
Abstract
Hyperlipidemia and oxidative stress are indispensable features of chronic kidney disease (CKD) that favor the development of atherogenic plaques and cardiovascular disease (CVD). A number of vasoactive mediators including proprotein convertase subtilisin–kexin type 9 (PCSK9), endothelin-1, nitric oxide, and angiotensin II have fundamental roles in the pathophysiology of atherosclerotic events; moreover, their levels are affected by dyslipidemia and oxidative stress due to renal dysfunction. Therefore, therapeutic measures aimed at correcting dyslipidemia and alleviating oxidative stress could potentially protect against CVD in CKD patients. In this review, we discuss the relation between dyslipidemia, oxidative stress, and vasoactive mediators as well as the available treatment options against these disturbances in CKD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperlipidemia and dyslipidemia are important risk factors for the development and progression of atherosclerosis [1] and hyperlipidemia could result in renal dysfunction and chronic kidney disease (CKD) in long term [2]. Moreover, lipid metabolism is often disturbed in CKD patients [2]. Increased incidence of cardiovascular events in CKD patients could be attributed to elevated production of reactive oxygen species (ROS), electrolyte imbalances, proteinuria, and inflammation [3].
It is now known that hypercholesterolemia induces oxidative stress in the endothelial cells [4], initiating the peroxidation of cell membranes and unsaturated free fatty acids [5]. Furthermore, free radicals alter the composition of lipoproteins, producing oxidized low-density lipoproteins (Ox-LDL) inside the blood vessels. Ox-LDL has multiple biological functions and favors increased expression of pro-inflammatory cytokines, adhesive molecules, and growth factors. Foam cell are formed by the accumulation of cholesterol inside the macrophages, favoring the development of atheromatous plaques which leads to glomerulosclerosis [5]. Atherogenic Ox-LDLs disturb the function of various vasoactive molecules such as nitric oxide (NO), endothelin 1 (ET1), angiotensin II, transforming growth factor beta (TGF-β), causing pathologic changes in the kidneys [6]. The impact of lipid accumulation in the kidneys could be observed both in glomeruli and in tubules, resulting in the development of glomerulosclerosis, fibrosis, tubular atrophy, and mesangial expansion [7].
Lipid abnormality in CKD
Dyslipidemia is generally observed in CKD patients. The pattern of abnormal lipid profile is different between various degrees and groups of CKD patients, depending on the renal function, underlying disease, and the severity of the proteinuria [8]. Elevations in apoprotein B (apoB), apoCIII, and apoE-containing lipoproteins, together with decreased levels of lipoproteins that contain apoAI, and apoAII like HDL-C are the most salient abnormalities in CKD patients [9]. HDL-C has anti-inflammatory and anti-oxidant properties, preventing the conversion of macrophages to foam cells by reverse transport of cholesterol to the liver [10]. Anti-oxidant actions of HDL-C are effected by the activity of paraoxonase 1 (PON1) and glutathione peroxidase (GPx); and both enzymes are reduced in CKD patients, leading to relentless oxidative changes in the proteins and lipids and formation of Ox-LDL [11]. In addition to the prevention of atherosclerosis, HDL-C is able to decrease the expression of anti-inflammatory molecules, including monocyte chemoattractant protein 1 (MCP1), E-selectin, and NF-kB expression, and thereby, reduces the infiltration of monocytes into the vessel walls [10]. Consequently, reduced levels of HDL-C contribute to the progression of atherosclerotic plaques in the subjects with renal disease. Albumin-associated transportation of free cholesterol is decreased due to hypoalbuminemia in patients with nephrotic syndrome or CKD. Urinary loss of LCAT impedes the conversion of HDL3 into HDL2 and the levels of apoA1 are reduced [12]. Engagement of HDL-C to the ATP-binding cassette transporter A-1 (ABCA1) is facilitated by apoA1; therefore, apoA1 deficiency favors the formation of foam cells and atherosclerotic plaques [13].
Total cholesterol levels might be higher than normal in CKD patients and its levels have inverse relation with mortality due to cardiovascular complications [10]. Highly atherogenic, small and dense LDL-C particles (sdLDL) are also formed in these patients [14]. By contrast, in ESRD patients undergoing dialysis hyperlipidemia is associated with improved survival that is termed “reverse epidemiology” or “risk factor paradox”. Short-term risk factors including malnutrition and inflammation are found to relate more to the poor outcomes in dialysis patients than conventional risk factors including obesity, hyperlipidemia, and hypertension [15].
CKD patients have higher levels of glycerolipids, free fatty acids, and glycerophospholipids, and as GFR decreases, the levels of saturated fatty acids are increased in these individuals [16]. Moreover, the levels of sulfatide, plasmenyl ethanol amine, ceramide and phosphatidyl choline are decreased in the LDL-C particles; alterations that increases the risk of CVD in CKD patients [17].
Lipoprotein lipase (LPL) has a unique role in transforming IDL-C to the LDL-C. In proteinuric patients, the enzymatic function of LPL is decreased due to increased ratio of apoCIII (LPL deactivator) to apoCII (LPL activator) [2]. Defects in LPL and hepatic lipase disrupt the clearance of IDL-C, VLDL-C, and chylomicrons, leading to the elevation of atherogenic IDL-C levels in serum [18]. Therefore, accurately evaluating lipid profile and fatty acids levels are important for determining the risk of atherosclerosis in CKD patients.
Oxidative stress in CKD
Hyperlipidemia-associated oxidative stress changes the proteins in the vascular walls, contributing to the development of atherosclerosis in CKD patients [19]. When encountered with ROS, the LDL-C particle turns into oxidized low-density lipoprotein (Ox-LDL) that itself favors the generation of ROS, intensifying oxidative stress [20]. Apart from promoting the motility and chemotactic activity of macrophages, Ox-LDL induces cholesterol accumulation inside macrophages, giving rise to the formation of foam cells and atherosclerotic plaques [21]. Additionally, Ox-LDL induces apoptosis in various cells including renal glomerular and tubular cells through increasing the expression of CHOP and the activity of C-Jun kinase-1 signaling pathway [22]. Moreover, Ox-LDL-induced inflammation in the kidneys causes glomerular sclerosis and renal fibrosis [23].
Nitric oxide
Nitric oxide (NO) is a vasodilator, generated from l-arginine by NO synthase (eNOS), and is secreted from renal macula densa and vascular cells. It modulates the function of glomerular and tubular function [24]. It seems that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase are the main sources of ROS generation in hypercholesterolemia and promote the formation of peroxynitrite [19, 25], that reduces the bioavailability of NO, a protective agent against endothelial atherosclerotic lesions [26]. This is a fundamental process that aids to the progression of endothelial dysfunction, increasing the risk of CVD in CKD patients [26]. Moreover, production of NO reduces in CKD patients [27] and these reductions in NO contribute to the pathologic changes in the kidneys including mesangial expansion, glomerulosclerosis, and renal fibrosis [28]. ADMA is an important biomarker for atherosclerosis that possibly favors endothelial dysfunction [26].
ADMA is a competitive inhibitor of eNOS and, therefore, reduces NO production [29]. Under pathologic states like dyslipidemia, and CKD, increased levels of ADMA lead to endothelial dysfunction [30]. ADMA levels might be increased due to reductions in the function of dimethyl arginine aminohydrolase (DDAH) which metabolizes ADMA [31]. Moreover, its levels might be elevated because of the increases in the function of protein-arginine methyl transferase (PRMT) that augments methylation of arginine residues and increases the production of ADMA [30].
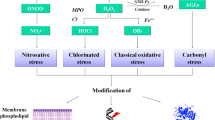
While ADMA levels are increased in CKD patients, it has no correlation with eGFR and Cr and only serves as the predictor of CVD [32, 33]. Zoccali et al. stated ADMA as a potent predictor factor for heart attack and death in CKD patients [34]. As a result, ADMA could be implemented as a novel biomarker in detection of CKD (Fig. 1).

Hyperlipidemia and Ox-LDL increase the production of vasoactive factors that induce renal injury by endothelial dysfunction
Angiotensin II
Angiotensin II (Ang II) has two receptors: Ang II receptor type 1 (AT1) and Ang II receptor type 2 (AT2). Many physiologic and pathophysiologic functions of Ang II are mediated via AT1 and the expression of AT1 is induced by hypercholesterolemia [35].
LDL-C and Ox-LDL increase Ang II levels and facilitate the progression of atherosclerosis [36]. Ang II induces the formation of superoxide anions by activating NADPH oxidase in the vascular smooth muscle cells (VSMC) and mesangial cells; as a result, the production of pro-fibrotic mediator TGF-β is increased, activating the intracellular Smad via ERK [37]. In addition to increasing the expression of plasminogen activator inhibitor-1 (PAI-1), platelet-derived growth factor (PDGF), vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1(ICAM-1) in the vascular walls, Ang II induces the expression of extracellular matrix proteins including fibronectin and type IV collagen, facilitating glomerular fibrosis in the kidneys [37].
Bianche et al. showed that the inhibition of Ang II could lower the progression of CKD and mitigate hypercholesterolemia and proteinuria [38].
Angiotensin-converting enzyme (ACE) inhibitors not only decelerate the progression of CKD but also alleviate hypercholesterolemia. Furthermore, these agents are effective therapeutic measures in reducing the risk of CVD [39].
Endothelin 1
Endothelin 1 is a vasoconstrictor synthesized in the glomerular and tubular cells [40]. In the kidneys, ET1 regulates local blood flow, podocyte function, and mesangial constriction [41]. Under hypercholesterolemic states, when the levels of LDL-C are increased and the levels of HDL-C are reduced, ET1 levels have demonstrated to be elevated which participate in the development of atherosclerosis [42]. Ox-LDL is also able to increase the production of ET1 [43]. ET1 acts as a chemoattractant molecule for macrophages and monocytes; it, moreover, functions as a mitogenic factor for the smooth muscle cells [41, 42]. In CKD patients with hypercholesterolemia, circulating levels of ET1 are increased [44]. ET1 has a major role in the development of proteinuria and renal fibrosis in CKD patients by disturbing the integrity of podocytes [45]. Furthermore, ET1 increases the production of other vasoconstrictors and growth factors such as angiotensin II [46]. Hence, ET1 blocked improves vascular endothelial and renal function in CKD patients [47].
Sterol regulatory element-binding proteins
Sterol regulatory element-binding proteins (SREBPs) are transcription factors that belong to the basic helix–loop–helix–leucine zipper family, with crucial roles in lipid homeostasis, acting as the main regulators of cholesterol and fatty acid synthesis in the liver [48]. SREBP-1a and -1c activate the genes involved in the synthesis of fatty acids including acetyl-CoA carboxylase and fatty acid synthase; by contrast, SREBPs-2 is involved in the induction of the expression of the genes that participate in cholesterol synthesis including HMG-CoA reductase, dihydroxymethyl glutaryl-CoA reductase, farnesyl diphosphate synthase, and squalene synthase [49]. Under high glucose levels of glucose in streptozotocin-induced diabetic mice, the expression of SREBPs-1a and -1c are increased in the renal cortex, elevating the expression of fatty acid synthase and acetyl-CoA carboxylase which culminated in the accumulation of triglycerides in the renal cells [50]. Ang II is also able to activate SREBP-1, augmenting extracellular matrix synthesis through upregulating TGF-β [51]. Elevations in the SREBP-1 and triglyceride content of the kidneys increase the expression of fibrotic factors like TGF-β and vascular endothelial growth factor (VEGF) [51]. TGF-β increases the production of extracellular matrix through activating ERK and Smad in the mesangial cells, aggravating renal fibrosis [52].
Proprotein convertase subtilisin–kexin type 9
Proprotein convertase subtilisin-kexin type 9 (PCSK9) is a serine protease produced in the kidneys [53]. PCSK9 binds to low-density lipoprotein receptor (LDL-R, aiding to the destruction of LDL-R inside lysosomes; and therefore, by reducing LDL-C catabolism elevates LDL-C levels [53]. PCSK9, moreover, increases the expression of oxidized low-density lipoprotein receptor-1 (LOX-1) in VSMCs [53]. PCSK9-associated reductions in low-density lipoprotein receptor-related protein 1(LRP-1) interferes with LPa catabolism, elevating its serum levels and the risk of atherosclerosis [54]. PCSK9 levels are increased in CKD patients that contribute to the development of dyslipidemia [55]. Moreover, PCSK9 serum levels are elevated in patients with hypercholesterolemic nephrotic syndrome [56]. No relation, however, has been observed between eGFR and PCSK9 levels in CKD patients [57]. Treatment with statins in CKD patients activates SREBP-2 that in addition to increasing the expression of PCSK9, elevates the expression of LDL-R, improving the clearance of LDL-C from serum [58]. Monoclonal antibodies targeted against PCSK9 have been found to be effective in reducing LDL-C levels in patients with hypercholesterolemia [59]. The outcome of ODYSSE1/COMBO clinical trial denoted that alirocumab significantly decreased LDL-C levels in serum compared with placebo, as well as ezetimibe plus statin. However, its efficacy in reducing serum triglyceride levels was controversial and further studies need to be conducted to assess its efficacy and safety in CKD patients [60]. Thereby, PCSK9 can use as a new biomarker for lipid metabolism and is a new medicine target for hypercholesterolemia. Inhibition of PCSK9 not only decreases LDL-C levels but also reduces the risk of cardiovascular diseases [59].
Treatment of dyslipidemia in CKD
Currently, statins are the most common agents used in the treatment of hyperlipidemia in patients with CKD. Statins inhibit HMG-CoA reductase and decrease hepatic production of cholesterol, reducing cholesterol levels by 20–50% [61]. Moreover, these agents possess anti-inflammatory properties and decrease the expression of TNF-α, IL-6, TGF-β, VCAM, and fibronectin in the renal mesangial cells. Apart from increasing NO synthesis, statins prevent LDL-C oxidation by alleviating oxidative stress. This anti-oxidant property of statins improves endothelial function [62]. Statins can ameliorate the outcomes of the CAD and CKD patients and reduce the rate of cardiovascular disease mortality [63]. The findings regarding statin effects on renal function are disparate. Some studies have shown that statin therapy can be effective in improving renal function and increasing eGFR in patients [64, 65]; however, another study has demonstrated that statin therapy has no beneficial effect on renal function [66]. Accordingly, more in-depth studies are required to clarify the exact benefit of statins in CKD patients (Table 1).
Most statins are mainly metabolized in the liver; therefore, dose adjustment in early CKD is typically not needed when eGFR is more than 30 ml/min. However, the maximum dose of these agents need to be restricted in more advanced CKD with eGFR less than 30 ml/min. Atorvastatin, fluvastatin, lovastatin, and simvastatin are metabolized almost exclusively by the hepatic routes; consequently, minimal dose restrictions would suffice in CKD patients. By contrast, pitavastatin, pravastatin, and rosuvastatin are metabolized both in the liver and in the kidney, making dose restrictions imperative in CKD patients [67].
Nicotinic acid derivatives including niacin reduced triglyceride synthesis by inhibiting diacylglycerol transferase and hormone-sensitive lipase. They, moreover, inhibit VLDL-C secretion and thus, reduce LDL-C production. Additionally, nicotinic acid is able to decrease Apo A1 clearance and increase HDL-C synthesis; overall, these agents reduce cardiovascular events [68].
Fibric acid derivatives such as fenofibrate and gemfibrozil are agonists of transcription factor peroxisome proliferator-activated receptor alpha (PPAR-alpha). Despite beneficial effects of fibrates and niacin in treating hyperlipidemia, their adverse effects in patients with renal dysfunction could be unfavorable [68]. Therefore, further studies are needed for their effects in patients with CKD and their side effects.
Literature is scant regarding the use of non-statin lipid-lowering agents in CKD patients. Niacin has equivalent lipid-lowering effect in CKD patients as compared to non-CKD individuals; moreover, its use has gained interest in CKD patients for its phosphorus-lowering properties [69]. By contrast, fibrates are metabolized in the kidneys and thus generally contraindicated in patients with CKD [67].
Bile acids including colesevelam, colestipol, cholestyramine and colestimide are involved in the cholesterol catabolism and have as important role in the absorption of the fats [61]. The adverse effects of these drugs in the gastrointestinal tract, however, limit their clinical utility [70].
Ezetimibe is the first member of the cholesterol absorption inhibitors that selectively prevent intestinal absorption of cholesterol and phytoesterols; therefore, reduce LDL-C levels without affecting lipid-soluble vitamins. Ezetimibe is used for the treatment of hyperlipidemia in patients who do not tolerate statin therapy [71].
Since oxidative stress has a substantial role in the progression of CKD, anti-oxidants could be regarded as novel therapeutic agents in these group of patients. Several lines of evidence have shown that anti-oxidants and vitamin E improve lipid profile in patients undergoing hemodialysis and have protective effects on cardiovascular complications [72, 73]; however, their effects in long term should be well investigated. CKD patients have high risks of developing cardiovascular events even in the early stages [74]. Therefore, identification and treatment of dyslipidemia prior to the development of ESRD is inevitable (Fig. 2).

Renal injury and fibrosis. Under hyperlipidemic states, the production of Ang II is increased with the formation of Ox-LDL particles, activating the inflammatory factors such as tumor necrosis factor-alpha and NF-kB. Moreover, the production of fibrotic factors such as VCAM, ICAM, PDGF, and TGF-B are increased that culminate in the accumulation of type IV collagen and fibronectin in the extracellular spaces and renal fibrosis
Conclusion
Cardiovascular complications are common causes of mortality in CKD patients. Decreased HDL-C levels in patients with CKD are an important contributing factor for the development of atherosclerosis. Moreover, hyperlipidemia-associated oxidative stress and inflammation have detrimental effects in various organs including the kidneys. Ox-LDL disturbs endothelial function and increases the risk of atherosclerosis. In addition, oxidative stress reduces NO synthesis and simultaneously promotes Ang II production. Under hyperlipidemic states, increased production of Ang II might aggravate renal injury. ET-1 levels are elevated due to hyperlipidemia in CKD patients, accelerating the progression of proteinuria and renal dysfunction. Evidences still denote that CVD is the main cause of mortality in CKD patients and collectively identification and management of lipid abnormalities in CKD patients is of vital value.
References
Argani H, Ghorbanihaghjo A, Vatankhahan H, Rashtchizadeh N, Raeisi S, Ilghami H (2016) The effect of red grape seed extract on serum paraoxonase activity in patients with mild to moderate hyperlipidemia. Sao Paulo Med J 134(3):234–239
Nitta K (2012) Clinical assessment and management of dyslipidemia in patients with chronic kidney disease. Clin Exp Nephrol 16(4):522–529
Kanbay M, Afsar B, Siriopol D, Unal HU, Karaman M, Saglam M, Gezer M, Taş A, Eyileten T, Guler AK (2016) Endostatin in chronic kidney disease: associations with inflammation, vascular abnormalities, cardiovascular events and survival. Eur J Int Med 33:81–87
Razavi S-M, Gholamin S, Eskandari A, Mohsenian N, Ghorbanihaghjo A, Delazar A, Rashtchizadeh N, Keshtkar-Jahromi M, Argani H (2013) Red grape seed extract improves lipid profiles and decreases oxidized low-density lipoprotein in patients with mild hyperlipidemia. J Med Food 16(3):255–258
Lee HS, Song CY (2009) Oxidized low-density lipoprotein and oxidative stress in the development of glomerulosclerosis. Am J Nephrol 29(1):62–70
Trevisan R, Dodesini AR, Lepore G (2006) Lipids and renal disease. J Am Soc Nephrol 17(4 suppl 2):S145–S147
Rüster C, Wolf G (2006) Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol 17(11):2985–2991
Massy ZA, De Zeeuw D (2013) LDL cholesterol in CKD—to treat or not to treat? Kidney Int 84(3):451–456
Meenakshi Sundaram SP, Nagarajan S, Manjula Devi AJ (2014) Chronic kidney disease—effect of oxidative stress. Chin J Biol 2014:216210
Vaziri ND (2014) Role of dyslipidemia in impairment of energy metabolism, oxidative stress, inflammation and cardiovascular disease in chronic kidney disease. Clin Exp Nephrol 18(2):265–268
Mohammed CJ, Xie Y, Brewster PS, Ghosh S, Dube P, Sarsour T, Kleinhenz AL, Crawford EL, Malhotra D, James RW (2019) Circulating lactonase activity but not protein level of PON-1 predicts adverse outcomes in subjects with chronic kidney disease. J Clin Med 8(7):1034
Vaziri ND (2016) HDL abnormalities in nephrotic syndrome and chronic kidney disease. Nat Rev Nephrol 12(1):37
Moradi H, Vaziri ND, Kashyap ML, Said HM, Kalantar-Zadeh K (2013) Role of HDL dysfunction in end-stage renal disease: a double-edged sword. J Ren Nutr 23(3):203–206
Kwan BC, Kronenberg F, Beddhu S, Cheung AK (2007) Lipoprotein metabolism and lipid management in chronic kidney disease. J Am Soc Nephrol 18(4):1246–1261
Kalantar-Zadeh K, Block G, Humphreys MH, Kopple JD (2003) Reverse epidemiology of cardiovascular risk factors in maintenance dialysis patients. Kidney Int 63(3):793–808
Chen H, Chen L, Liu D, Chen D-Q, Vaziri ND, Yu X-Y, Zhang L, Su W, Bai X, Zhao Y-Y (2017) Combined clinical phenotype and lipidomic analysis reveals the impact of chronic kidney disease on lipid metabolism. J Proteome Res 16(4):1566–1578
Reis A, Rudnitskaya A, Chariyavilaskul P, Dhaun N, Melville V, Goddard J, Webb DJ, Pitt AR, Spickett CM (2015) Top-down lipidomics of low density lipoprotein reveal altered lipid profiles in advanced chronic kidney disease. J Lipid Res 56(2):413–422
Vaziri ND (2016) Disorders of lipid metabolism in nephrotic syndrome: mechanisms and consequences. Kidney Int 90(1):41–52
Csonka C, Sárközy M, Pipicz M, Dux L, Csont T (2016) Modulation of hypercholesterolemia-induced oxidative/nitrative stress in the heart. Oxid Med Cell Longev 2016:3863726
Vona R, Gambardella L, Cittadini C, Straface E, Pietraforte D (2019) Biomarkers of oxidative stress in metabolic syndrome and associated diseases. Oxid Med Cell Longev 2019:8267234
Kachhawa K, Agrawal D, Rath B, Kumar S (2017) Association of lipid abnormalities and oxidative stress with diabetic nephropathy. J Integr Nephrol Androl 4(1):3
Yu S, Zhou X, Hou B, Tang B, Li J, Zhang B (2016) Protective effect of rosuvastatin treatment by regulating oxidized low-density lipoprotein expression in a rat model of liver fibrosis. Biomed Rep 5(3):311–316
Kose E, An T, Kikkawa A, Matsumoto Y, Hayashi H (2014) Effects on serum uric acid by difference of the renal protective effects with atorvastatin and rosuvastatin in chronic kidney disease patients. Biol Pharm Bull 37(2):226–231
Baylis C (2006) Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Rev Nephrol 2(4):209
Dorighello GG, Paim BA, Kiihl SF, Ferreira MS, Catharino RR, Vercesi AE, Oliveira HC (2016) Correlation between mitochondrial reactive oxygen and severity of atherosclerosis. Oxid Med Cell Longev 2016:7843685
Duni A, Liakopoulos V, Rapsomanikis K-P, Dounousi E (2017) Chronic kidney disease and disproportionally increased cardiovascular damage: does oxidative stress explain the burden? Oxid Med Cell Longev 2017:9036450
Reddy Y, Kiranmayi V, Bitla A, Krishna G, Rao PS, Sivakumar V (2015) Nitric oxide status in patients with chronic kidney disease. Indian J Nephrol 25(5):287
Girardi JM, Farias RE, Ferreira AP, Raposo NRB (2011) Rosuvastatin prevents proteinuria and renal inflammation in nitric oxide-deficient rats. Clinics 66(8):1457–1462
Rysz J, Gluba-Brzózka A, Franczyk B, Jabłonowski Z, Ciałkowska-Rysz A (2017) Novel biomarkers in the diagnosis of chronic kidney disease and the prediction of its outcome. Int J Mol Sci 18(8):1702
Cvetković T, Pavlović R, Đorđević V, Stojanović I, Veličković-Radovanović R, Ignjatović A, Stefanović N, Živanović S, Đorđević V (2012) Dimethylarginine–biomarkers in progression of kidney disease/Dimetilarginini–biomarkeri u progresiji bubrežnih oboljenja. J Med Biochem 31(4):301–308
Ignjatović A, Cvetković T, Pavlović R, Đorđević V, Milošević Z, Đorđević V, Pavlović D, Stojanović I, Živanović S (2013) ADMA and C-reactive protein as mortality predictors in dialysis patients. Open Med 8(3):346–353
Schmidt RJ, Baylis C (2000) Total nitric oxide production is low in patients with chronic renal disease. Kidney Int 58(3):1261–1266
Kielstein JT, Böger RH, Bode-Böger SM, Frölich JC, Haller H, Ritz E, Fliser D (2002) Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J Am Soc Nephrol 13(1):170–176
Zoccali C, Bode-Böger SM, Mallamaci F, Benedetto FA, Tripepi G, Malatino LS, Cataliotti A, Bellanuova I, Fermo I, Frölich JC (2001) Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 358(9299):2113–2117
Andreadou I, Iliodromitis EK, Lazou A, Görbe A, Giricz Z, Schulz R, Ferdinandy P (2017) Effect of hypercholesterolaemia on myocardial function, ischaemia–reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br J Pharmacol 174(12):1555–1569
Pizoń T, Rajzer M, Wojciechowska W, Wach-Pizoń M, Drożdż T, Wróbel K, Gruszka K, Rojek M, Kameczura T, Jurczyszyn A, Kąkol J, Czarnecka D (2018) The relationship between plasma renin activity and serum lipid profiles in patients with primary arterial hypertension. J Renin Angiotensin Aldosterone Syst. https://doi.org/10.1177/1470320318810022
Nogueira A, Pires MJ, Oliveira PA (2017) Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo 31(1):1–22
Bianchi S, Bigazzi R, Caiazza A, Campese VM (2003) A controlled, prospective study of the effects of atorvastatin on proteinuria and progression of kidney disease. Am J Kidney Dis 41(3):565–570
Ougaard M, Jensen H, Thuen I, Petersen E, Kvist P (2018) Inhibitors of the renin-angiotensin system ameliorates clinical and pathological aspects of experimentally induced nephrotoxic serum nephritis. Ren Fail 40(1):640–648
De Miguel C, Speed JS, Kasztan M, Gohar EY, Pollock DM (2016) Endothelin-1 and the kidney: new perspectives and recent findings. Curr Opin Nephrol Hypertens 25(1):35
Czopek A, Moorhouse R, Webb DJ, Dhaun N (2015) Therapeutic potential of endothelin receptor antagonism in kidney disease. Am J Physiol Regul Integr Comp Physiol 310(5):R388–R397
Al-Dujaili ANG, Al-Shemeri MK (2016) Effect of silver nanoparticles and rosuvastatin on endothelin and obestatin in rats induced by high fat-diet. Res J Pharm Biol Chem Sci 7(3):1022–1030
Sudano I, Spieker LE, Hermann F, Flammer A, Corti R, Noll G, Lüscher TF (2006) Protection of endothelial function: targets for nutritional and pharmacological interventions. J Cardiovasc Pharmacol 47:S136–S150
Larivière R, Lebel M (2003) Endothelin-1 in chronic renal failure and hypertension. Can J Physiol Pharmacol 81(6):607–621
Vaneckova I, Hojna S, Kadlecova M, Vernerova Z, Kopkan L, Cervenka L, Zicha J (2018) Renoprotective effects of ETA receptor antagonists therapy in experimental non-diabetic chronic kidney disease: is there still hope for the future? Physiol Res 67:S55–S67
Kohan DE, Barton M (2014) Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86(5):896–904
Schneider MP, Mann JF (2014) Endothelin antagonism for patients with chronic kidney disease: still a hope for the future. Nephrol Dial Transplant 29(suppl_1):i69–i73
Moslehi A, Hamidi-zad Z (2018) Role of SREBPs in liver diseases: a mini-review. J Clin Transl Hepatol 6(3):332
DeBose-Boyd RA, Ye J (2018) SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci 43(5):358–368
Sun L, Halaihel N, Zhang W, Rogers T, Levi M (2002) Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem 277(21):18919–18927
Wang TN, Chen X, Li R, Gao B, Mohammed-Ali Z, Lu C, Yum V, Dickhout JG, Krepinsky JC (2015) SREBP-1 mediates angiotensin II-induced TGF-β1 upregulation and glomerular fibrosis. J Am Soc Nephrol 26(8):1839–1854
Mustafa M, Wang TN, Chen X, Gao B, Krepinsky JC (2016) SREBP inhibition ameliorates renal injury after unilateral ureteral obstruction. Am J Physiol Ren Physiol 311(3):F614–F625
Bandyopadhyay D, Ashish K, Hajra A, Qureshi A, Ghosh RK (2018) Cardiovascular outcomes of PCSK9 inhibitors: with special emphasis on its effect beyond LDL-cholesterol lowering. J Lipids 2018:3179201
Canuel M, Sun X, Asselin M-C, Paramithiotis E, Prat A, Seidah NG (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8(5):e64145
Kwakernaak AJ, Lambert G, Slagman MC, Waanders F, Laverman GD, Petrides F, Dikkeschei BD, Navis G, Dullaart RP (2013) Proprotein convertase subtilisin–kexin type 9 is elevated in proteinuric subjects: relationship with lipoprotein response to antiproteinuric treatment. Atherosclerosis 226(2):459–465
Haas ME, Levenson AE, Sun X, Liao W-H, Rutkowski JM, Network NSS, de Ferranti SD, Schumacher VA, Scherer PE, Salant DJ (2016) The role of proprotein convertase subtilisin/kexin type 9 in nephrotic syndrome-associated hypercholesterolemia. Circulation 134(1):61–72
Rogacev KS, Heine GH, Silbernagel G, Kleber ME, Seiler S, Emrich I, Lennartz S, Werner C, Zawada AM, Fliser D (2016) PCSK9 plasma concentrations are independent of GFR and do not predict cardiovascular events in patients with decreased GFR. PLoS One 11(1):e0146920
Shrestha P, van de Sluis B, Dullaart RP, van den Born J (2019) Novel aspects of PCSK9 and lipoprotein receptors in renal disease-related dyslipidemia. Cell Signal 55:53–64
Toth PP, Dwyer JP, Cannon CP, Colhoun HM, Rader DJ, Upadhyay A, Louie MJ, Koren A, Letierce A, Mandel J (2018) Efficacy and safety of lipid lowering by alirocumab in chronic kidney disease. Kidney Int 93(6):1397–1408
Zheng-Lin B, Ortiz A (2018) Lipid management in chronic kidney disease: systematic review of PCSK9 targeting. Drugs 78(2):215–229
Reiner Ž, Catapano AL, De Backer G, Graham I, Taskinen M-R, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P (2011) ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 32(14):1769–1818
Sironi L, Gianazza E, Gelosa P, Guerrini U, Nobili E, Gianella A, Cremonesi B, Paoletti R, Tremoli E (2005) Rosuvastatin, but not simvastatin, provides end-organ protection in stroke-prone rats by antiinflammatory effects. Arterioscler Thromb Vasc Biol 25(3):598–603
Shen H, Chen X, Lu J, Yang H, Xu Y, Zhu A, Zhang X, Ye F, Gu Y (2018) Effects of statin therapy on chronic kidney disease patients with coronary artery disease. Lipids Health Dis 17(1):84
Tonelli M, Moyé L, Sacks FM, Cole T, Curhan GC (2003) Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 14(6):1605–1613
Wu Y, Wang Y, An C, Dong Z, Liu H, Zhang Y, Zhang M, An F (2012) Effects of rosuvastatin and atorvastatin on renal function. Circ J 76(5):1259–1266
Baigent C (2005) Cholesterol Treatment Trialists’ (CTT) Collaborators: efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 366:1267–1278
Mikolasevic I, Žutelija M, Mavrinac V, Orlic L (2017) Dyslipidemia in patients with chronic kidney disease: etiology and management. Int J Nephrol Renovasc Dis 10:35–45
Agrawal S, Zaritsky JJ, Fornoni A, Smoyer WE (2018) Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat Rev Nephrol 14(1):57
Park CW (2013) Niacin in patients with chronic kidney disease: is it effective and safe? Kidney Res Clin Pract 32(1):1
Omran J, Al-Dadah A, Dellsperger KC (2013) Dyslipidemia in patients with chronic and end-stage kidney disease. Cardiorenal Med 3(3):165–177
Shattat GF (2015) A review article on hyperlipidemia: types, treatments and new drug targets. Biomed Pharmacol J 7(1):399–409
Boaz M, Smetana S, Weinstein T, Matas Z, Gafter U, Iaina A, Knecht A, Weissgarten Y, Brunner D, Fainaru M (2000) Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet 356(9237):1213–1218
Islam KN, O’Byrne D, Devaraj S, Palmer B, Grundy SM, Jialal I (2000) Alpha-tocopherol supplementation decreases the oxidative susceptibility of LDL in renal failure patients on dialysis therapy. Atherosclerosis 150(1):217–224
Obialo C, Ofili E, Norris K (2018) Statins and cardiovascular disease outcomes in chronic kidney disease: reaffirmation vs. repudiation. Int J Environ Res Public Health 15(12):2733
Acknowledgements
This work was supported by the Biotechnology Research Center, Tabriz University of Medical Sciences (Grant number: 95.5-9.16).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jabarpour, M., Rashtchizadeh, N., Argani, H. et al. The impact of dyslipidemia and oxidative stress on vasoactive mediators in patients with renal dysfunction. Int Urol Nephrol 51, 2235–2242 (2019). https://doi.org/10.1007/s11255-019-02319-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-019-02319-7