
Abstract
Adult Mesenchymal Stem Cells (MSC) are cells that can be defined as multipotent cells able to differentiate into diverse lineages, under appropriate conditions. These cells have been widely used in regenerative medicine, both in preclinical and clinical settings. Initially discovered in bone marrow, MSC can now be isolated from a wide spectrum of adult and foetal tissues. Studies to evaluate the therapeutic potential of these cells are based on their ability to arrive to damaged tissues. In this paper we have done a comparative study analyzing proliferation, surface markers and OCT4, SOX9, RUNX2, PPARG genes expression in MSC cells from Bone marrow (BMMSC) and Adipose tissue (ASC). We also analyzed the role of Stem Cell Factor (SCF) on MSC proliferation and on ASCs metalloproteinases MMP-2, MMP-9 secretion. Healthy dogs were used as BMMSC donors, and ASC were collected from omentum during elective ovariohysterectomy surgery. Both cell types were cultured in IMDM medium with or without SCF, 10% Dog Serum (DS), and incubated at 38 °C with 5% CO2. Growth of BMMSCs and ASCs was exponential until 25–30 days. Flow citometry of MSCs revealed positive results for CD90 and negative for CD34, CD45 and MCH-II. Genes were evaluated by RT-PCR and metalloproteinases by zymografy. Our findings indicate morphological and immunological similarities as well as expression of genes from both origins on analyzed cells. Furthermore, SCF did not affect proliferation of MSCs, however it up-regulated MMP-2 and MMP-9 secretion in ASCs. These results suggest that metalloproteinases are possibly essential molecules pivoting migration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular therapy has evolved quickly over the last decade both at the level of in vitro and in vivo preclinical research and in clinical trials. Mesenchymal stem cells (MSCs), have generated a great amount of interest in the field of regenerative medicine due to their biological properties (Hoffman and Dow 2016; Bajek et al. 2016).
MSCs were described mostly derived from bone marrow (BMMSCs) but, in recent years, adipose-derived mesenchymal stem cells (ASCs) have also been widely studied since they can be easily obtained (Frese et al. 2016).
MSCs secrete soluble factors which stimulate migration, mitosis and differentiation of local stem cells, enhance angiogenesis and modulate immunoreactions, and all of these facts make them interesting tools for tissue engineering and regeneration (Fu et al. 2017; Kyurkchiev et al. 2014).
These cells have a remarkable capacity of extensive in vitro expansion which allows them to rapidly reach the desired number for in vivo therapy. In 2006, the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT) defined MSCs by the following three criteria: MSCs must be adherent to plastic under standard tissue culture conditions; MSCs must express certain cell surface markers such as CD73, CD90, and CD105, and lack expression of other markers including CD45, CD34, CD14, or CD11b, CD79alpha or CD19 and HLA-DR surface molecules; MSCs must have the capacity to differentiate into osteoblasts, adipocytes, and chondroblasts under in vitro conditions; consequently these cells express OCT4 SOX9, RUNX2, PPARG genes as stem and early differentiation markers (Aubin et al. 2002). These cell markers have been described by Neupane et al. 2008 and Reich et al., 2012. Currently, studies have confirmed the existence of MSCs in canine adipose tissue as well as in bone marrow (Alipour et al. 2015; Csaki et al. 2009; Guercio et al. 2013; Reich et al. 2012). In veterinary medicine, MSCs are notably used in therapy of various disease states (Quintanilha et al. 2014; Marx et al. 2015; Murphy et al. 2003; Wang and Cao 2014).
MSCs are also capable of migrating to, and engraft in sites of inflammation after systemic administration and exert local functional effects in the resident tissue. Various studies have demonstrated that under a variety of pathologic conditions, MSCs selectively home to sites of injury, irrespective of the tissue (Lapidot et al. 2005; Ortiz et al. 2003; Wang et al. 2012). Homing is a multistep process in which growth factors promote a significant up-regulation in homing-related molecules (Janowska-Wieczorek et al. 1999; Lapidot et al. 2005; Zheng et al. 2003). It has been described that MSCs produce numerous growth factors and cytokines facilitating endogenous repair.
In the context of therapeutic use of Mesenchymal Stem Cells, it is mandatory to guaranty that cells home to damaged tissues and it is essential to find out the mechanism leading to migration. Homing depends on chemokine receptor CXCR4, and the expression of this receptor on cultured MSCs is induced by many factors (Shi et al. 2007; Sohni and Verfaillie 2013).
On the other hand, matrix metalloproteases (MMPs) have been demonstrated to play a role in MSC migration. Also, expression of MMPs in MSCs is influenced by several factors (Ries et al. 2007; Sohni and Verfaillie 2013).
Matrix metalloproteinases (MMPs) are required to break extracellular proteins, which are essential for any individual cell to interact with their surrounding environment. MMPs are subject to strict control. In 1971 it was demonstrated that MMPs are synthesized as inactive zymogens that require activation by proteolysis (Harper and Bloch 1971; Sternlicht and Werb 2001).
These proteins are the major group of enzymes regulating cell-matrix composition. MMPs genes show a highly conserved structure. Ample evidence exists on the role of MMPs in normal and pathological processes (Deruyina and Quigley 2006).
The extracellular matrix (ECM) is composed of a complex mixture of molecules; MMP-2 and MMP-9 (gelatinases) can degrade nearly all extracellular matrix components (Janowska-Wieczorek et al. 2000).
MMP-2 is secreted in a latent form with a molecular weight of 72 kDa becoming 59 to 62 kDa upon proteolytic activation. On the other hand, MMP-9 is secreted with a molecular weight of 92 kDa in a latent form and is converted to its active form (82 to 68 kDa). The use of MSCs as cellular therapy requires migration and engrafting in resident tissues, processes which are correlated with matrix metalloproteinases expression (Ho et al. 2009).
Herein, we have treated our MSCs with SCF, as a model to mimic damage regeneration. We have done this study on canine bone marrow and adipose tissue, comparing their phenotype, cell doubling time and surface markers. We also analyzed the expression of OCT4, SOX9, RUNX2, PPARG genes. Interestingly, our results show that SCF stimulates Metalloproteinases MMP-2 and MMP-9 secretion on MSCs from adipose tissue, which may have a crucial role in facilitating migration and homing of MSCs to the injured tissue. This finding can be of great interest for potential therapeutical applications.
Methods
Animals
Bone marrow: 8 healthy 2-year-old female dogs were used as bone marrow (BM) donors. BM from the iliac crest and wing of the ilium, approximately 10 ml, was collected in a syringe with 2 ml CPDA as anticoagulant, using a sterile technique under intravenous anesthesia with medetomidine (18 μg/kg) and diazepam (0.4 mg/kg); butorphanol (0.2 mg/kg) was used as analgesic. A second dose of butorphanol was given 6 h later, and further doses were administered subsequently every 6 h if pain was observed (values higher than 6/24 using the Glasgow Composite Measure Pain Scale, short form, CMPS-SF) (Reid et al. 2007). Furthermore, the dogs used in this study were treated according to the Spanish (Real Decreto 53/2013) and European Community legislation for the care and welfare of experimental animals, with approval from the Experimentation Animals Welfare Committee of the Veterinary Faculty, Complutense University of Madrid (approval reference number 052/PI).
Adipose tissue: 20 healthy 1-4-years-old female dogs were used as adipose tissue donors. Peritoneal fat (5 g) was obtained from the omentum of clinically healthy dogs at the time of elective ovariohysterectomy. All dogs were clinically healthy and presented no abnormalities in hematological and serum chemistry testing.
Both samples were processed between 1 and 24 h after collection. The adipose sample was kept at 4ªC in PBS containing 2% Penicillin/Streptomycin (P/S).
All dog owners signed an informed consent form allowing the use of removed tissue for research purposes.
BMMSC: Bone marrow samples were diluted 1/2 with PBS and mononuclear cells (MNC) were obtained by density gradient centrifugation using Ficoll-Paque Plus (d = 1,077 g/cm3, Bio-Science AB). Samples were washed once and centrifuged for 10 min at 1500 rpm. After that, cells were plated in culture wells (2 × 106/well) in Iscove’s Modified Dulbecco’s Medium (IMDM, Sigma–Aldrich St. Louis MO USA), with 10% Beagle dog serum (serum was derived from pooled batches obtained from whole blood collected in tubes free of anticoagulant, centrifuged at 2000 rpm for 10 min and filtered through 0.2 µm); cells were incubated at 38 °C, 5% CO2 and 90% air humidity. After an overnight, wells were washed 2–3 times with PBS and medium was recharged. After 10 days, cultures were established.
ADMSC: Samples were washed two times to eliminate blood clots, and homogenized manually. Tissues were digested with collagenase I 1 mg/ml (Sigma–Aldrich St. Louis MO USA) in Phosphate Buffered Saline (PBS) for an hour at 38 °C, 5% CO2 and 90% air humidity until an opaque solution was observed. Collagenase reaction was stopped by adding the same volume of IMDM medium. Thereafter, it was filtrated through a cell strainer of 100 μm, and was centrifuged in conical tube at 800 g/5 min. Supernatant was removed, and the cell fraction was washed in PBS and centrifuged at 800 g/ 5 min; supernatant was removed, and cell fraction was carefully collected and transferred to a 6-well plate with medium (IMDM, 10% DS, 1% Penicillin–Streptomycin, Sigma). Cells were cultured in humidified incubator at 38 °C, 5%CO2. After 3 days, wells were washed 2–3 times with PBS and medium was recharged. After 7 days, cultures were established.
Establishment of cell culture: the procedure was similar for both types of samples. Cells were detached using Trypsin–EDTA (0.05% trypsin-0.5 mM EDTA, Sigma–Aldrich St. Louis MO USA) for 3 min, neutralized with the same volume of IMDM medium and centrifuged at 800 rpm for 5 min. Both BMMSCs and ASCs growing processes were carried out with or without 100 ng/ml stem cell factor (SCF).
Cells were platted at 7000 cells/cm2 concentration in IMDM, with 10% allogeneic dog serum at 38 °C, 5% CO2 and 90% air humidity. The procedure was repeated for every passage. Along the different passages, cells were counted to measure in vitro growth and doubling time with or without SCF. Only cellular suspensions with > 95% of viable cells were used. Viability of cells was measured by Trypan Blue staining (Sigma–Aldrich St. Louis MO USA).
Cell-conditioned medium
500.000 cells were incubated in 200 µl of IMDM free of serum, at 38 °C, 5% CO2 and 90% air humidity for 18 h with and without 100 ng/ml of human recombinant SCF (R&D systems). The solution was centrifuged and conditioned medium was collected in non-reducing sample buffer (62.5 mM Tris–HCl, 5% SDS, 0.05% bromophenol blue and 20% glycerol, all from Sigma–Aldrich St. Louis MO USA). Cells and conditioned medium were store at − 20 °C until use.
Granulocytes
Granulocytes were obtained by density gradient centrifugation of dog blood using Ficoll-Paque Plus (d = 1,077 g/cm3, Bio-Science AB). After that, a 15 min lysis using RBC Lysis Buffer (Bio-Science AB) was performed. Granulocytes were stored at − 20 °C until use.
Colony-forming assays
Bone marrow MNC (1 × 105 cells/ml) and BMMSC from passages 2nd and 3rd (1 × 105 cells/ml) were assayed for colony-forming unit (CFU) in 0.33% agar as previously described (Fermín et al. 2004). These cells were stimulated with 100 ng/ml of human recombinant SCF (Sigma –Aldrich), 100 ng/ml of canine recombinant GM CSF (R&D systems), and 1 U/ml human erythropoietin (Roche Diagnostics, Indianapolis, USA). Cells were cultured at 38 °C in 5% CO2 in a humidified atmosphere. Hematopoietic colonies (CFCs) containing granulocytes, macrophages or both, were counted after 9–11 days.
RT-PCR analysis
Total RNA was extracted from a minimum of 106 BMMSCs and ASCs, using Pure Link RNA mini kit (Ambion) according to the manufacturer’s protocol. Specimens were adjusted to 40 mg/μl RNA and treated using High Capacity cDNA to RNA kit (Applied Biosystems) according to the manufacturer’s protocol. Minus RT sample were included. PCR was conducted using 2 μl cDNA, AmpliTaq Gold® DNA Polymerase with Gold Buffer (Applied Biosystems), MgCl2 (Applied Biosystems) and dNTPs (Applied Biosystems). All nucleotides were purchased from Metabion (Taper group). A set of primers was designed to amplify canine OCT4, RUNX2, SOX9 and PPARG genes. Primers designs were carried out using the Primer3 program (Koressaar and Remm 2007) and the National Centre for Biotechnology Information Blast Search Program (http://www.ncbi.nlm.nih.gov/).
Primers were as follows
-
GAPDH
-
Forward: 5′-CATCAACGGGAAGTCCATCT-3′
-
Reverse: 5′-CGACATCAAGAAGGTAGTGAAGC-3′
-
OCT-4
-
Forward: 5′-CAATTTGCCAAGCTCCTGA-3′
-
Reverse: 5′-CGCCAGAGGAAAGGATAC-3′
-
PPARG
-
Forward: 5′-TGGCAAAGAGCTGAGAGGAC-3′
-
Reverse 5′-AAAATCAAGTTCAAACACATCACC-3′.
-
RUNX2
-
Forward: 5′-GAGCACCGAAGAACAACTG-3′,
-
Reverse: 5′-CCACAGAAAACCCATCCAGA-3′;
-
SOX9
-
Forward: 5′-AACGGCTCGAGCAAGAAC-3′
-
Reverse: 5′-GAGTGCACCTCGTTCATGC-3′
Cycling conditions were as follows: 94 °C for 5 min, following 35 cycles of 94 °C for 30 s, 58ªC for 30 s for OCT4 and 60 °C for 30 s for others, 72 °C for 30 s and finally 72 °C for 10 min. PCR products were separated using a 2% agarose gel electrophoresis and visualized by Sybr green (Thermo Fischer). To check the specificity of the primers used, amplicons obtained were sequenced and compared with the target gene sequences available in the GenBank/ EMBL databases using the Blast software (http://www.ncbi.nlm.nih.gov/BLAST).
Flow cytometry immunophenotyping
Non-conjugated monoclonal antibodies: anti-canine CD90 (provided by Dr. Elisabeth Kremmer), anti-canine MHC-II (Bio-Rad) and anti-canine CD34 (Ostronoff et al. 2008) were used. Two hundred microliter of BMMSCs or ASCs suspensions, containing 200.000 cells, were placed in 5 mL sterile plastic tubes and incubated for 30 min at 4 °C with 10 µL of monoclonal antibodies. Cells were washed with MACS buffer medium (300 g, 5 min) and then suspended in 50 µL of this buffer. After 15 min of incubation at 4 °C in the dark, with fluorescein isothiocyanate (FITC, AbD Serotec Kidlington UK) or phycoerythrin (PE, AbD Serotec Kidlington UK), cells were washed with MACS buffer medium (Miltenyi Biotec) for 5 min under 300×g and suspended in 600 µL of PBS.
Conjugated monoclonal antibody: Anti-canine CD45 (AbD Serotec Kidlington UK conjugated with PE) was used. Two hundred microliter of BMMSCs or ASCs suspension containing 200.000 cells was mixed with 10 µL of the antibody in a 5 mL plastic tube and incubated in the dark at room temperature for 30 min. Then, cells were washed with MACS buffer medium (300 g, 5 min) and resuspended in 600 µL of PBS.
Cells were analyzed immediately after being marked or 48 h after using a fixing solution (Bio-Science AB). In both cases results were similar.
Flow cytometry acquisition was performed using a FACScan (Becton Dikinson, San Jose CA, USA). A minimum of 10,000 events were acquired for each sample. Flow cytometry data were analyzed using FacsDiva 6.0 software (Becton Dikinson, San Jose CA, USA).
Gelatin zymography
Gelatinolytic activities were assessed under no-reducing conditions in 10% polyacrylamide gel co-polymerized with 0.1% pork skin gelatin. Neutrophils (Granulocytes) described to secrete MMP were used as a control (León et al. 2005). Cells and conditioned medium with and without 100 ng/ml SCF were analyzed. After electrophoresis, gels were washed with 2.5% Triton X-100 for 1 h to remove SDS and allow proteins to renature before being incubated in digestion buffer (Tris–HCl pH 7.5 50 mM; CaCl2 5 mM; NaCl 200 mM; ZnCl2 1 μM) for 24 h. Zymograms were stained with coomasie blue 0.5% and distained in a methanol/acetic acid solution. Enzyme activity was analyzed by looking to the clear zones of gelatin lysis against a blue background. Parallel gels were incubated in the presence of 5 mM EDTA to determine if zones of lysis were produced by metalloproteinases. All reagents come from Sigma–Aldrich St. Louis MO USA. Laser densitometric scanning determined the intensity of bands.
Statistics
Results were analyzed using the software program Graph Pad Prism version 6. Student’s t-test was used to investigate the significance of the differences among groups. Unpaired two-tailed t-test was performed for comparisons between two groups of non-paired data. Normal distribution of data was assessed using Kolmogorov–Smirnov and Shapiro–Wilk tests. The p value < 0.05 was considered to be significant.
Results
Cell growth
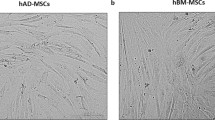
Microscopic direct observation of BMMSCs and ASCs showed adhesive cell populations with spindle-shaped, fibroblastic cell morphology along the plate (Fig. 1). Flow cytometry represented the size and complexity of the mesenchymal stem cells from two different sources, (A) from bone marrow-derived and (B) from adipose-derived cells (Fig. 1) Size and complexity of those two populations were almost identical, with slightly larger ADMSC, being hard to distinguish the origin unless it was known in advance.

Morphology of canine MSCs. a BMMSCs microscopic Leitz Labovert FS with EF L20/0.32 lens direct observation. b ASCs microscopic Leitz Labovert FS with EF L20/0.32 lens direct observation. c and d Flow cytometry of both BMMSC and ADMSC cell types in terms of size, as measured by the forward scattered light (FSC), and granularity, measured by the sideward scattered light (SSC)
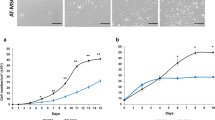
In vitro BMMSCs growth was exponential until 30 days, and doubling time of all samples studied remained constant throughout 1-3th passages (Fig. 2). ASCs growth was also exponential. BMMSCs showed a longer lag period than ASCs, approximately 6 days.

Expansion process. Comparative study of total number of cells versus number of passages for BMMSCs and ASCs with or without SCF. BMMSCs and ASCs growth with or without SCF are exponential. (n = 3–8 for each experimental point). Data were normalized / 10 cm2
BMMSC doubling time was 3.34 ± 0.75 days without SCF and 3.48 ± 0.93 days with SCF (P = 0.825). On the other hand ASCs doubling time was 1.08 ± 0.22 days without SCF and 1.07 ± 0.21 days with SCF (P = 0.563). Thus, there were not statistically significant differences between the input groups. Doubling time along different passages was measured by exponential regression with total number of cells and days data using an online cell calculation (Roth 2006).
Colony-forming assays
The number of CFC colonies obtained from bone marrow mononuclear cells was 129 ± 28 (n = 8); however, as expected, in the 2nd and 3rd passages of BMMSC, no growth of CFC colonies took place.
Flow cytometry
Using a panel of representative markers for Mesenchymal Stem Cells, we tested if the Mesenchymal stem cells obtained from (A) bone marrow and (B) adipose-tissue-derived were able to express the described markers. In both populations (BMMSCs and ASCs), on the 2nd and 3rd passages, cells were CD90 positive whereas expression of CD45, CD34 and CDMHCII was negative. (Fig. 3 and Table 1).

Cell surface markers. Representative dot plots of stained BMMSCs and ASCs labeled with CD90, CD34, CD45 and CDMHC. Both BMMSCs and ASCs have positive results for CD90 and negative results for CD34, CD45 and CDMHCII
Expression of pluripotency and early differentiation markers
To demonstrate the genes expression in the BMMSCs and ASCs samples, total RNA was extracted, and PCR products obtained from the cDNA were separated on 2% agarose gel Fig. 4. shows the expression of stem cell markers OCT4, SOX9, RUNX2, PPARG. PCR fragments of the expected size (OCT4: 504-bp; RUNX2: 450-bp; PPARG: 520- bp; SOX9: 383-pb and GAPDH: 587- bp) generated with the designed forward and reverse primers were found for the target locus in the assessed samples.

Pluripotency marker expression on BMMSCs and ASCs. Expression of stem cell markers OCT4, SOX9, RUNX2, PPARG were detected in BMMSCs and ASCs. Line 1, indicated molecular weight markers; lines 2 and 8, GAPDH served as housekeeping gene. Line 3 and 9, OCT4 gene. Lines 4 and 10, SOX9 gene; lines 5 and 11, RUNX2gene; lines 6 and 12, PPARG gene. Lines 7 and 13 correspond to the RT negative control. Data are allocated respectively for BMMSCs and ADSCs. Expression genes were detected in at least three different specimens for bone marrow and adipose tissue. Data shown were obtained from the same donor
Line 1 shows molecular weight markers; lines 2 and 8, GAPDH used as housekeeping gene; line 3 and 9, OCT4 gene; lines 4 and 10, RUNX2 gene; lines 5 and 11, SOX9 gene; lines 6 and 12, PPARG gene and lines 7 and 13 correspond to the RT negative control. Data are allocated respectively for BMMSCs and ASCs.
Expression genes were detected in at least three different specimens for bone marrow and adipose tissue. Data shown were obtained from the same donor.
To check the specificity of the primers, PCR fragments were sequenced and compared to the target gene sequences available in the GenBank/ EMBL databases displaying a 99% match.
Results showed that canine OCT4, RUNX2, SOX9 and PPARG gene were expressed in both BMMSCs and ASCs, indicating their capacity to differentiate into particular lineages under in vitro conditions, in accordance with Han et al. 2014 and Reich et al. 2012.
Gelatin zymography
Zymography results (Fig. 5), indicate expression of metalloproteinases in canine ASCs. In line 3, we can observe Proenzymes MMP-2 (72 kDa) and MMP-9 (92 kDa). The active forms of MMP-2 (59–62 KDa) and MMP-9 (82 − 68 kDa) (lines 4, 5) metalloproteinases were found in cell conditioned medium obtained with or without SCF; these forms presented a higher relative electrophoretic migration than those observed in the cells. Gelatin digestion was completely abolished when gels were incubated in the presence of 5 mM EDTA (+ EDTA line 6). Interestingly, SCF had a stimulatory effect on ADMSC, increasing secretion of active forms of MMP-9 and MMP-2 after 18 h of incubation; in fact, there was a 1. Sixfold increase in intensity of gelatinase bands in the presence of SCF. Line 1 represents granulocyte cells used as positive control.

Gelatin zimography. Fresh ASCs cells and conditioned media were assayed for gelatin zimography on 10% acrilamide gel contained 0.1% gelatin. Cells were incubated in the absence (-SCF) and presence (+ SCF) of hCSF for 18 h. After zimography gel was stained with Coomase Blue. Line 1 indicated molecular weight markers. In line 2, 15,000 Granulocytes were loaded as a positive control. In line 3, 25,000 ASCs were loaded. Line 4 corresponds to conditioned medium obtained from 500,000 ASCs. Line 5 corresponds to conditioned medium obtained from 500,000 cells + 100 ng/ml SCF and Line 6 cells with EDTA. Optical density for line 4 was 0.03795 and for line 5 0.05304 (1.6 X), n = 2. A representative zymogram of two independent experiments is shown
Discussion
Mesenchymal Stem Cells have become an important tool for a number of therapeutic applications. In this paper we studied and compared biological characteristics from two sources, bone marrow and adipose-derived mesenchymal stem cells (BMMSCs and ASCs), to establish optimal conditions for their therapeutical use. In this study, we have added SCF to the cell cultures, in an attempt to mimic damage regeneration conditions found in vivo.
Our data confirm the high morphological resemblance of canine MSCs of both origins, finding highly similar spindle-shaped, fibroblastic cell morphology independently of SCF presence. Additionally, flow cytometry confirmed similarity of both cell types in terms of size, as measured by forward scattered light (FSC), and granularity measured by sideward scattered light (SSC) (Fig. 1a,b). These results are in accordance with those found by Reich et al. 2012.
Expansion data indicate that cells from both origins can be expanded in vitro maintaining constant their doubling time until 3th passage. As we can see in Fig. 2, ASCs proliferation capacity is higher than that of BMMSCs, probably due to their doubling time value.
Shihua et al. 2012, conducted the first stem cell clinical trial approved from SFDA in China and their standard procedure requires that the optimal passage number should be less than six passages. On the other hand, SCF does not affect proliferation capacity neither of BMMSC nor ASC (Fig. 2a,b). We observed a longer initial lag phase in BMMSC than in ASC, which is in accordance with data described in humans, where BMMSCs also have a prolonged lag phase followed by fast division (Chamberlain et al. 2007). CD90, a widely accepted surface marker on MSCs in humans, (Dominici et al. 2006) was expressed by over 90% of the cells of both origins at all passages analyzed (Fig. 3 and Table 1). These findings are in accordance with those of Csaki et al. 2010. MSCs from both origins are immune privileged, because they do not express MHCII (Vieira et al. 2010); this characteristic enables transplantation of allogeneic MSCs without inducing immune rejection. The lack of expression in BMMSC for CD34 and CD45 indicated that cells of hematopoietic origin had been excluded during the cell expansion process; this was also corroborated by the lack of haematopoietic CFCs growing from cells during 2nd and 3rd passages. On the other hand, OCT4, SOX9, RUNX2, PPARG genes were expressed in both types of MSCs, an essential fact as it allows cells to differentiate (Fig. 4).
Considering how complicated it is to obtain BMMSCs, it is preferable to work with ASCs. This is the case in Veterinary medicine and especially in small breeds, where the amount of bone marrow that can be harvested is limited.
For these reasons, ASC have been utilized in the majority of trials in companion animals, such as treatment of Keratoconjunctivitis Sicca, Inflammatory Bowel Diseases, Atopic Dermatitis, Dilated Cardiomyopathy among others (Hoffman and Dow 2016).
Our results support this possibility since similar morphological behavior and markers as well as differentiation marker genes expression were observed in cells from the two origins, (Figs. 1, 2, 3, 4 and Table 1) as other authors have already described (Trivanović et al. 2015; Zhu et al. 2012).
In this respect, it is important to point out that SCF did not have any effect on the parameters cited above in canine MSCs.
As it is well known, during an injury, host cells release different growth factors that have a positive influence on homing MSCs. Homing relies mainly on the chemokine receptor CXCR4, and the expression of this receptor on MSCs is stimulated by many growth factors. (Sohni and Verfaillie 2013; Vieira et al. 2010). Pretreatment of cultured MSCs with cytokines increased expression of chemokine receptors (CXCR4) (Shi et al. 2007). Similarly, matrix metalloproteases (MMPs), have been demonstrated to play an important role in MSC migration (De Becker et al. 2007).
With the aim to evaluate if SCF has an effect on metalloproteinases expression, we analyzed the constitutive expression of MMP-2 and MMP-9 in ASCs, as well as the extracellular release of MMPs active forms. These endopeptidases are zinc-dependent and capable of degrading nearly all components of intracellular matrix, and are expressed by many cells (Almalki and Agrawal 2016; Lapidot et al. 2005) We have demonstrated expression of MMP-2 and MMP-9 in ASCs as well as their active forms (59 to 62 kDa and 82 to 68 kDa) in the conditioned medium we have developed, with and without SCF.
We suggest that Pro metalloproteinases are activated by cleavage of its amino terminal propeptide by some proteinase secreted by ASCs. As far as we know this is the first time that these metalloproteinases are described to be expressed constitutively in canine ASCs, as well as released in their active forms to the cellular surrounding. In this context, it is important to point out the finding that SCF produced a 1.6 fold increase on expression of the active forms (Fig. 5, line 5 respect line 6).
The use of MSCs as cellular therapy implies migration and engrafting in resident tissues, processes which are correlated with matrix metalloproteinases expression. In order to gain insight in the mechanisms regulating this process, we have treated mesenchymal stem cells from canine bone marrow and adipose tissue with SCF, and have compared their phenotype, cell doubling time and surface markers. We also have assessed differentiation marker genes in cells from both origins. On the other hand, we tested out expression of metalloproteinases MMP-2 and MMP-9 in adipose derived mesenchymal stem cells, analyzing the role of SCF on its secretion. Based on our results, we describe, for the first time, that these metalloproteinases are constitutively expressed in canine ASCs. We can also conclude that SCF has an important role up-regulating secretion of these metalloproteinases, possibly essential molecules pivoting migration. Further investigations on canine MSCs differentiation into specific tissues and migration assays may prove the interest of MMPs and lead to the development of novel therapeutic strategies to target specific diseases.
Our findings allow us to propose the use of ASC and SCF simultaneously, to provide fast and efficient cell migration to damaged tissues.
References
Alipour F, Parham A, Kazemi Mehrjerdi H, Dehghani H (2015) Equine adipose-derived mesenchymal stem cells: phenotype and growth characteristics, gene expression profile and differentiation potentials. Cell J 16(4):456–465. https://doi.org/10.22074/cellj.2015.491
Almalki SG, Agrawal DK (2016) Effects of matrix metalloproteinases on the fate of mesenchymal stem cells. Stem Cell Res Ther 7(1):129. https://doi.org/10.1186/s13287-016-0393-1
Aubin JE, Liu F, Candeliere GA (2002) PCR methods for studying stem cells and progenitors. Methods Mol Biol 185:403–415
Bajek A, Gurtowska N, Olkowska J, Kazmierski L, Maj M, Drewa T (2016) Adipose-derived stem cells as a tool in cell-based therapies. Arch Immunol Ther Exp (Warsz) 64(6):443–454. https://doi.org/10.1007/s00005-016-0394-x
Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T (2003) Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 253(1–2):269–285
Chamberlain G, Fox J, Ashton B, Middleton J (2007) Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 25(11):2739–2749
Csaki C, Matis U, Mobasheri A, Shakibaei M (2009) Co-culture of canine mesenchymal stem cells with primary bone-derived osteoblasts promotes osteogenic differentiation. Histochem Cell Biol 131(2):251–266. https://doi.org/10.1007/s00418-008-0524-6
De Becker A, Van Hummelen P, Bakkus M, Vande Broek I, De Wever JD, Waele M, Van Riet I (2007) Migration of culture-expanded human mesenchymal stem cells hrough bone marrow endothelium is regulated by matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-3. Haematologica 92(4): 440–449; https://doi.org/10.3324/haematol.10475
Deruyina E, Quigley J (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25(1):9–34
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D et al (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4):315–317
Fermín ML, Gaitán S, Fragío C, Léon LG, Ostronoff LK, Kremmer E, Kolb HJ, Tejero C (2004) Canine long-term bone marrow culture neutrophil production and functionality. Acta Haematol 111(4):196–204
Frese L, Dijkman PE, Hoerstrup SP (2016) Adipose tissue-derived stem cells in regenerative medicine. Transfus Med Hemother 43(4):268–274
Fu Y, Karbaat L, Wu L, Leijten J, Both SK, Karperien M (2017) Trophic effects of mesenchymal stem cells in tissue regeneration. Tissue Eng Part B Rev 23(6):515–528. https://doi.org/10.1089/ten.TEB.2016.0365
Guercio A, Di Bella S, Casella S, Di Marco P, Russo C, Piccione G (2013) Canine mesenchymal stem cells (MSCs): characterization in relation to donor age and adipose tissue-harvesting site. Cell Biol Int 37(8):789–798. https://doi.org/10.1002/cbin.10090
Han SH, Jang G, Bae BK, Han SM, Koh YR, Ahn JO, Jung WS, Kang SK, Ra JC, Lee HW, Youn HY (2014) Effect of ectopic OCT4 expression on canine adipose tissue-derived mesenchymal stem cell proliferation. Cell Biol Int 38(10):1163–1173. https://doi.org/10.1002/cbin.10295
Harper E, Bloch KJ (1971) The zymogen of tadpole collagenase. Biochemistry 10(16):3055–3041
Ho IA, Chan KY, Ng WH, Guo CM, Hui KM, Cheang P, Lam PY (2009) Matrix metalloproteinase 1 is necesary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells 27(6):1366–1375
Hoffman AM, Dow SW (2016) Concise review: stem cell trials using companion animal disease models. Stem cells 34(7):1709–1729. https://doi.org/10.1002/stem.2377
Janowska-Wieczorek A, Marquez LA, Nabholtz JM, Cabuhat ML, Montaño J, Chang H, Rozmus J, Russell JA, Edwards DR, Turner AR (1999) Growth factors and cytokines upregulate gelatinase expression in bone marrow CD34 + cells and their transmigration through reconstituted basement membrane. Blood 93(10):3379–3390
Janowska-Wieczorek A, Marquez LA, Dobrowsky A, Ratajczak MZ, Cabuhat ML (2000) Differential MMP and TIMP production by human marrow and peripheral blood CD34 + cells in response to chemokines. Exp Hematol 28(11):1274–1285
Koressaar T, Remm M (2007) Enhancements and modifications of primer design program Primer3. Bioinformatics 23(10):1289–1291
Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, Belemezova K, Kyurkchiev S (2014) Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells 6(5):552–570. https://doi.org/10.4252/wjsc.v6.i5.552
Lapidot T, Dar A, Kollet O (2005) How do stem cells find their way home? Blood 106(6):901–910
León LG, Ostronoff LK, Fermín ML, Fragío C, Kremmer E, Tejero C (2005) In vitro generation of mature neutrophils from canine Lin- bone marrow cells. Vet Immunol Inmunopathol 107(1–2):41–50
Marx C, Silveira MD, Beyer Nardi N (2015) Adipose-derived stem cells in veterinary medicine: characterization and therapeutic applications. Stem Cells Dev 24(7):803–813. https://doi.org/10.1089/scd.2014.0407
Murphy JM, Fink DJ, Hunziker EB, Barry FP (2003) Stem cell therapy in a caprine model of osteoarthritis. Arthritis Rheum 48(12):3464–3474
Neupane M, Chang C, Kiupel M, Yuzbasiyan-Gurkan V (2008) Isolation and characterization of canine adipose-derived mesenchymal stem cells. Tissue Eng Part A 14(6):1007–1015. https://doi.org/10.1089/tea.2007.0207
Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG (2003) Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci USA 100(14):8407–8411
Ostronoff LK, Kremmer E, Fermín ML, Fragío C, Mysliwietz J, Kolb HJ, Tejero C (2008) Canine stem cell factor augments expression of matrix metalloproteinase-9 by CD34 cells. Cytotherapy 10(2):193–102. https://doi.org/10.1080/14653240701827407
Quintanilha LF, Takami T, Hirose Y, Fujisawa K, Murata Y, Yamamoto N, Goldenberg RC, Terai S, Sakaida I (2014) Canine mesenchymal stem cells show antioxidant properties against thioacetamide-induced liver injury in vitro and in vivo. Hepatol Res 44(10):E206–E217. https://doi.org/10.1111/hepr.12204
Reich CM, Raabe O, Wenisch S, Bridger P, Kramer M, Arnhold S (2012) Isolation, culture and chondrogenic differentiation of canine adipose tissue and bone marrow-derived mesenchymal stem cells–a comparative study. Vet Res Commun 36(2):139–148. https://doi.org/10.1007/s11259-012-9523-0
Reid J, Nolan AM, Hughes JML, Lascelles D, Pawson P, Scott EM (2007) Development of the short-form Glasgow Composite Measure Pain Scale (CMPS-SF) and derivation of an analgesic intervention score. Anim Welf 16(S):97–104
Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P (2007) MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood 109(9):4055–4063
Roth V (2006) http://www.doubling-time.com/compute.php
Shi M, Li J, Liao L, Chen B, Li B, Chen L, Jia H, Zhao RC (2007) Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica 92(7):897–904
Sohni A, Verfaillie CM (2013) Mesenchymal stem cells migration homing and tracking. Stem Cells Int 2013:130763. https://doi.org/10.1155/2013/130763
Sternlicht MD, Werb Z (2001) How matrix metalloproteinases regulate cell behabior. Annu Rev Cell Dev Biol 17:463–516
Trivanović D, Jauković A, Popović B, Krstić J, Mojsilović S, Okić-Djordjević I, Kukolj T, Obradović H, Santibanez JF, Bugarski D (2015) Mesenchymal stem cells of different origin: comparative evaluation of proliferative capacity, telomere length and pluripotency marker expression. Life Sci 141:61–73. https://doi.org/10.1016/j.lfs.2015.09.019
Vieira NM, Brandalise V, Zucconi E, Secco M, Strauss BE, Zatz M (2010) Isolation, characterization, and differentiation potential of canine adipose-derived stem cells. Cell Transplant 19(3):279–289. https://doi.org/10.3727/096368909X481764
Wang W, Cao W (2014) Treatment of osteoarthritis with mesenchymal stem cells. Sci China Life Sci 57(6):586–595. https://doi.org/10.1007/s11427-014-4673-7
Wang S, Qu X, Zhao RC (2012) Clinical applications of mesenchymal stem cells. J Hematol Oncol 5:19–28. https://doi.org/10.1186/1756-8722-5-19
Zheng Y, Watanabe N, Nagamura-Inoue T, Igura K, Nagayama H, Tojo A, Tanosaki R, Takaue Y, Okamoto S, Takahashi TA (2003) Ex vivo manipulation of umbilical cord blood-derived hematopoietic stem/progenitor cells with recombinant human stem cell factor can up-regulate levels of homing-essential molecules to increase their transmigratory potential. Exp Hematol 31(12):1237–1246
Zhu X, Shi W, Tai W, Liu F (2012) The comparition of biological characteristics and multilineage differentiation of bone marrow and adipose derived mesenchymal stem cells. Cell Tissue Res 350(2):277–287. https://doi.org/10.1007/s00441-012-1453-1
Acknowledgements
We are very thankful to Dra. Carmen Fajardo for her scientific assistance. We thank Flow Cytometry, Genomics and Proteomics UCM facilities, for their technical support. Leticia G. León care Marie Curie grant. Authors would like to thank Elisabeth Salva for their technical assistance. Some studies reported in this manuscript were supported by a grant from the Deutsche José Carreras Leukämie Stiftung e.V. (R01/R13.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure of interest
The authors have no commercial, proprietary, or financial interest in the products or companies described in this article.
Additional information
Highlights
Standard phenotype from bone marrow BMMSCs and adipose tissue ASCs was confirmed.
We verified OCT4, SOX9, RUNX2, and PPARG genes expression in MSC from both origins.
ASCs constitutively express MMP-2 and MMP-9, being up-regulated by SCF.
Rights and permissions
About this article

Cite this article
Enciso, N., Ostronoff, L.L.K., Mejías, G. et al. Stem cell factor supports migration in canine mesenchymal stem cells. Vet Res Commun 42, 29–38 (2018). https://doi.org/10.1007/s11259-017-9705-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-017-9705-x