLeod A (2003) Tick-Borne Diseases and Poverty. The Impact of Ticks and Tick-Borne Diseases on the Livelihood of Small-Scale and Marginal Livestock Owners in India and Eastern and Southern Africa. UK: Research Report, DFID Animal Health Programme, Centre for Tropical Veterinary Medicine, University of Edinburgh 116
MoAIWD, (2022) Agricultural production estimates survey. Ministry of Agriculture Irrigation and Water Development, Lilongwe, Malawi
Mohanta UK, Chikufenji B, Galon EM, Ji S, Ma Z, El-Sayed SAES, Amer MM, Do TT, Xuan X (2023) Molecular characterization and phylogeny of Anaplasma marginale, A. Phagocytophilum and A. Bovis in livestock of Bangladesh. Parasitol Int 97:102790. https://doi.org/10.1016/j.parint.2023.102790
Mohanta UK, Chikufenji B, Galon EM, Ji S, Ma Z, El-Sayed SAES, Ringo AE, Do TT, Xuan X (2023) Molecular Detection and Phylogenetic Analyses of Babesia spp. and Theileria spp. in Livestock in Bangladesh. Microorganisms 11:1563. https://doi.org/10.3390/microorganisms11061563
Mtshali K, Khumalo ZTH, Nakao R, Grab DJ, Sugimoto C, Thekisoe OMM (2016) Molecular detection of zoonotic tick-borne pathogens from ticks collected from ruminants in four South African provinces. J Vet Med Sci 77:1573–1579. https://doi.org/10.1292/jvms.15-0170
Ocaido M, Muwazi RT, Opuda JA (2009) Economic impact of ticks and tick-borne diseases on cattle production systems around Lake Mburo National Park in South Western Uganda. Trop Anim Health Prod 41:731–739. https://doi.org/10.1007/S11250-008-9245-Z
Odongo DO, Sunter JD, Kiara HK, Skilton RA, Bishop RP (2010) A nested PCR assay exhibits enhanced sensitivity for detection of Theileria parva infections in bovine blood samples from carrier animals. Parasitol Res 106:357–365. https://doi.org/10.1007/s00436-009-1670-z
Olds CL, Mason KL, Scoles GA (2018) Rhipicephalus appendiculatus ticks transmit Theileria parva from persistently infected cattle in the absence of detectable parasitemia: Implications for East Coast fever epidemiology. Parasit Vectors 11:126. https://doi.org/10.1186/s13071-018-2727-6
Ota N, Mizuno D, Kuboki N, Igarashi I, Nakamura Y, Yamashina H, Hanzaike T, Fujii K, Onoe S, Hata H, Kondo S, Matsui S, Koga M, Matsumoto K, Inokuma H, Yokoyama N (2009) Epidemiological Survey of Theileria orientalis Infection in Grazing Cattle in the Eastern Part of Hokkaido, Japan. J Vet Med Sci 71:937–944
Otgonsuren D, Sivakumar T, Amgalanbaatar T, Narantsatsral S, Tuvshintulga B, Zoljargal M, Munkhgerel D, Davkharbayar B, Baatarjargal P, Myagmarsuren P, Battsetseg B, Battur B, Yokoyama N (2020) Molecular epidemiological survey of Babesia bovis, Babesia bigemina, and Babesia sp. Mymensingh infections in Mongolian cattle. Parasitol Int 77:102107. https://doi.org/10.1016/j.parint.2020.102107
Parola P, Raoult D (2001) Ticks and tick-borne bacterial diseases in humans: an emerging infectious threat. Ticks Tick-Borne Dis 32:897–928. https://doi.org/10.1086/319347
Ringo AE, Adjou Moumouni PF, Lee SH, Liu M, Khamis YH, Gao Y, Guo H, Zheng W, Efstratiou A, Galon EM, Li J, Tiwananthagorn S, Inoue N, Suzuki H, Thekisoe O, Xuan X (2018) Molecular detection and characterization of tick-borne protozoan and rickettsial pathogens isolated from cattle on Pemba Island, Tanzania. Ticks Tick-Borne Dis 9:1437–1445. https://doi.org/10.1016/j.ttbdis.2018.06.014
Ringo AE, Nonga HE, Galon EM, Ji S, Rizk MA, El-Sayed SAES, Mohanta UK, Ma Z, Chikufenji B, Do TT, Xuan X (2022) Molecular Investigation of Tick-Borne Haemoparasites Isolated from Indigenous Zebu Cattle in the Tanga Region Tanzania. Animals 12:3171. https://doi.org/10.3390/ani12223171
Simuunza M, Weir W, Courcier E, Tait A, Shiels B (2011) Epidemiological analysis of tick-borne diseases in Zambia. Vet Parasitol 175:331–342. https://doi.org/10.1016/j.vetpar.2010.09.027
Sivakumar T, Tuvshintulga B, Kothalawala H, Silva SSP, Lan DTB, Long PT, Ybañez AP, Ybañez RHD, Benitez DF, Tayebwa DS, de Macedo ACC, Schnittger L, Yokoyama N (2020) Host range and geographical distribution of Babesia sp. Mymensingh. Trans Emerg Dis 67:2233–2239. https://doi.org/10.1111/tbed.13546
Tamura K, Stecher G, Kumar S (2021) MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Tembo S, Collins NE, Sibeko-Matjila KP, Troskie M, Vorster I, Byaruhanga C, Oosthuizen MC (2018) Occurrence of tick-borne haemoparasites in cattle in the Mungwi district, Northern Province, Zambia. Ticks Tick Borne Dis 9:707–717. https://doi.org/10.1016/j.ttbdis.2018.02.004
Terkawi MA, Huyen NX, Shinuo C, Inpankaew T, Maklon K, Aboulaila M, Ueno A, Goo YK, Yokoyama N, Jittapalapong S, Xuan X, Igarashi I (2011) Molecular and serological prevalence of Babesia bovis and Babesia bigemina in water buffaloes in the northeast region of Thailand. Vet Parasitol 178:201–207. https://doi.org/10.1016/j.vetpar.2011.01.041
Teshale S, Geysen D, Ameni G, Dorny P, Berkvens D (2018) Survey of Anaplasma phagocytophilum and Anaplasma sp. “Omatjenne” infection in cattle in Africa with special reference to Ethiopia. Parasit Vectors 11:162. https://doi.org/10.1186/s13071-018-2633-y
Uilenberg G (1992) Veterinary Significance of Ticks and Tick-Borne Diseases. Tick Vect Biol. https://doi.org/10.1007/978-3-642-76643-5_2
Walker AR, Bouattour A, Camicas JL, Estrada-Pena A, Horak I, Latif A, Pegram RG, Preston PM (2003) Ticks of domestic animals in Africa: a guide to identification of species Bioscience Reports, Edinburgh, UK
Woldehiwet Z (2009) The natural history of Anaplasma phagocytophilum. Vet Parasitol 167:108–122. https://doi.org/10.1016/j.vetpar.2009.09.013
Woodford JD, Jones TW, Rae PF, Boid R, Bell-Sakyi L (1990) Seroepidemiological studies of bovine babesiaosis on Pemba Island, Tanzania. Vet Parasitol 37:175–184. https://doi.org/10.1016/0304-4017(90)90001-R
Ybañez AP, Ybañez RH, Claveria FG, Cruz-Flores MJ, Xuan X, Yokoyama N, Inokuma H (2014) High genetic diversity of Anaplasma marginale detected from Philippine cattle. J Vet Med Sci 76:1009–1014
Acknowledgements
We thank Madalitso Jessie Nkhata, Vincent Kachisi, Joe Magombo, and the animal owners for the various roles played in this study.
Funding
This study was funded by the grant-in-Aid for Scientific Research (18KK0188), JSPS Core-to-Core program, and Strategic International Collaborative Research Project (JPJ008837) sponsored by the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Author information
Authors and Affiliations
Contributions
Conceptualization, formal analysis, data curation, investigation, visualisation, methodology, writing original draft: Boniface Chikufenji. Writing-review and editing the original draft: Elisha Chatanga and Kyoko Hayashida. Methodology and Writing-review and editing: Uday Kumar Mohanta. Methodology and logistics: Nathan Kamanga, Eloiza May Galon, Aaron Ringo and Zhuowei Ma. Conceptualization, funding acquisition, resources, supervision, writing -review and editing: Xuenan Xuan.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
Authorization (permission ID number: DAHLD 002/2022) for sampling from cattle in the study locations was obtained from the Ministry of Agriculture Irrigation and Water Development (MoAIWD) through the Department of Animal Health and Livestock Development (DAHLD). Before sample collection, cattle owners were briefed on the significance of the activity and freely accepted to take part. Blood was collected by licensed veterinarians by following the ethical guidelines of Obihiro University of Agriculture and Veterinary Medicine, Hokkaido, Japan (animal experiment approval ID numbers: 22–23).
Consent for publication
All authors have read and approved the final version of this paper and have agreed for its publication.
Competing interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Chikufenji, B., Mohanta, U.K., Hayashida, K. et al. Molecular detection and phylogenetic analysis of tick-borne pathogens in cattle from southern Malawi. Vet Res Commun 48, 2753–2760 (2024). https://doi.org/10.1007/s11259-024-10395-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-024-10395-z