
Abstract
Restoration interventions such as revegetation are globally-important to combat biodiversity declines and land degradation. However, restoration projects are generally poorly monitored because current approaches to monitoring are limited in their ability to assess important components of biodiversity, such as belowground microbial diversity. Since soil bacterial communities mediate many belowground ecosystems processes and represent substantial biodiversity in their own right, bacteria are important components to monitor during ecosystem restoration. High-throughput amplicon sequencing (DNA metabarcoding) has been put forward as a potential cost-effective, scalable and easy-to-standardise partial solution to restoration’s monitoring problem. However, its application to restoration projects has to date been limited. Here, we used DNA metabarcoding of bacterial 16S rRNA gene from soil DNA to explore community differences across a 16-year restoration chronosequence. The bacterial composition in the oldest revegetation sites was comparable to the remnant sites. Proteobacteria and Acidobacteria were significantly higher in relative sequence abundance, while Actinobacteria was significantly lower, with time since revegetation. Classes Alphaproteobacteria and Acidobacteria were indicative of remnant and the oldest revegetation sites, while Deltaproteobacteria and Rubrobacteria were characteristic of younger revegetation sites. Changes in the soil physical and chemical characteristics associated with revegetation appear to shape bacterial community structure and composition. These findings provide evidence that revegetation can have positive effects on belowground microbial communities, and help demonstrate that the soil bacterial community can be restored towards its native state by revegetation, which may be useful in restoration monitoring.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Terrestrial coastal ecosystems often have high conservation value and provide many ecosystem services (Reyers et al. 2013; You et al. 2018). However, they are also sensitive to land degradation (e.g. via erosion) (Temmerman et al. 2013), following overexploitation, habitat transformation and pollution, which have impacted their ecological value (Lotze et al. 2006). In response, coastal ecosystems are increasingly targeted for ecological restoration in order to return native biodiversity and ecosystem services (Bullock et al. 2011; Liu et al. 2008). Indeed, in some circumstances, coastal ecosystem restoration can provide a more sustainable, cost-effective and ecologically sound alternative to coastal engineering solutions, such as installing sea walls (Temmerman et al. 2013).
Terrestrial ecosystems, including coastal areas, consist of aboveground and belowground ecological components that interact to shape communities (Collen and Nicholson 2014; Delgado-Baquerizo et al. 2017; Wardle et al. 2004). Plant-soil feedback processes influence plant and microbial communities and play important roles in determining their structure and successional dynamics (Callaway et al. 2004; Herrera et al. 2016; Mangan et al. 2010). Belowground microbiota influence plant community composition and productivity through microbe-mediated organic matter decomposition and nutrient cycling. In turn, low plant biomass may decrease the nutrients available to microbiota, subsequently decreasing their diversity (Qi et al. 2011; Yao et al. 2018).
The diversity and community dynamics of soil bacteria are often linked to soil physical, chemical and biological properties (e.g. soil age, pH, nutrient levels) (Guo et al. 2016; Moon et al. 2016; Trivedi et al. 2016; Zechmeister-Boltenstern et al. 2011). Revegetation has been shown to lead to changes in soil properties, such as pH, organic matter, carbon-to-nitrogen ratio and texture (An et al. 2009; Gellie et al. 2017; Guo et al. 2018; Yan et al. 2018). However, studies of the links between revegetation and soil properties, including both abiotic and biotic, in a coastal context are limited. For example, bacterial diversity has been shown to increase over a 3-year revegetation chronosequence in a coastal context (Banning et al. 2011b), and bacterial community structure has been associated with abiotic soil properties in coastal zones (Hua et al. 2017; Steenwerth et al. 2002). It is clear that revegetation can increase soil bacterial biomass, activity, and diversity in a variety of ecosystems (Nemergut et al. 2007; Sigler and Zeyer 2002). However, the extent to which these trends are maintained in a coastal context remains unclear.
In this study, we test the hypothesis that replanting the native plant community at a degraded coastal area associates with similarities found in the ecological reference site. We used high-throughput amplicon sequencing to explore the soil bacterial community across a 16-year restoration chronosequence, including samples from remnant sites. We analysed these samples to address the following questions: (i) Do soil physicochemical parameters correspond to time since revegetation, and do these physicochemical parameters associate with the bacterial community? (ii) Does the soil bacterial community differ more between remnant sites and young revegetation sites or between remnant sites and older revegetation sites? (iii) Which bacterial taxa are indicators of the different ages of revegetation?
Materials and methods
Site description and sampling
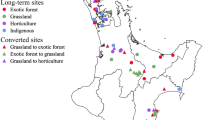
Our study system was an active coastal restoration site at Fishery Beach (35.63° S, 138.12° E), 4 km from Cape Jervis in South Australia (Fig. 1). This site was dominated by pasture grasses used for sheep grazing until a land management decision for revegetation was made in the late 1990s (i.e. revegetation for land management purposes, and not for experiments). Revegetation was done in a consistent way between 11 and 16 years ago, and included the combined use of the same site preparation method (i.e. shallow surface rip), site maintenance (i.e. fencing to exclude stock and woody weed removal) and replanting the native dominant tree Allocasuarina verticillata (drooping sheoak), plus low density native shrubs (e.g. Dodonaea viscosa) at all sites. A. verticillata is a nitrogen fixing tree species native to southeastern Australia, including the restoration site. The remnant, reference sites were approximately 5 km east of the restoration site and were also dominated by A. verticillata. The remnant sites were protected from grazing and have had minimal human impact since 1971.

Location and area of revegetation and remnant sites at Fishery Beach, South Australia. The revegetation at sites included in our study was undertaken 16, 15, 14, 13, 12 and 11 years before sampling. Remnant sites were approximately 5 km from the 16 years ago site along the coast
Sampling was conducted as part of the Biomes of Australian Soil Environments (BASE) project according to the methods described in Bissett et al. (2016). In December 2014, we sampled soil from three randomly selected 25 × 25 m quadrats at each of six revegetation sites, including sites restored 11, 12, 13, 14, 15 and 16 years before sampling. In addition, four randomly selected 25 × 25 m quadrats were sampled in remnant reference sites, giving a total of 22 quadrats. Soil was sampled from the 0–10 and 20–30 cm soil horizons at each quadrat, which is the standard protocol for BASE. Nine soil samples per quadrat were pooled into a sterile plastic bag, transported to the laboratory immediately after collection in sterile 50 mL falcon tubes on dry ice, stored at − 20 °C for DNA analysis—known hereafter as the replicates (n = 44). 300 g of homogenised soil was also sampled for soil physicochemical analysis, including soil moisture, ammonium, nitrate, available phosphorus, sulphur, organic carbon, conductivity and soil pH (H20). Soil chemical and physical attributes were determined at CSBP Laboratories (Perth, Western Australia), using the methods described in Bissett et al. (2016). DNA extraction and sequence analyses were conducted according to the methods described in Bissett et al. (2016). Briefly, soil DNA was extracted and then pooled from 3 × 0.25 g soil samples per replicate at the Australian Genome Research Facility (AGRF, Adelaide, Australia) using MoBio powersoil DNA extraction kits according to manufacturer’s instructions. We amplified the bacterial 16S ribosomal DNA with PCR for each replicate using the 16S rRNA gene primers 27F and 519R (Lane 1991). Sequences (300 bp PE) were then produced using the Illumina MiSEQ platform.
Sequence data and bioinformatic analysis
Sequence data used for this work were generated from the BASE database workflow (Bissett et al. 2016), and were downloaded as OTU abundance tables from the BASE download portal (https://data.bioplatforms.com/bpa/otu/; samples 102.100.100/192237–19354; 102.100.100/192273–19280) using the taxonomic filters: Amplicon = “27f519r_Bacteria, Kingdom = “Bacteria”, Phylum ≠ “Bacteria_unclassified” on 30th May, 2018 (OTU table is available at Figshare under DOI https://doi.org/10.25909/5d54a4dbe417c.25909/5d54a4dbe417c). Sequence data have been deposited with the NCBI sequence read archive under Bioproject number PRJNA317932.
Bioinformatic analysis was carried out according to Bissett et al. (2016). Briefly, read merging was done with FLASH (Magoč and Salzberg 2011), and merged reads < 400 bp and those containing Ns or homopolymer runs > 8 bp were removed using MOTHUR v1.34.1 (Schloss et al. 2009). OTU's at 97% sequence similarity were picked from remaining reads as follows: (1) demultiplex reads; (2) make an OTU FASTA mapping file split into chimeric and non-chimeric reads by sorting reads by abundance, remove reads with < 6 copies, cluster reads into OTUs of ≥ 97% similarity and identifying chimeras using UPARSE (Edgar 2013) (3) map original reads from (1) to the OTU representative sequences to compile a read abundance (OTU) table (Rideout et al. 2014).
OTUs were identified using Greengenes (DeSantis et al. 2006), using the RDP naïve Bayesian classifier (Wang et al. 2007) in MOTHER at a 60% read similarity cut-off. At the time data download BASE protocols also discarded OTUs not identified as belonging to bacteria, unidentified at the phylum level. Further details of our molecular methods can be found in Bissett et al. (2016).
Statistics
R v 3.5.1 (R Core Team 2018) was used to run all statistics. Except for alpha diversity, we discarded the OTUs with less than 100 reads across all 44 samples before further data analysis, because their abundances are likely to be poorly estimated and as such could not be attributed to a treatment effect. The number of reads was rarefied to the replicate with the lowest number of reads (94,867 and 108,543 reads, in 0–10 and 20–30 cm soil samples, respectively) with the rarefy function in vegan v 2.5-1 (Oksanen et al. 2018), with subsampling without replacement. OTU richness was estimated using the Chao 1 nonparametric richness estimator. Diversity was estimated as the effective number of species (Jost 2006) using the Shannon–Wiener index (H) and the Gini-Simpson index (D), where the Shannon–Wiener index and Gini-Simpson index were transformed by using the formula exp(H) and 1/(1 − D), respectively, to estimate the effective number of species.
Distance based redundancy analyses (db-RDA) based on Bray–Curtis distance matrices of bacterial 97% OTUs were used to visualise the effect of restoration on bacterial composition, and the relationships between soil physical and chemical variables and bacterial community composition. Prior to the ordination, the soil physicochemical variables were normalised using the decostand function in vegan. Permutation test (9999 permutations) with the permutest function in vegan was introduced after db-RDA to measure the significant association between bacterial community composition and soil physical and chemical variables.
Bacterial OTUs that significantly associated with the revegetation chronosequence pairwise combinations were identified through indicator species analysis (Dufrêne and Legendre 1997), using the indicator value as the statistical index (De Cáceres et al. 2010; Egidi et al. 2019; Rime et al. 2015). Indicator species analysis was run using the multipatt function implemented in the indicspecies package with 99,999 permutations. P value correction for multiple testing was run using the fdrtool function implemented in the fdrtool package (Strimmer 2008) with a false discovery rate of 10% (q < 0.10). Heatmaps were generated using hierarchical clustering methods with the plot_heatmap function in phyloseq v 1.22.3 (McMurdie and Holmes 2013).
Differences in relative sequence abundance, OTU richness, diversity indices, phyla, classes and soil characteristics across the restoration chronosequence, soil depths and the interaction between revegetation site and soil depths were analysed using permuted analysis of variance with the aovp function implemented in lmPerm 2.1.0 package (Wheeler and Torchiano 2016) with 5000 permutations.
The Adonis method for permutational multivariate analysis of variance (Anderson 2001) was used to test significant differences in bacterial community composition across the restoration chronosequence at the 0–10 and 20–30 cm soil depths. The Tukey HSD test was used to determine if the relationship between two revegetation sites was statistically significant or not.
Results
Bacterial community changes
We generated a total of 5,917,424 raw 16S rRNA gene reads across the 44 samples. Significant changes in effective species number based on Shannon’s diversity were observed, but, no significant changes were seen in richness of the bacterial community across the restoration chronosequence (Table 1). A total of 3,817,735 bacterial sequences and 10,322 bacterial 97% OTUs (4631 ± 971 SD per sample) remained for further analysis after quality filtering. Distance based redundancy analysis (db-RDA) showed distinct bacterial communities across the restoration chronosequence (Fig. 2), where sites revegetated between 11 to 15 years before sampling had similar bacterial communities, and showed significant differences to remnant sites and sites revegetated 16 years ago (Table A5). The permuted analysis of variance test also showed that significant differences were observed between soil depths (P = 0.047), but no significant interaction was observed (P = 0.223).

Distance based redundancy analysis (db-RDA) of soil physicochemical variables and bacterial community. db-RDA was based on Bray–Curtis distance matrices of bacterial 16S rRNA OTUs at a 0–10 cm and b 20–30 cm soil depths across the restoration chronosequence. The variables in red indicate significant effects on the bacterial community
The phylum proteobacteria had the greatest relative sequence abundance and richness (24.47% and 23.46%, 2326 and 2343 OTUs, at 0–10 and 20–30 cm, respectively), followed by Actinobacteria (23.81% and 23.27%; 2107 and 2113 OTUs, at 0–10 and 20–30 cm, respectively) and Acidobacteria (19.09% and 18.63%; 1931 and 1941 OTUs, at 0–10 and 20–30 cm, respectively) (Fig. 3, Table A1). All dominant phyla (each with > 1.5% of the total number of sequences in all samples) varied significantly in relative sequence abundance and varied significantly in Shannon’s diversity (except for Firmicutes) across the restoration chronosequence (Tables 2, A1; Fig. 3). Among the dominant phyla, Proteobacteria and Acidobacteria showed a trend of increasing relative sequence abundance at the 0–10 cm soil depth with time since revegetation, while Actinobacteria was significantly lower in relative sequence abundance at the 20–30 cm soil depth. The phyla Nitrospirae and WPS-2 had small relative sequence abundances and were higher in relative sequence abundances at 20–30 cm than 0–10 cm soil depths. Nitrospirae was significantly lower in 16-year old revegetation sites and remnant sites, while WPS-2 only appeared in 16-year old revegetation sites and remnant sites (Table A1; Fig. 3).

Stackplot showing changes in relative sequence abundance of bacterial phyla. Relative sequence abundances of the dominant bacterial phyla and rare bacteria at a 0–10 cm and b 20–30 cm depths across the restoration chronosequence
Overall, the most abundant classes among the Proteobacteria, Actinobacteria and Acidobacteria were Alphaproteobacteria, Rubrobacteria and Acidobacteria, respectively (Table A2). The classes Acidobacteria-6 (Acidobacteria), Deltaproteobacteria (Proteobacteria) and Rubrobacteria (Actinobacteria) decreased significantly in relative sequence abundance with time since revegetation, and Alphaproteobacteria (Proteobacteria) and Acidobacteriia (Acidobacteria) showed the opposite trend, where these differences were most pronounced between sites revegetated 11–15 years ago vs. 16-year old revegetation sites and remnant sites (Tables 2, A2). These five classes were the most abundant across the restoration chronosequence (Tables 2, A2; Fig. 4).

Stackplot showing changes in relative sequence abundance of bacterial classes. Relative sequence abundances of the dominant bacterial classes and rare bacteria at a 0–10 cm and b 20–30 cm depths across the restoration chronosequence
Indicator OTUs
Indicator species analysis was used to identify bacterial OTUs indicative of the different restoration stages. We observed 67 and 76 bacterial indicator OTUs at 0–10 and 20–30 cm soil depths, respectively (each with > 0.1% of the total number of sequences in all samples; q < 0.10; Figs. A1, A2). Indicator OTUs from the remnant and 16-year old revegetation sites were similar and were different from the other revegetation sites. Furthermore, the remnant and 16-year old revegetation sites had more indicator OTUs with larger indicator values (> 0.50), than the sites revegetated between 11 to 15 years before sampling (Figs. A1, A2).
Heatmaps based on the cluster analysis were run to visualise differences in relative sequence abundances of indicators across the restoration chronosequence. These plots showed the indicators associated with the restoration chronosequence, and these OTUs largely formed distinct assemblages across the restoration ages (Figs. 5, 6). Indicator analysis also revealed that the OTUs in the classes Alphaproteobacteria and Acidobacteria were generally associated with remnant and 16-year old revegetated sites, while those from the Deltaproteobacteria and Rubrobacteria were characteristic of sites revegetated between 11 to 15 years before sampling (Figs. 5, 6, A1, A2).

Heatmap with dendrograms based on the indicator OTU abundance across the restoration chronosequence. The 67 indicator OTUs (each having > 0.1% total relative sequence abundance) at 0–10 cm soil depths

Heatmap with dendrograms based on the indicator OTU abundance across the restoration chronosequence. The 76 indicator OTUs (each having > 0.1% total relative sequence abundance) at 20–30 cm soil depths
Bacterial community associations with soil abiotic properties
Notable differences in soil physical and chemical characteristics were observed across the restoration chronosequence and soil depths (Tables A3, A4). Differences with time since restoration included lower soil pH (P < 0.001) and higher sulphur (P = 0.030). Nitrate, phosphorous, organic carbon, conductivity and sulphur were significantly higher in 0–10 cm compared to 20–30 cm soil depths, while pH was significantly lower in 0–10 cm compared to 20–30 cm soil depths (Tables A3, A4).
Distance based redundancy analysis (db-RDA) showed that the proportion of variation in the bacterial community explained by the first two axes respectively was 53.07% and 13.70%, and 58.62% and 13.02% at 0–10 cm and 20–30 cm soil depths, respectively. A distinct bacterial community structure across the restoration chronosequence was observed (Figs. 2, A3). To further understand the relationships between bacterial community composition and soil properties, their Pearson correlation coefficients were calculated using Permutation tests. The bacterial community strongly associated with soil physical and chemical variables based on Bray–Curtis dissimilarity and Permutation tests at 0–10 (F6,15 = 4.019, P = 0.001) and 20–30 cm (F6,15 = 5.050, P < 0.001) (Figs. 2, A3). The factors with the highest correlation with the first two axes at 0–10 cm soil depth were soil moisture (R2 = 0.440, P = 0.004), and at 20–30 cm soil depth were conductivity (R2 = 0.475, P = 0.002), soil moisture (R2 = 0.431, P = 0.006), nitrate (R2 = 0.377, P = 0.014) and sulphur (R2 = 0.307, P = 0.028) (Fig. 2, Table A6).
Discussion
We used high-throughput 16S rRNA amplicon sequencing to explore differences in soil bacterial community composition across a 16-year coastal restoration chronosequence. The restoration intervention was revegetation of the native and dominant tree Allocasuarina verticillata. We observed that sites revegetated between 11 to 15 years before sampling had similar bacterial communities and were distinct from older revegetated (16 year) and remnant sites. Our study showed that changes on soil bacterial communities were associated with variation in soil moisture, nitrate, sulphur and phosphorus across the restoration chronosequence.
Our sampling design was not ideal to make space-for-time proxy conclusions on the effect of time since revegetation on the bacterial community. Our youngest revegetation site was 11 years old when sampled, and our sampling sites suffer from spatial autocorrelation of treatments (De Palma et al. 2018). Furthermore, we observed a large split in bacterial community structure between the sites revegetated 15 and 16 years prior to sampling, where the 15 years old site clustered with the sites sampled 11 to 14 years prior to sampling, and the 16 years old site clustered with the remnant vegetation site. Explaining such a divide requires additional investigation (e.g. longitudinal sampling), as it could potentially be explained not by the time since revegetation, but rather by unsampled site-specific features (e.g. groundwater). In spite of this, our results indicate the potential for this coastal revegetation intervention to have had a positive effect on the sub-surface soil bacterial community.
Most previous high-throughput ecological studies on bacterial community changes have focused on natural ecological processes, such as natural regeneration (Guo et al. 2018) or soil development (Jesus et al. 2009; Rime et al. 2015), and only few studies have explored bacterial responses to active ecological restoration (Banning et al. 2011a; Cavagnaro et al. 2016; Xue et al. 2017), with none in coastal contexts to our knowledge. Our work further contributes to the growing evidence that soil bacterial communities differ between varying plant communities—including those differing in age. Such differences imply a positive response to aboveground ecological restoration interventions, such as revegetation (Gellie et al. 2017).
Bacterial community differences
There is a common assumption in restoration that ‘if you plant it, they will come’ (Harris 2009). However, it is generally not well understood how long ecological recovery takes to occur, particularly for non-target taxa, such as bacteria. Two previous studies have shown considerable bacterial community recovery after 8 (Gellie et al. 2017) and 14 years of revegetation (Banning et al. 2011a), but in other studies, the bacterial community has been found not to respond, after 4 years of reduced grazing for example (Hamonts et al. 2017). As such, further work is required to more fully understand how and when microbial ecosystem components respond to traditional restoration interventions, such as revegetation.
Our study highlights the opportunity for using molecular tools to identify and assess bacterial groups during restoration, which may serve as potential ecological indicators of restoration processes. Proteobacteria, Actinobacteria and Acidobacteria were the phyla that exhibited the greatest differences in relative abundance between the revegetated sites in our study. Proteobacteria and Acidobacteria showed a significantly higher proportion of reads, while Actinobacteria were significantly lower, with time since revegetation. These associations are similar to a previous study on bacterial community changes in a woodland restoration system (Gellie et al. 2017). Indeed, the relative abundance of several Acidobacteria taxa seemed to show an increasing trend with time since revegetation, which is not surprising as these taxa tend to be more abundant in forests and less so in pastures (Fierer et al. 2007; Huang et al. 2015).
Bacteria are important foundations of ecosystem functions (e.g. nutrient cycling, symbioses, substrate formation) and supply valuable ecosystem services (e.g. carbon storage, nutrient availability for plants). In our study, we observed the bacterial phylum Nitrospirae to be significantly lower in relative abundance in older revegetation sites, which is consistent with previous work in a degraded steppe ecosystem (Yao et al. 2018). Nitrospirae are involved in the nitrogen cycle and performs nitrite oxidation in nitrification (Koch et al. 2015; Luecker et al. 2010). While further work is needed to confirm whether Nitrospirae was performing this function in our study system, its presence provides indirect evidence that it had the potential to do so.
We observed clear differences in bacterial OTU Shannon diversity across our samples, which is consistent with previous studies on soil bacteria (Zheng et al. 2017); such as rainforest-to-agriculture land conversion (Rodrigues et al. 2013). Other studies have found that land-use change does not alter soil bacterial diversity, even after 20-years of restoration (Liao et al. 2018), or conversion of long-standing primary forest to pasture or croplands (Jesus et al. 2009).
Overall, there is a great need for longitudinal, spatially-independent studies to further the development of molecular tools to identify and track microbial changes and indicators of restoration success. Previous studies, and ours, have sampled sites that differ in the time since restoration, but were each sampled at a single point in time. Such designs are not ideal to determine whether restoration was the primary cause of change, or whether changes occurred at all. Such design limitations reduce the strength of inferences about changes that may have occurred due to restoration and the identification of indicator taxa that could, with further development, be representatives of restoration success.
Potential bacterial indicator taxa of ecosystem restoration are useful as they make candidates for focussed, follow-up work that aims to better understand their function (Breed et al. 2019; Liddicoat et al. 2019). The species Burkholderia bryophila (phylum Proteobacteria) was a significant indicator for remnant and 16-year old revegetation sites, and has previously been reported in native grassland ecosystems (Vandamme et al. 2007). Two species in the genus Rubrobacter (phylum Actinobacteria) had significant indicator values for sites revegetated between 11 to 15 years before sampling, which has been reported to have an active role in efflorescence formation and mineral precipitation, which can contribute to biodeterioration processes (Mihajlovski et al. 2017).
Associations between soil abiotic properties and bacteria
Soil conditions have strong effects on the structure of microbial communities (Hanson et al. 2012; Singh and Gupta 2018; Yao et al. 2018). Soil moisture, nitrate, sulphur and conductivity at the 20–30 cm soil depth were each significantly correlated with the bacterial community across the restoration chronosequence. Such associations between soil abiotic conditions and bacterial community structure are expected (Liao et al. 2018; Singh and Gupta 2018). However, our findings are important in a restoration context because even though it is clear that strong associations exist between above and belowground biota (Wardle et al. 2004), excess soil nitrogen—often present pre-restoration, particularly in post-agricultural landscapes—can inhibit restoration (e.g. it can widen the environmental filter for invasive species; Funk 2008). Further work is needed to address the feedback loops between the bacterial community, plant community and soil conditions as they are generally not well understood.
We observed differences in the bacterial community with soil depth, which is likely contributed to by differences in resource inputs. Surface soils are expected to have greater detritus accumulation and availability of organic carbon and macronutrients than sub-surface soils (Allison et al. 2007). Soil depth had no overall effect on bacterial diversity and relative abundance. At the phylum level, soil depth had a significant effect on many taxa and most showed greater diversity and abundance in surface soils—as expected. A notable exception was the phylum Acidobacteria, which showed greater relative sequence abundance in deeper soils, which is counter to expectations (Fierer et al. 2007). More work is clearly needed to understand the impact of depth gradients and correlated environmental variables on the soil bacterial community, particularly in a restoration context.
Monitoring restoration of microbiota with high-throughput sequencing approaches
Monitoring changes to microbiota during ecosystem restoration is important because these microbiota have key functional ecological roles and represent substantial biodiversity in their own right (An et al. 2009; Breed et al. 2019). Consequently, a cost-effective quantification approach for microbiota is required if these microbial communities are to be successfully integrated into restoration monitoring and assessment. Microbial community cultivation-based approaches are methods that can be used to characterise microbial communities (Balint et al. 2016), however the majority of microorganisms are uncultivable by standard techniques, which leaves many taxa unmonitored (Epstein 2013). Soil microbial biomass (used to, for example, derive fungal to bacterial ratios), microbial enzyme activity and soil respiration are also established approaches to quantify microbial communities (Baldrian 2019; Kuzyakov and Blagodatskaya 2015). Data derived from these approaches can be proxies of or actual measurements of biochemical processes (Bandick and Dick 1999; Ros et al. 2003), and they have been used in restoration contexts (Li et al. 2018; Ross et al. 1982). While these methods are good at characterising general microbial patterns, and importantly can provide a picture of microbial function, they lack the fine-scale resolution that can be generated with high-throughput sequencing based approaches.
High-throughput sequencing has facilitated the emergence of DNA metabarcoding by sequencing a particular amplicon (or barcode) from an environmental DNA sample—an approach generally called metabarcoding or high-throughput amplicon sequencing (Gomez-Escribano et al. 2016; Lindahl et al. 2013; Taberlet et al. 2012). This approach can identify the biological sources of genetic material with reasonable accuracy and precision (Barnes and Turner 2015; Corlett 2017). High-throughput amplicon sequencing has been put forward as a cost-effective, efficient and easy-to-standardise approach that can be used to survey and monitor cryptic biodiversity (Ji et al. 2013), including in a restoration context (Breed et al. 2019). Such high-throughput sequencing approaches are by no means a panacea to monitoring microbiota, as these approaches are often limited by poor taxonomic databases and inaccessibility of functional attributes of some techniques (Egidi et al. 2019; Ji et al. 2013). Furthermore, false positives may occur for multiple reasons, such as contamination during sampling or during laboratory work, and PCR or sequencing errors (Ficetola et al. 2015). We emphasise that further work is required to improve high-throughput sequencing approaches, such as DNA metabarcoding, as a useful tool for monitoring and assessing the restoration of microbiota.
Conclusions
The development of high-throughput amplicon sequencing methods offers extraordinary scientific and practical opportunities for better understanding the roles of microbial communities in restoration contexts; for example, opening opportunities to develop targeted microbiome inoculations. However, the full potential and limitations of these methods to monitor restoration projects require more work to understand how consistent soil bacterial communities are in responding to revegetation and how they respond to other restoration interventions (Bulman et al. 2018; Cowart et al. 2015). Most restoration projects assess their success based either on simple input metrics (e.g. number of trees planted) or aim for outcomes that are difficult to quantify (e.g. improve ecosystem integrity) (Collen and Nicholson 2014; Harris 2003; Moreno et al. 2017). Our findings provide further encouraging evidence for establishing a DNA metabarcoding method as a scalable and comprehensive tool to monitor the microbiome of restoration interventions. With further development, high-throughput amplicon sequencing has great potential to be an effective tool used to monitor the efficacy of restoration interventions, as has been shown in other allied areas of ecology (Thomsen and Willerslev 2015; Valentini et al. 2016).
References
Allison VJ, Yermakov Z, Miller RM, Jastrow JD, Matamala R (2007) Using landscape and depth gradients to decouple the impact of correlated environmental variables on soil microbial community composition. Soil Biol Biochem 39:505–516
An S, Huang Y, Zheng F (2009) Evaluation of soil microbial indices along a revegetation chronosequence in grassland soils on the Loess Plateau, Northwest China. Appl Soil Ecol 41:286–292
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
Baldrian P (2019) The known and the unknown in soil microbial ecology. FEMS Microbiol Ecol 95:9
Balint M, Bahram M, Eren AM, Faust K, Fuhrman JA, Lindahl B, O'Hara RB, Opik M, Sogin ML, Unterseher M, Tedersoo L (2016) Millions of reads, thousands of taxa: microbial community structure and associations analyzed via marker genes. FEMS Microbiol Rev 40:686–700
Bandick AK, Dick RP (1999) Field management effects on soil enzyme activities. Soil Biol Biochem 31:1471–1479
Banning NC, Gleeson DB, Grigg AH, Grant CD, Andersen GL, Brodie EL, Murphy D (2011a) Soil microbial community successional patterns during forest ecosystem restoration. Appl Environ Microbiol 77:6158–6164
Banning NC, Phillips IR, Jones DL, Murphy DV (2011b) Development of microbial diversity and functional potential in Bauxite Residue Sand under rhabilitation. Restor Ecol 19:78–87
Barnes MA, Turner CR (2015) The ecology of environmental DNA and implications for conservation genetics. Conserv Genet 17:1–17
Bissett A, Fitzgerald A, Meintjes T, Mele PM, Reith F, Dennis PG, Breed MF, Brown B, Brown MV, Brugger J, Byrne M, Caddy-Retalic S, Carmody B, Coates DJ, Correa C, Ferrari BC, Gupta VV, Hamonts K, Haslem A, Hugenholtz P, Karan M, Koval J, Lowe AJ, Macdonald S, McGrath L, Martin D, Morgan M, North KI, Paungfoo-Lonhienne C, Pendall E, Phillips L, Pirzl R, Powell JR, Ragan MA, Schmidt S, Seymour N, Snape I, Stephen JR, Stevens M, Tinning M, Williams K, Yeoh YK, Zammit CM, Young A (2016) Introducing BASE: the biomes of Australian soil environments soil microbial diversity database. Gigascience 5:1–11
Breed MF, Harrison PA, Blyth C, Byrne M, Gaget V, Gellie NJC, Groom SVC, Hodgson R, Mills JG, Prowse TAA, Steane DA, Mohr JJ (2019) The potential of genomics for restoring ecosystems and biodiversity. Nat Rev Genet 20(10):615–628
Bullock JM, Aronson J, Newton AC, Pywell RF, Rey-Benayas JM (2011) Restoration of ecosystem services and biodiversity: conflicts and opportunities. Trends Ecol Evol 26:541–549
Bulman SR, McDougal RL, Hill K, Lear G (2018) Opportunities and limitations for DNA metabarcoding in Australasian plant-pathogen biosecurity. Australas Plant Pathol 47:467–474
Callaway RM, Thelen GC, Rodriguez A, Holben WE (2004) Soil biota and exotic plant invasion. Nature 427:731–733
Cavagnaro T, Cunningham S, Fitzpatrick S (2016) Pastures to woodlands: changes in soil microbial communities and carbon following reforestation. Appl Soil Ecol 107:24–32
Collen B, Nicholson E (2014) Taking the measure of change. Science 346:166–167
Corlett RT (2017) A bigger toolbox: biotechnology in biodiversity conservation. Trends Biotechnol 35:55–65
Cowart DA, Pinheiro M, Mouchel O, Maguer M, Grall J, Mine J, Arnaud-Haond S (2015) Metabarcoding is powerful yet still blind: a comparative analysis of morphological and molecular surveys of seagrass communities. PLoS ONE 10:e0117562
De Cáceres M, Legendre P, Moretti M (2010) Improving indicator species analysis by combining groups of sites. Oikos 119:1674–1684
De Palma A, Sanchez-Ortiz K, Martin PA, Chadwick A, Gilbert G, Bates AE, Börger L, Contu S, Hill SLL, Purvis A (2018) Challenges with inferring how land-use affects terrestrial biodiversity: study design, time, space and synthesis. In: Bohan DA, Dumbrell AJ, Woodward G, Jackson M (eds) Advances in ecological research. Academic Press, Cambridge, pp 163–199
Delgado-Baquerizo M, Powell JR, Hamonts K, Reith F, Mele P, Brown MV, Dennis PG, Ferrari BC, Fitzgerald A, Young A, Singh BK, Bissett A (2017) Circular linkages between soil biodiversity, fertility and plant productivity are limited to topsoil at the continental scale. New Phytol 215(3):1186–1196
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072
Dufrêne M, Legendre P (1997) Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr 67:345–366
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996
Egidi E, Wood JL, Celestina C, May TW, Mele P, Edwards J, Powell J, Bissett A, Franks AE (2019) Delving into the dark ecology: a continent-wide assessment of patterns of composition in soil fungal communities from Australian tussock grasslands. Fung Ecol 39:356–370
Epstein S (2013) The phenomenon of microbial uncultivability. Curr Opin Microbiol 16:636–642
Ficetola GF, Pansu J, Bonin A, Coissac E, Giguet-Covex C, De Barba M, Gielly L, Lopes CM, Boyer F, Pompanon F, Rayé G, Taberlet P (2015) Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol Ecol Resour 15:543–556
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Funk JL (2008) Differences in plasticity between invasive and native plants from a low resource environment. J Ecol 96:1162–1173
Gellie NJC, Mills JG, Breed MF, Lowe AJ (2017) Revegetation rewilds the soil bacterial microbiome of an old field. Mol Ecol 26:2895–2904
Gomez-Escribano JP, Alt S, Bibb MJ (2016) Next generation sequencing of Actinobacteria for the discovery of novel natural products. Mar Drugs 14:78
Guo XP, Chen HYH, Meng MJ, Biswas SR, Ye LX, Zhang JC (2016) Effects of land use change on the composition of soil microbial communities in a managed subtropical forest. For Ecol Manag 373:93–99
Guo Y, Chen X, Wu Y, Zhang L, Cheng J, Wei G, Lin Y (2018) Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci Tot Environ 635:598–606
Hamonts K, Bissett A, Macdonald BCT, Barton PS, Manning AD, Young A (2017) Effects of ecological restoration on soil microbial diversity in a temperate grassy woodland. Appl Soil Ecol 117–118:117–128
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH (2012) Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10:497
Harris JA (2003) Measurements of the soil microbial community for estimating the success of restoration. Eur J Soil Sci 54:801–808
Harris J (2009) Soil microbial communities and restoration ecology: facilitators or followers? Science 325:573–574
Herrera PS, Lebeis SL, Bailey JK (2016) Giving back to the community: microbial mechanisms of plant-soil interactions. Funct Ecol 30:1043–1052
Hua J, Feng Y, Jiang Q, Bao X, Yin Y (2017) Shift of bacterial community structure along different coastal reclamation histories in Jiangsu, Eastern China. Sci Rep 7:10096
Huang X, Liu L, Wen T, Zhu R, Zhang J, Cai Z (2015) Illumina MiSeq investigations on the changes of microbial community in the Fusarium oxysporum f.sp cubense infected soil during and after reductive soil disinfestation. Microbiol Res 181:33–42
Jesus EC, Marsh TL, Tiedje JM, de Moreira FMS (2009) Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J 3:1004–1011
Ji Y, Ashton L, Pedley SM, Edwards DP, Tang Y, Nakamura A, Kitching R, Dolman PM, Woodcock P, Edwards FA, Larsen TH, Hsu WW, Benedick S, Hamer KC, Wilcove DS, Bruce C, Wang X, Levi T, Lott M, Emerson BC, Yu DW (2013) Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol Lett 16:1245–1257
Jost L (2006) Entropy and diversity. Oikos 113:363–375
Koch H, Luecker S, Albertsen M, Kitzinger K, Herbold C, Spieck E, Nielsen PH, Wagner M, Daims H (2015) Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira. Proc Natl Acad Sci USA 112:11371–11376
Kuzyakov Y, Blagodatskaya E (2015) Microbial hotspots and hot moments in soil: concept & review. Soil Biol Biochem 83:184–199
Lane D (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, New York, pp 125–175
Li J, Tong X, Awasthi MK, Wu F, Ha S, Ma J, Sun X, He C (2018) Dynamics of soil microbial biomass and enzyme activities along a chronosequence of desertified land revegetation. Ecol Eng 111:22–30
Liao H, Zheng C, Li J, Long J (2018) Dynamics of soil microbial recovery from cropland to orchard along a 20-year chronosequence in a degraded karst ecosystem. Sci Tot Environ 639:1051–1059
Liddicoat C, Weinstein P, Bissett A, Gellie NJ, Mills JG, Waycott M, Breed MF (2019) Can bacterial indicators of a grassy woodland restoration inform ecosystem assessment and microbiota-mediated human health? Environ Int 129:105–117
Lindahl BD, Nilsson RH, Tedersoo L, Abarenkov K, Carlsen T, Kjoller R, Koljalg U, Pennanen T, Rosendahl S, Stenlid J, Kauserud H (2013) Fungal community analysis by high-throughput sequencing of amplified markers—a user's guide. New Phytol 199:288–299
Liu J, Li S, Ouyang Z, Tam C, Chen X (2008) Ecological and socioeconomic effects of China's policies for ecosystem services. Proc Natl Acad Sci USA 105:9477–9482
Lotze HK, Lenihan HS, Bourque BJ, Bradbury RH, Cooke RG, Kay MC, Kidwell SM, Kirby MX, Peterson CH, Jackson JBC (2006) Depletion, degradation, and recovery potential of estuaries and coastal seas. Science 312:1806–1809
Luecker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, Rattei T, Damste JSS, Spieck E, Le Paslier D, Daims H (2010) A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Natl Acad Sci USA 107:13479–13484
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Mangan SA, Schnitzer SA, Herre EA, Mack KML, Valencia MC, Sanchez EI, Bever JD (2010) Negative plant-soil feedback predicts tree-species relative abundance in a tropical forest. Nature 466:752–755
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217
Mihajlovski A, Gabarre A, Seyer D, Bousta F, Di Martino P (2017) Bacterial diversity on rock surface of the ruined part of a French historic monument: the Chaalis abbey. Int Biodeterior Biodegrad 120:161–169
Moon JB, Wardrop DH, Bruns MAV, Miller RM, Naithani KJ (2016) Land-use and land-cover effects on soil microbial community abundance and composition in headwater riparian wetlands. Soil Biol Biochem 97:215–233
Moreno M, de-Bashan LE, Hernandez J-P, Lopez BR, Bashan Y (2017) Success of long-term restoration of degraded arid land using native trees planted 11 years earlier. Plant Soil 421:83–92
Nemergut DR, Anderson SP, Cleveland CC, Martin AP, Miller AE, Seimon A, Schmidt SK (2007) Microbial community succession in an unvegetated, recently deglaciated soil. Microb Ecol 53:110–122
Oksanen J., Blanchet F.G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpson G.L., Solymos P., Stevens M.H.S., Szoecs E. and Wagner H. 2018. Vegan: community ecology package. R package version 2.5-1
Qi S, Zheng H, Lin Q, Li G, Xi Z, Zhao X (2011) Effects of livestock grazing intensity on soil biota in a semiarid steppe of Inner Mongolia. Plant Soil 340:117–126
R Core Team (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Reyers B, Biggs R, Cumming GS, Elmqvist T, Hejnowicz AP, Polasky S (2013) Getting the measure of ecosystem services: a social-ecological approach. Front Ecol Environ 11:268–273
Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, Chase J, McDonald D, Gonzalez A, Robbins-Pianka A (2014) Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2:e545
Rime T, Hartmann M, Brunner I, Widmer F, Zeyer J, Frey B (2015) Vertical distribution of the soil microbiota along a successional gradient in a glacier forefield. Mol Ecol 24:1091–1108
Rodrigues JLM, Pellizari VH, Mueller R, Baek K, Jesus EC, Paula FS, Mirza B, Hamaoui GS Jr, Tsai SM, Feigl B, Tiedje JM, Bohannan BJ, Nuesslein K (2013) Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc Natl Acad Sci USA 110:988–993
Ros M, Hernandez MT, García C (2003) Soil microbial activity after restoration of a semiarid soil by organic amendments. Soil Biol Biochem 35:463–469
Ross D, Speir T, Tate K, Cairns A, Meyrick KF, Pansier EA (1982) Restoration of pasture after topsoil removal: effects on soil carbon and nitrogen mineralization, microbial biomass and enzyme activities. Soil Biol Biochem 14:575–581
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Sigler WV, Zeyer J (2002) Microbial diversity and activity along the forefields of two receding glaciers. Microb Ecol 43:397–407
Singh JS, Gupta VK (2018) Soil microbial biomass: a key soil driver in management of ecosystem functioning. Sci Tot Environ 634:497–500
Steenwerth KL, Jackson LE, Calderón FJ, Stromberg MR, Scow KM (2002) Soil microbial community composition and land use history in cultivated and grassland ecosystems of coastal California. Soil Biol Biochem 34:1599–1611
Strimmer K (2008) fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24:1461–1462
Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E (2012) Towards next-generation biodiversity assessment using DNA metabarcoding. Mol Ecol 21:2045–2050
Temmerman S, Meire P, Bouma TJ, Herman PMJ, Ysebaert T, De Vriend HJ (2013) Ecosystem-based coastal defence in the face of global change. Nature 504:79–83
Thomsen PF, Willerslev E (2015) Environmental DNA—an emerging tool in conservation for monitoring past and present biodiversity. Biol Conserv 183:4–18
Trivedi P, Delgado-Baquerizo M, Anderson IC, Singh BK (2016) Response of soil properties and microbial communities to agriculture: implications for primary productivity and soil health indicators. Front Plant Sci 7:990
Valentini A, Taberlet P, Miaud C, Civade R, Herder J, Thomsen PF, Bellemain E, Besnard A, Coissac E, Boyer F (2016) Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol 25:929–942
Vandamme P, Opelt K, Knoechel N, Berg C, Schoenmann S, De Brandt E, Eberl L, Falsen E, Berg G (2007) Burkholderia bryophila sp nov and Burkholderia megapolitana sp nov., moss-associated species with antifungal and plant-growth-promoting properties. Int J Syst Evol Microbiol 57:2228–2235
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wardle DA, Bardgett RD, Klironomos JN, Setala H, van der Putten WH, Wall DH (2004) Ecological linkages between aboveground and belowground biota. Science 304:1629–1633
Wheeler B, Torchiano M (2016) lmPerm: permutation tests for linear models. R package version 2.1.0
Xue L, Ren H, Li S, Leng X, Yao X (2017) Soil bacterial community structure and co-occurrence pattern during vegetation restoration in Karst rocky desertification area. Front Microbiol 8:2377
Yan D, Mills JG, Gellie NJC, Bissett A, Lowe AJ, Breed MF (2018) High-throughput eDNA monitoring of fungi to track functional recovery in ecological restoration. Biol Conserv 217:113–120
Yao M, Rui J, Li J, Wang J, Cao W, Li X (2018) Soil bacterial community shifts driven by restoration time and steppe types in the degraded steppe of Inner Mongolia. CATENA 165:228–236
You S, Kim M, Lee J, Chon J (2018) Coastal landscape planning for improving the value of ecosystem services in coastal areas: using system dynamics model. Environ Pollut 242:2040–2050
Zechmeister-Boltenstern S, Michel K, Pfeffer M (2011) Soil microbial community structure in European forests in relation to forest type and atmospheric nitrogen deposition. Plant Soil 343:37–50
Zheng J, Chen J, Pan G, Wang G, Liu X, Zhang X, Li L, Bian R, Cheng K, Zheng J (2017) A long-term hybrid poplar plantation on cropland reduces soil organic carbon mineralization and shifts microbial community abundance and composition. Appl Soil Ecol 111:94–104
Acknowledgements
This work was supported by funding from the China Scholarship Council (201408410176 awarded to DY). We thank L. Blake, M. Durant, I. Fox, F. Hutchings, C. Jackson, M. Laws, J. McDonald for technical and field assistance. We are grateful for the contribution of the Biomes of Australian Soil Environments (BASE) consortium (https://data.bioplatforms.com/organization/pages/bpa-base/acknowledgements) in the generation of data used in this publication. The BASE project is supported by funding from Bioplatforms Australia through the Australian Government National Collaborative Research Infrastructure Strategy.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Eleonora Egidi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yan, D., Bissett, A., Gellie, N. et al. Soil bacterial community differences along a coastal restoration chronosequence. Plant Ecol 221, 795–811 (2020). https://doi.org/10.1007/s11258-019-00979-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11258-019-00979-0