
Abstract
Hyponatremia has complex pathophysiology, is frequent and has potentially severe clinical manifestations, and its treatment is associated with high risks. Hyponatremia can be hypertonic, isotonic or hypotonic. Hypotonic hyponatremia has multiple etiologies, but only two general mechanisms of development, defective water excretion, usually because of elevated serum vasopressin levels, or excessive fluid intake. The acute treatment of symptomatic hypotonic hyponatremia requires understanding of its targets and risks and requires continuous monitoring of the patient’s clinical status and relevant serum biochemical values. The principles of fluid restriction, which is the mainstay of management of all types of hypotonic hyponatremia, should be clearly understood and followed. Treatment methods specific to various categories of hyponatremia are available. The indications and risks of these treatments should also be well understood. Rapid correction of chronic hypotonic hyponatremia may lead to osmotic demyelination syndrome, which has severe clinical manifestations, and may lead to permanent neurological disability or death. Prevention of this syndrome should be a prime concern of the treatment of hypotonic hyponatremia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyponatremia is defined as a serum sodium concentration ([Na]S) less than 135 mEq/L. It is the most common electrolyte disorder with a prevalence of 20–30 % in hospitalized patients [1] and 7.7 % in outpatients with electrolyte disorders [2]. Hyponatremia is associated with severe illnesses and is the cause of symptoms ranging from mild and difficult to detect to life threatening [3, 4]. Management and prevention of hyponatremia require assessment of its severity and elucidation of its pathogenetic mechanism [3, 4].
The first step in understanding hyponatremia is clarification of the information provided by [Na]S. The information sought in [Na]S and serum osmolality is whether the serum is hypertonic, isotonic or hypotonic [4]. Tonicity, or effective osmolality, of a solution is its property to cause shrinking or swelling of cells suspended in it through water loss or gain, respectively. The water transfers are driven by differences in osmotic pressure between the intracellular and extracellular compartments. Hypertonic solutions cause water exit from cells; hypotonic solutions cause water entry into cells; and isotonic solutions do not cause net water movement into or out of the cells.
Disturbances in serum tonicity may cause severe neurological manifestations. Information on serum tonicity provided by [Na]S is simple for hypernatremia, but complex for hyponatremia. Without exception, hypernatremia denotes a hypertonic state. Hyponatremia is an accurate marker of hypotonicity in the absence of excesses of extracellular solutes other than sodium salts. When there is gain of such solutes, hyponatremia is associated with hypertonicity. Finally, hyponatremia is associated with isotonicity when there is a decrease in the aqueous fraction of the serum or an extracellular gain of an isotonic solution with a solute that is not a sodium salt. Differentiating between the three states of tonicity is critical for the treatment of patients presenting with hyponatremia. Recent developments have increased the number and complexity of therapeutic options for hypotonic hyponatremia. The purpose of this review is to present the diagnosis and management of all categories of hyponatremia with emphasis on treatment of hypotonic hyponatremia and prevention of treatment complications.
Hypertonic hyponatremia
Hypertonic hyponatremia occurs when there is an excess of extracellular solutes, other than sodium salts, encountering difficulty in entering into cells. Gain in these solutes causes osmotic shift of intracellular water into the extracellular compartment. The fluid entering the extracellular compartment, which is poor in sodium, dilutes the extracellular sodium and leads to hyponatremia.
Serum osmolality is elevated, above 295 mOsm/kg, in hypertonic hyponatremia. The clinical manifestations of hypertonicity in this state are usually severe at effective serum osmolality levels above 320 mOsm/kg. Hyperglycemia is the prototype of hypertonic hyponatremia. Mannitol, sucrose, maltose and low molecular weight dextrans are exogenous solutes that can cause hypertonic hyponatremia. Gain in solutes that cross cell membranes and are distributed in total body water, e.g., urea or various alcohols, causes an increase in serum osmolality, but will not cause hypertonicity and, therefore, will not affect [Na]S [4]. In hyperglycemia, serum tonicity (effective osmolality) is estimated as:
where serum glucose concentration ([Glucose]S) is in mg/dL.
In hyperglycemia, [Na]S and serum tonicity receive a second influence from osmotic diuresis, which leads to water loss in excess of the loss of electrolytes and causes a rise in [Na]S, whereas, as noted, osmotic water exit from the cells as a consequence of extracellular glucose gain causes a decrease in [Na]S. Depending on which influence is larger, [Na]S at presentation with hyperglycemia can be low, normal or high [5].
Replacement of the excessive loss of water caused by severe hyperglycemia requires large volumes of hypotonic solutions. Formula 1, which represents serum tonicity during hyperglycemia, is not a suitable guide for the tonicity of the replacement solutions, because glucose metabolism after insulin administration results in loss of body solute and decrease in tonicity in the absence of any further loss or gain of fluids [6, 7]. The estimate of serum tonicity that is used as a guide of the tonicity of the replacements solutions is the corrected [Na]S. [Na]S corrected to a [Glucose]S of 100 mg/dL is calculated as follows [8, 9]:
The tonicity value corresponding to the corrected [Na]S is lower than the value computed from Formula 1 [7]. For example, if [Na]S is 125.6 mEq/L and [Glucose]S is 1,000 mg/dL, serum tonicity from Formula 1 will be 306.8 mOsm/L. Based on this calculation, the replacement solution should be hypotonic. Formula 2 calculates that after correction of hyperglycemia [Na]S will be 140 mEq/L, indicating the need for isotonic fluid replacement, which should be used initially in this case. Formula 2 does not account for fluid and solute losses during treatment. Because of the unpredictable magnitude of these losses, frequent monitoring of clinical signs, urinary losses of water, sodium and potassium, and serum chemistries including [Na]S, [Glucose]S, and serum potassium concentration is critical for directing changes in the volume and composition of the replacement solutions [7].
Isotonic hyponatremia
Isotonic hyponatremia is associated with the absence of clinical manifestations of dystonicity and normal serum osmolality levels. Isotonic hyponatremia can be caused by pseudohyponatremia or nonconductive irrigant solutions.
Pseudohyponatremia
Pseudohyponatremia is a laboratory artifact [10]. The serum has aqueous and nonaqueous fractions. The nonaqueous fraction is composed mainly of proteins and lipids. Sodium is located in the aqueous fraction only. Under normal circumstances, the aqueous fraction represents 93 % of the total serum volume. Nowadays, [Na]S is measured using the principle of ion-selective electrode (ISE) where an electrode is immersed into blood or serum and the sodium activity is measured as a function of the electrical potential difference across the electrode. ISE does not measure sodium concentration per se but sodium activity. However, for practical purposes, activity and concentration are considered to be equivalent. Two ISE methods are used in clinical laboratories: direct ISE and indirect ISE.
Direct ISE methods (e.g., arterial blood gas machine) measure sodium activity in plasma water of undiluted specimens. Sodium in plasma water normally has an activity equivalent to a concentration of 150 mEq/L. This value is independent of the volume of the nonaqueous fraction. Most instruments using direct ISE have built-in conversion algorithms that give results in total serum: A sodium concentration of 150 mEq/L in serum water corresponds to a [Na]S of 139.5 mEq/L.
Indirect ISE methods measure sodium concentration in total serum. They require a fixed volume of diluent to be added to the serum sample before measurement occurs (≈30-fold dilution). Then, [Na]S is calculated based on the assumption that serum is 93 % water and 7 % proteins and lipids. Most laboratories still use the indirect ISE method to measure [Na]S on a routine basis. When the serum contains unusually high levels of proteins (e.g., multiple myeloma, use of intravenous immunoglobulins) or lipids (e.g., hypertriglyceridemia, lipoprotein X in cholestasis), the volume of the nonaqueous fraction increases and displaces the water fraction so that serum contains less water and therefore less sodium per unit volume. Under these pathological circumstances, use of the indirect ISE method leads to a predictable error: Dilution by a fixed volume of diluent will cause a falsely low [Na]S measurement. This error does not occur with the direct ISE methods.
Nonconductive irrigant solutions
A variety of transurethral (e.g., transurethral prostatectomy or TURP), hysteroscopic and other percutaneous procedures utilize large volumes of irrigating solutions in an effort to dilate the operating field and to wash away debris and blood. Nonconductive irrigant solutions were preferred because they are electrolyte free and do not produce sparks when monopolar cauteries are used. The nonconductive solutions are isotonic (mannitol) or slightly hypotonic (sorbitol and glycine) compared with normal plasma. Glycine is the most widely used solution.
Irrigant solutions are absorbed directly into the vascular system when a vein is severed during the surgical procedure. Osmolar gap is the difference between serum osmolality directly measured by a colligative property, usually depression of the serum freezing point, and serum osmolarity that is calculated as the sum of the osmotic contributions of sodium salts, glucose and urea in the serum. The serum osmolarity [Osm]S is calculated by the following formula:
where serum urea ([Urea]S) is in mg/dL. When urea is measured not as the total molecule, but as blood urea nitrogen ([BUN]S), the term [BUN]S/2.8 is substituted for [Urea]S/6 in Formula 3.
Initially, the solute of nonconductive irrigant solutions entering the blood compartment is distributed in the extracellular compartment increasing the serum osmolar gap. Water that was absorbed with the solute remains extracellular as well and dilutes the extracellular sodium causing hyponatremia. Depending on the osmolality of the solution, plasma osmolality remains unchanged (mannitol) or falls slightly (sorbitol and glycine) initially, hence the term isotonic hyponatremia. Shortly after surgery, [Na]S starts to increase. The mechanisms of this increase in [Na]S differ: [Na]S increases as mannitol, an osmotic diuretic, is excreted in the urine with water. After sucrose use, [Na]S increases as sucrose, also an osmotic diuretic, is excreted in the urine with water. In addition, some of the absorbed sucrose is metabolized to carbon dioxide and water leading to decrease in plasma osmolality. A part of the post-absorptive increase in [Na]S in this case is caused by osmotic transfer of water from the extracellular compartment into the cells. Glycine diffuses slowly into the intracellular compartment carrying water with it and improving hyponatremia. Glycine is then metabolized to urea, another solute that is measured by the osmometer; therefore, the plasma osmolality remains stable.
A syndrome characterized by cardiovascular (i.e., bradycardia, hypotension and chest pain) and neurological manifestations (e.g., confusion, anxiety, paresthesias, visual disturbances) developed in a number of patients after transurethral prostatectomy (TURP) or other operations using glycine solutions as irrigant. This syndrome is known as the “TURP syndrome.” It is unlikely that the neurological manifestations of this syndrome are caused by hyponatremia alone since there is only a modest reduction in serum osmolality. What is more, brain edema is only minimal in these patients. It is thought now that these symptoms are due to the accumulation of toxic metabolites of glycine such as ammonia [11].
A postoperative syndrome of inappropriate vasopressin (ADH) secretion (SIADH) and, in the case of glycine solutions, direct ADH secretion stimulated by glycine may prolong water retention in these patients and therefore the duration of hyponatremia. Isotonic hyponatremia is now less commonly seen in patients undergoing urological and gynecological procedures with the development of the bipolar electrocautery, which permits the use of normal saline as irrigant.
Hypotonic hyponatremia
Hypotonic (true) hyponatremia is typically associated with serum osmolality values less than 275 mOsm/kg. However, hypotonic hyponatremia in the presence of high serum concentrations of solutes with total body water distribution (e.g., urea, ethyl alcohol) may be associated with normal or even elevated values of serum osmolality [4]. The clinical manifestations of hypotonic hyponatremia are, by and large, the results of increased intracranial pressure caused by brain cell swelling [12].
General mechanisms of hypotonic hyponatremia
In a seminal publication, Edelman and collaborators demonstrated that in the absence of solutes with extracellular distribution, other than sodium salts, the only determinants of [Na]S are as follows: total exchangeable sodium (Nae), total exchangeable potassium (Ke) and total body water (TBW) [13]. This relationship is represented in the Edelman equation, which is considered the master dysnatremia equation. A simplified version of this equation is shown below [14]:
Formula 4 should be used as the basis of analyses of dysnatremia. Total body sodium content can be measured by isotope dilution using radioactive sodium (e.g., 24Na). Using this technique, total body sodium can be divided into exchangeable and nonexchangeable pools. The exchangeable pool represents 70 % of total body sodium and is the fraction of total body sodium that exerts an osmotic effect. Total exchangeable sodium approximately constitutes 40 mEq/kg of body weight, i.e., a 70 kg man will have 2,800 mEq of total exchangeable sodium. Bone is a big reservoir of sodium with one-third of total body sodium residing there. 40 % of the bone sodium is exchangeable [15]. Total body potassium can be measured in a similar manner as sodium. Approximately 95 % of total body potassium is exchangeable.
Mathematically, hyponatremia will occur only if the fraction shown in Formula 4 decreases. There are five different theoretical scenarios where this can occur:
-
1.
Unchanged total exchangeable cations with increased TBW.
-
2.
Decreased total exchangeable cations with unchanged TBW.
-
3.
Increased total exchangeable cations with a greater increase in TBW.
-
4.
Decreased total exchangeable cations with a lesser decrease in TBW.
-
5.
Decreased total exchangeable cations with increased TBW.
Table 1 shows clinical scenarios associated with each of the five combinations. Hyponatremia caused by an isolated decrease in the numerator (Nae + Ke) as in scenario 2 above is of interest, because not only a decrease in total exchangeable sodium, but also a decrease in total exchangeable potassium, often manifested with hypokalemia, can cause hyponatremia. Large external losses of potassium imply exit of potassium from the cells. Although there is a commensurate entrance of sodium into the cell, the major cause of hyponatremia is no change or relatively smaller loss of water than potassium (Formula 4). During potassium repletion, [Na]S increases in the absence of administered sodium, as dictated by Formula 4. Figure 1 presents a scheme of the classification of hyponatremia. Most hypotonic hyponatremias are due to increased total body water caused by increased water intake, decreased water excretion or a combination of the two.

Classification of hyponatremia. [Na+]s serum sodium concentration, POsm plasma osmolality, UOsm urine osmolality, EABV effective arterial blood volume, ECFV extracellular fluid volume and SIADH syndrome of inappropriate antidiuresis. EABV may be normal or elevated in SIADH
Increased water intake
Although the kidneys have normally an extraordinary capacity to excrete large amounts of water, there is a limit to this capacity. Hyponatremia develops when this limit is exceeded. The volume of water needed to exceed the renal maximal capacity for water excretion can be estimated from the formula expressing urine osmolarity (U Osm):
By rearranging Formula 5, the urine volume for any given urine solute load and urine osmolality level is determined in the following way:
The usual loads of solute excreted in the urine are between 600 and 900 mOsm of solute per day. Formula 6 computes the maximal urine volume allowed by a given solute load when U Osm is at its potentially minimal value (50 mOsm/kg). A load of fluid exceeding this maximal urine volume will result in water retention and hyponatremia.
Decreased water excretion
The kidneys are the main organs responsible for water excretion. Three different renal mechanisms lead to decreased renal water excretion, increased ADH activity, which is the most common mechanism of true hyponatremia [16], decreased glomerular filtration rate (GFR) and decreased solute intake and excretion.
The contribution of solute excretion to the urinary water excretion can be illustrated using Formula 6. Solutes relevant to water diuresis are freely filtered in the glomeruli and are reabsorbed by the tubules with varying degrees of difficulty. The main solutes are urea and electrolytes (e.g., salt). In steady state conditions, solute intake is equal to urine solute load. If U Osm is held constant, the urine solute load is the only determinant of the urine volume and hence of water excretion (Formula 6). Maximal urine volume, calculated by this formula when U Osm is at its potentially lowest value of 50 mOsm/kg, is 2 L/day if the solute load is 100 mOsm/day, 4 L/day if solute load is 200 mOsm/day and 18 L/day if solute load is 900 mOsm/day.
Etiology of hypotonic hyponatremia
Table 2 shows clinical entities causing hyponatremia. The classification in Table 2 shows the predominant pathophysiologic abnormality. However, patients classified in one category may present with varying volume and ADH states and a combination of mechanisms causing hyponatremia. For example, beer potomania routinely exhibits low urinary solute excretion rate in addition to a relatively large water intake [17]. Although effective arterial blood volume (EABV) is increased in most patients with nephrotic syndrome, it is decreased in nephrotic patients developing hyponatremia. This decrease in EABV causes ADH secretion. Lack of aldosterone in primary adrenal insufficiency causes sodium loss and hypovolemia, while lack of cortisol in secondary and tertiary adrenal insufficiency causes excess serum ADH levels because cortisol suppresses ADH [4]. Severe hypothyroidism is associated with increased ADH levels but also with low GFR [4]. Finally, patients with SIADH and water gain also exhibit urinary sodium loss during the phase of development of the syndrome.
Peripheral renin–angiotensin system (RAS) blocking agents inhibit the synthesis or actions of angiotensin II in peripheral tissues but not in the brain because they cannot cross the blood brain barrier (BBB). Peripheral RAS blockade leads to elevated levels of angiotensin I in the circulation. Angiotensin I crosses the BBB and is converted to angiotensin II centrally. Brain angiotensin II is a potent stimulus for ADH release and thirst, with the potential complication of hyponatremia. The noncentrally active angiotensin-converting enzyme (ACE) inhibitors enalapril, benazepril, moexipril and quinapril do not cross the BBB. All angiotensin receptor blockers (ARBs) except telmisartan are unable to cross the BBB. Finally, certain central RAS blockers such as lisinopril are unable to block central ACE at lower doses and can potentially also cause hyponatremia [18].
The renal prostaglandin PGE2 has multiple actions on the mechanism of urinary concentration, including decrease in the ADH-mediated trafficking of aquaporin-2; inhibition of the countercurrent multiplication by prevention of the recycling of the Na+–K+–2-Cl- cotransporter (NKCC2) in the thick ascending limb of the loop of Henle; and decrease in hyaluronan production by papillary interstitial cells [19]. Hyaluronan has a large capacity for binding water, altering the papillary interstitial matrix and causing resistance to water flow. Nonsteroidal anti-inflammatory agents (NSAIDs), which block the synthesis of PGE2, can contribute to the development of hyponatremia. However, it appears unlikely that NSAIDs can be the sole cause of hyponatremia.
SIADH is the prototype of hypotonic hyponatremia. Two stages of SIADH, the stage of development and the stage of maintenance, are recognized. In the stage of development, water retention induced by ADH leads to gain in body weight. This water gain is not clinically detectable, because edema is usually absent. The initial volume expansion in SIADH leads to natriuresis that serves to regulate ECF volume toward normal. According to Formula 4, hyponatremia in SIADH is due to both decrease in the numerator (sodium loss) and increase in the denominator (water gain). In experimental studies by Leaf and coworkers done in healthy volunteers in the 1950s, response to a continuous infusion of arginine vasopressin was blunted after 7 days. The manifestations of this blunted response include increase in urine volume and decreases in urine sodium excretion and osmolality [20]. This phenomenon is called escape from antidiuresis. Decrease in proximal tubular reabsorption of sodium and water [21] and downregulation of aquaporin 2 [22] have been identified as mechanisms of escape from antidiuresis. The stage of maintenance of SIADH is determined by escape from antidiuresis. If intake of water and sodium is kept steady in this stage, body weight and [Na]S are also steady and urinary sodium excretion is roughly equal to dietary sodium intake.
Brain adaptation in hypotonic hyponatremia
Under steady state conditions, intracellular and extracellular osmolality are equal and net water movement into or out of cells does not occur. The decrease in plasma effective osmolality that occurs in hypotonic hyponatremia causes water movement into the brain cells along osmotic gradients, producing brain edema [12]. Astrocytes, which are the most common nonneuronal cell type in the CNS and represent about 50 % of the human brain volume, have an important role in brain water handling. They selectively swell after hypotonic stress, whereas neurons do not. Water moves into astrocytes likely via aquaporin 4 [22, 23]. Immediate adaptation to brain swelling includes movement of fluid from the brain cells into the cerebrospinal fluid (CSF). The water movement is driven by a hydrostatic pressure gradient created by increased intracranial pressure from cerebral edema. This is a limited adaptive mechanism. The main adaptation of the brain to swelling is by loss of solutes. This mechanism occurs in astrocytes.
The astrocyte adaptation to hyponatremia is called regulatory volume decrease (RVD). RVD consists of solute exit from the intracellular compartment in an effort to reestablish normal cellular volume. Initially, astrocytes lose electrolytes. Astrocyte swelling triggers extrusion of potassium and chloride ions likely via so far unidentified chloride channels, a variety of potassium channels, and potassium chloride cotransporters KCC-1 and KCC-3. Electrolyte losses account for 70 % of total brain solute losses. The loss of potassium is modulated by the action of the Na+–K+-ATPase pump, which plays an essential role in cell volume regulation. Estrogens inhibit this pump and this may be the reason why premenopausal woman are at higher risk of developing hyponatremic encephalopathy [12].
Electrolyte loss from the cells peaks at 3 h after initial brain swelling and is completed after 6 or 7 h. Altered ion gradients across the cell membrane interfere with cell function. This limits the loss of electrolytes during osmoregulation. Further reduction in cell volume is achieved by loss of organic osmolytes from the intracellular compartment. The main organic osmolytes lost during cell adaptation are glycerophosphorylcholine, phosphocreatine, creatine, glutamate, glutamine, taurine and myoinositol. The latter is the main contributor to volume control in hyponatremia. Full adaptation occurs by 48 h [24].
Clinical manifestations of hypotonic hyponatremia
The brain is the primary target of hyponatremia. The degree of brain swelling determines the increase in intracranial pressure and, by and large, the clinical manifestations of hyponatremia. In general, the greater and more rapid the fall in [Na]S is, the greater the increase in brain swelling will be. Acute hyponatremia is defined as hyponatremia that develops in less than 48 h. In acute hyponatremia, there is no time for full adaptation. Chronic hyponatremia is defined as hyponatremia that develops gradually, usually over more than 48 h. In chronic hyponatremia, the brain has time to fully adapt to the hypotonic state.
Severe neurological manifestations of hyponatremia include seizures, stupor, coma and death from brain herniation. This constellation of symptoms, known as hyponatremic encephalopathy, is encountered along with a significant degree of cerebral edema. Certain types of acute hyponatremia create a high risk of hyponatremic encephalopathy. These include hyponatremia occurring in water intoxication (i.e., psychogenic polydipsia, marathon runners, ecstasy use), postoperative patients, young women and patients with hypoxia or preexisting central nervous system disease. In addition to the neurological symptoms, severe hyponatremia can also cause noncardiogenic pulmonary edema or Ayus–Arieff syndrome [25].
Moderate symptoms of hyponatremia include lethargy, disorientation and confusion. Mild symptoms include fatigue, nausea and headache. Whether hyponatremia, even at moderate levels, is completely asymptomatic, has been disputed [26]. Recent evidence suggests that even mild hyponatremias ([Na]S 130–135 mEq/L), which appear to be asymptomatic, are the cause of subtle symptoms that are not detectable by routine clinical examination and require special neurological testing for their detection [26]. Acute hyponatremia is usually associated with moderate or severe symptoms. Chronic hyponatremia, which is usually associated with minimal symptoms, may cause moderate or severe symptoms when [Na]S is below 120 mEq/L.
Chronic hyponatremia, even mild, has been associated with increased mortality [27]. However, it may be difficult to ascertain whether mortality in hyponatremia occurring in the course of severe chronic illness is the result of hyponatremia or of the underlying illness. Chronic hyponatremia has also been associated with increased morbidity, including attention deficit, gait disturbances, osteoporosis and increased risk of falls and bone fractures [28, 29]. Glutamate is one of the osmolytes that brain cells lose during regulatory volume decrease. Chronic hyponatremia leads to brain glutamate deficiency. Glutamate is a neurotransmitter, which is involved in cerebellar function, and its deficiency has been shown to produce ataxia [29]. Gait disturbances associated with chronic hyponatremia may be related to brain glutamate depletion. In young Sprague–Dawley rats, chronic hyponatremia has been shown to play a role in the development of osteoporosis apparently by increasing bone resorption to mobilize sodium into the circulation where it is needed [29]. The association of gait disturbances and osteoporosis causes a high risk of bone fractures in chronic hyponatremic patients.
Treatment of hypotonic hyponatremia
The treatment of hyponatremia includes general measures that are applicable to all or most hyponatremias and measures that are applied to specific groups of hyponatremias [3, 4, 30–32].
General measures for the management of hyponatremia
Severe hyponatremia: hypertonic saline infusion
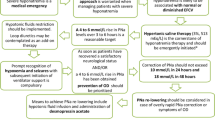
Severe hyponatremia is a medical emergency. Regardless of its mechanism, the initial treatment of severe hyponatremia is directed toward prevention or management of severe neurological manifestations and consists of intravenous infusion of hypertonic saline (3 % NaCl, sodium concentration 513 mEq/L). The goal is to increase [Na]S by 6 mEq/L in the first 6 h and postpone any further correction for the next day. However, if manifestations of severe hyponatremia persist, the rise in [Na]S can exceed 6 mEq/L in the first 6 h.
Treatment should start immediately upon the diagnosis of hyponatremia with infusion of 100–150 mL bolus of 3 % NaCl [3, 4]. At the same time, the total dose of hypertonic saline that is required to raise [Na]S by 6 mEq/L should be calculated and the mechanism of hyponatremia should be investigated. In order to minimize the risks of both overshooting and undershooting, the required total volume of hypertonic saline should be carefully calculated. This volume is calculated by published formulas [30, 32]. The Adrogué–Madias formula [30], which has been used extensively for this purpose, calculates the change in [Na]S (Δ[Na]S = [Na]S Final − [Na]S Initial) that will result from infusion of 1 L of saline with a known sodium concentration ([Na]Infusate) as follows:
The advantage of the Adrogué–Madias formula over previously used formulas is that it takes into account all main determinants of the change in [Na]S during nonisotonic saline infusion, including the volume of the infused saline [32]. A general expression applying the same principles as the Adrogué–Madias formula calculates the volume of saline (V Infusate) with a given [Na]Infusate that is needed for a desired increase in [Na]S (Δ[Na]S) as follows [32]:
Formulas 7 and 8 require an estimate of total body water at the initiation of treatment (TBWInitial). An estimate of the body weight at the normal state, when there was no hydration abnormality (WeightNormal), and measurement of body weight at presentation with hyponatremia (WeightInitial) can provide a reasonable approximation of TBWInitial, as follows: Body water at the normal state (TBWNormal) can be computed by applying WeightNormal in published anthropometric formulas estimating body water under normal hydration conditions [33–35]. TBWInitial is then calculated as follows [32]:
The effect of the estimate of TBWInitial on the required volume of hypertonic saline for a desired Δ[Na]S can be illustrated by the following example: Formula 8 computes that the volume of 3 % NaCl required to raise [Na]S from 100 to 106 mmol/L is 0.234 L if TBWInitial is 20 L and 1.179 L if TBWInitial is 80 L.
The accuracy of Formulas 7 and 8 is affected by the estimates of TBW, which can differ substantially from actual body water and by urinary or other losses of water, sodium and potassium during treatment [32]. For these reasons, extreme caution is required during infusion of hypertonic saline. A safer way to achieve the desired increase in [Na]S is to infuse one-half of the calculated volume of hypertonic saline, measure [Na]S soon after the end of this infusion and adjust the subsequent boluses of hypertonic saline after the second [Na]S measurement. Formulas 7 and 8 have two uses: (a) they provide an estimate of the required volume of hypertonic saline that is tailored to the body size of the patient and (b) they provide a means of comparing the observed and predicted change in [Na]S midway through the infusion [32].
The critical measure during treatment of any hyponatremia with infusion of hypertonic or isotonic saline is monitoring of clinical status, [Na]S, urine volume and, in selected cases, urinary sodium and potassium excretion [32]. The patient must be transferred to an intensive care unit bed as soon as the situation allows it and [Na]S should be checked every 2 h at a minimum.
Moderate hyponatremia: hypertonic saline infusion
Moderately symptomatic hyponatremia is also considered a medical emergency. Slow intravenous infusion of NaCl 3 % is usually the treatment of choice. During infusion, the patient should be monitored in an intensive care unit bed and [Na]S should be measured every 2 h at a minimum. The volume of the infused saline can be calculated by the formulas mentioned above. Like in severe hyponatremia, the infusion rate should be titrated based on results of subsequent [Na]S measurements. The goal is to increase [Na]S by 6 mEq/L in 24 h.
The general indications for hypertonic saline (NaCl 3 %) infusion include acute hyponatremia, symptomatic hyponatremia (acute or chronic) and hyponatremia associated with intracranial pathology already at risk of cerebral edema (intracranial hemorrhage, neurosurgery, etc.).
Mild hyponatremia
Mildly symptomatic or “asymptomatic” hyponatremia is not a medical emergency. Therefore, there is more time to target the underlying pathophysiology of hyponatremia. Patients usually can be treated in a general medical ward. [Na]S should be checked at least every 4–6 h. The goal is to increase [Na]S by 6 mEq/L in 24 h. The treatment includes targeting the primary pathophysiology of hyponatremia.
Restriction of fluid intake
Restriction of fluid intake is fundamental in the management of hyponatremia. Because of the importance of this measure, the principles guiding fluid restriction will be presented in some detail. The goal of fluid restriction is net loss of water. This is achieved by limiting water gain so it becomes less than water loss. Fluid restriction includes all fluids ingested by the patient (e.g., juice and soup) and not just water. The volume of ingested fluid that is required to create a state of negative water balance is less than the sum of urine and insensible losses. In the worst-case scenario, when the urine contains very little free water (e.g., in SIADH) [36], fluid restriction should be less than insensible losses (i.e., <800 mL/day).
A more accurate way to estimate the amount of fluid restriction is by using the 24-h electrolyte-free water clearance formula \( ({\text{C}}_{{{\text{eH}}_{ 2} {\text{O}}}} ) \) [37], which represents the amount of free water excreted by the kidneys over a 24-h period:
where V U = Urine volume in 24 h, [Na]U = Urine sodium concentration and [K]U = Urine potassium concentration.
The relation between \( {\text{C}}_{{{\text{eH}}_{ 2} {\text{O}}}} \) and V U determines the changes in [Na]S as a consequence of urine losses of water, sodium and potassium [32]. If information about urine volume is unavailable, one still can predict whether the electrolyte-free water clearance is positive or negative and estimate the required fluid restriction by calculating the urine/serum electrolyte ratio: ([Na]U +[K]U)/[Na]S. Similar calculations can be made for water and electrolyte losses through the gastrointestinal tract. Of note, the sum of sodium and potassium concentration in most instances of gastrointestinal fluid loss is lower than the normal [Na]S [38]. This should lead to hypernatremia. Patients with protracted vomiting or diarrhea develop hyponatremia because of thirst and fluid consumption. Table 3 shows the effects of urine/serum electrolyte ratio on \( {\text{C}}_{{{\text{eH}}_{ 2} {\text{O}}}} \) and [Na]S.
The management of certain types of hyponatremia with fluid restriction alone exhibits difficulties. For example, patients with SIADH usually have a urine/serum electrolyte ratio >1, i.e., they have a negative \( {\text{C}}_{{{\text{eH}}_{ 2} {\text{O}}}} \). In some patients with SIADH, fluid restriction by itself does not result in improvement in [Na]S and additional therapies are needed (e.g., salt tablets and/or loop diuretics).
Loop diuretics
Loop diuretics can increase water excretion in hyponatremia with negative \( {\text{C}}_{{{\text{eH}}_{ 2} {\text{O}}}} \) (e.g., SIADH, hypervolemic hyponatremia) [32]. Water is reabsorbed in excess of solute in the inner medullary collecting duct. The driving force for water reabsorption is the hypertonicity of the medullary interstitium, which normally has an osmolality of 1,200 mOsm/kg at the papillary tip. Sodium chloride contributes to roughly 50 % of the medullary osmolality and urea contributes the remaining 50 %. Sodium chloride moves from the tubular lumen into the medullary interstitium. The first step in this sodium chloride transport is via the NKCC2 cotransporter, which is located in the apical membrane of the cells of the thick ascending limb of the loop of Henle. Loop diuretics inhibit NKCC2 and therefore reduce the amount of sodium chloride delivered to the medulla. This action abolishes the medullary gradient necessary for water reabsorption and therefore increases free water excretion. Loop diuretics hence usually produce hypotonic urine. In euvolemic patients, loop diuretics are usually administered with isotonic saline to avoid hypovolemia and the creation of an extra stimulus for ADH release.
Solute administration
Increasing urinary solute excretion by various means obligates renal water excretion as indicated by Formula 6 [39, 40]. Sodium tablets and urea tablets have been used for this purpose. Urea has been shown to have a protective effect against osmotic myelinolysis. Urea apparently promotes rapid reaccumulation of brain organic osmolytes [41]. This could explain why osmotic demyelination syndrome (ODS) occurs less frequently in chronic hemodialysis patients.
Specific measures for the management of hyponatremia
The management of hyponatremias caused by increased ADH activity targets the cause of the increased ADH. Hypovolemic hyponatremia is treated by isotonic saline infusion. This treatment carries a high risk of overcorrection. The factors that determine the rise in [Na]S during isotonic saline infusion and contribute to this high risk are as follows: (a) the value of the presenting body water that is entered in Formulas 7 or 8. This value represents at times an overestimate of the actual body water in hypovolemia, (b) the difference between the concentrations of sodium in the infusate and the serum of the patient and (c) water diuresis that follows removal of the volume stimulus for ADH release when hypovolemia is corrected [32]. Monitoring of the treatment of hypovolemic hyponatremia requires intensity similar to that of severe hyponatremia regardless of whether the symptoms of hypotonicity are severe, moderate or mild [32].
ADH release in hypervolemic hyponatremia is usually persistent because its two main causes, advanced congestive heart failure and advanced liver cirrhosis, are irreversible. Until recently, the management or prevention of mild hyponatremia in these conditions was based on water restriction and the use of loop diuretics. The development of vasopressin (V2) receptor blockers, which are discussed later, offers the promise of blocking the action of ADH in hypervolemic hyponatremia.
SIADH is the prototype euvolemic hyponatremia. The complete set of criteria for its diagnosis includes, in addition to hyponatremia, clinical euvolemia, U Osm > 100 mOsm/kg, [Na]U > 30 mEq/L on normal salt intake, serum uric acid <4 mg/dL, normal renal, thyroid and adrenal function and absence of diuretic use (particularly thiazides) [4, 36]. The diagnosis of SIADH is often based on few of these criteria, particularly in the presence of one of the conditions known to cause SIADH. In all cases, however, U Osm must be relatively high. Severe or moderate hyponatremia from SIADH should be treated as described above. If a reversible cause of ADH secretion is found (e.g., medications, lung or CNS infections, pain, nausea), the treatment should address this cause. Specific treatment is also used for other types of euvolemic hyponatremia, e.g., glucocorticoid administration in cases of secondary adrenal insufficiency and thyroid replacement in severe hypothyroidism.
Vaptans and desmopressin
Two therapeutic modalities for hyponatremia, V2 receptor antagonists (vaptans) and desmopressin infusion [42–44], have directly opposite effects on urine osmolality, and opposite indications and contraindications. Vaptans block the effect of ADH on the collecting duct and cause water diuresis. They are effective in hyponatremia associated with high serum ADH levels and high urine osmolality and ineffective in hyponatremia associated with dilute urine.
The main indications for vaptan use are hypervolemic hyponatremia due to congestive heart failure and euvolemic hyponatremia due to SIADH. Demeclocycline has been used for the same indications, particularly in SIADH. However, the effect of demeclocycline on serum sodium concentration has a delayed onset of action, is modest at best and is associated with significant nephrotoxicity especially in patients with liver cirrhosis [3, 4]. Current guidelines recommend against its use [4]. Vaptans must be initiated and reinitiated in an inpatient setting with frequent [Na]S monitoring. Fluid restriction should be avoided in the first 24 h. Contraindications for vaptan use include symptomatic hyponatremia requiring infusion of hypertonic saline, hypovolemic hyponatremia, known hypersensitivity to vaptans, and use of strong CYP3A4 inhibitors (e.g., ketoconazole). Poor response of hyponatremia to vaptans may be encountered in SIADH with very high circulating ADH levels, low glomerular filtration rate, large water intake and use of nonsteroidal anti-inflammatory agents. Other limitations of the clinical application of vaptans include the risk of overcorrection of hyponatremia, absence of evidence of their effects on morbidity or mortality, high costs and the potential of hepatotoxicity.
Desmopressin infusion causes production of urine with high osmolality and low volume. Desmopressin is ineffective in hyponatremias with high serum ADH levels and high urine osmolality that are sustained during treatment. Desmopressin infusion, usually in conjunction with hypertonic or isotonic saline infusion, is indicated when it is anticipated that treatment of hyponatremia will result in water diuresis and dangerously rapid rise in [Na]S. Hypovolemic hyponatremia constitutes the primary indication for desmopressin infusion [43, 44]. In addition, treatment of hyponatremia in primary polydipsia, beer potomania and low solute intake carries the risk of overcorrection that can be prevented by desmopressin infusion [44]. Table 4 shows specific therapies for frequently encountered hyponatremias.
Goals of correction of hypotonic hyponatremia
The specific goals of treatment of hyponatremia are summarized in this section [45]:
Severely symptomatic hyponatremia
Raise [Na]S by 6 mEq/L and re-evaluate. Symptomatic hyponatremia usually requires infusion of hypertonic saline (3 % NaCl).
Moderately symptomatic, mildly symptomatic and “asymptomatic” hyponatremia
Raise [Na]S by 6 mEq/L in any 24 h period.
Limits of correction
Maximal limits for [Na]S increase, aiming to prevent osmotic demyelination, are not correction goals but targets that should not be crossed. Current recommendations dictate that the limits of correction of [Na]S should not exceed 10–12 mEq/L in any 24-h period in patients at low risk of ODS and no more than 8 mEq/L in any 24-h period in patients at high risk of ODS (i.e., [Na]s <105 mEq/L, liver cirrhosis, malnutrition, alcohol dependence and hypokalemia) [3]. The absolute magnitude of correction is more important than the rate of correction. For example, chronic hyponatremia with [Na]S 115 mEq/L theoretically can be corrected up to 125 mEq/L in the first hour of correction as long as [Na]S remains at 125 mEq/L for the next 23 h and the total rise of [Na]S in a 24-hour period does not exceed 10 mEq/L.
Complications of rapid correction of chronic hyponatremia
Osmotic demyelination syndrome is a dreaded complication of correction of hyponatremia. ODS is also known as central pontine myelinolysis (CPM) because when first described the demyelinating lesions involved the pons. However, later on, it was found that the lesions could also affect the basal ganglia, the thalamus and other sites. The exact mechanism of demyelination is not clear [46–48]. One of the current hypotheses proposes that acute brain shrinking caused by rapid correction of hyponatremia causes astrocyte death and loss of cell-to-cell interactions between astrocytes and oligodendrocytes. Dying astrocytes are suggested to promote demyelination by enhancing the release of cytokines and other inflammatory mediators, and recruitment of T cells, microglia and macrophages [48].
Conditions that increase the risk of ODS include profound hyponatremia ([Na]S <105 mEq/L), alcoholism, malnutrition, advanced liver disease, liver transplantation and hypokalemia. The clinical manifestations of ODS can be devastating. The onset of symptoms typically is delayed several days (up to 1 week) after overcorrection of hyponatremia. The manifestations include altered mental status, quadriparesis, dysphagia and dysarthria. The diagnosis of ODS is usually based on the development of new-onset neurological symptoms in a patient with a recent overcorrection of hyponatremia. Magnetic resonance imaging of the brain can detect demyelinating lesions; however, it may not be positive for as long as 4 weeks after onset of symptoms. Approximately 40 % of the patients with ODS have full recovery and 25 % are left with persistent neurological deficits. Mortality from ODS is around 6 % [47].
There is no effective treatment. However, based on anecdotal information, there could be a benefit in prevention of ODS from relowering [Na]S, administration of corticosteroids or plasmapheresis. Prevention of overcorrection of hyponatremia reduces substantially the incidence of ODS. The most frequent cause of overcorrection of hyponatremia is rapid water diuresis [32]. This could be recognized by an increase in urine output or the development of a low urine osmolality. Lack of counting potassium loads administered to patients with hyponatremia and potassium deficits as parts of the total cation load, that increases the numerator in Formula 4, may also lead to rapid overcorrection and ODS [49].
The risk of overcorrection is high in hyponatremia caused by hypovolemia, primary polydipsia, low solute intake, beer potomania, cortisol deficiency and transient SIADH (e.g., drugs, postoperative state, pain, nausea). Anticipation of overcorrection by recognizing the risk factors is far more effective than treating established overcorrection. In patients who present with hyponatremia and hypokalemia, it is very important to account for the effects of repleting potassium on [Na]S. [Na]S should be measured frequently (at least every 2 h) during saline infusion or during the course of certain other treatments (e.g., restriction of fluid intake in primary polydipsia) to increase the chances of detecting early overcorrection.
If overcorrection is detected, relowering of [Na]S should be attempted. Animal studies and case series have shown that relowering [Na]S below the maximal limits of correction is an effective strategy that decreases the chances of complications [50]. Infusion for 1 h of 3 mL/kg 5 % dextrose in water and repeated measuring of [Na]S is the first corrective step. Rates of dextrose infusion greater than 250–300 mL/h can cause significant hyperglycemia even in nondiabetic patients. This can produce free water loss due to osmotic diuresis with a subsequent increase in [Na]S. Intravenous infusion of desmopressin in a dose of 2–4 mcg every 8 h should be considered in those cases since it prevents overcorrection and is safe [50].
Conclusions
Understanding the principles of diagnosis and management of hyponatremia can lead to significant improvement in its outcomes. Monitoring of the patient’s clinical status, [Na]s and other relevant serum and urine biochemical parameters is critical in both proper correction of [Na]S and prevention of ODS.
References
Upadhyay A, Jaber BL, Madias NE (2006) Incidence and prevalence of hyponatremia. Am J Med 119(7 Suppl. 1):S30–S35
Liamis G, Rodenburg EM, Hofman A et al (2013) Electrolyte disorders in community subjects: prevalence and risk factors. Am J Med 126(3):256–263
Verbalis JG, Goldsmith SR, Greenberg A et al (2013) Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med 126(10 Suppl 1):S1–S42
Spasovski G, Vamholder R, Allolio B et al (2014) Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J Endocrinol 170(3):G1–G47
Arieff AI, Carroll HJ (1972) Nonketotic hyperosmolar come with hyperglycemia: clinical features, pathophysiology, renal function, acid-base balance, plasma-cerebrospinal fluid equilibria and the effects of therapy in 37 cases. Medicine (Baltimore) 51(2):73–94
Tomkins AM, Dormandy TL (1971) Osmolality pattern during recovery from diabetic coma. Lancet 2(7731):952–955
Tzamaloukas AH, Sun Y, Konstantinov NK et al (2013) Principles of quantitative fluid and cation replacement in extreme hyperglycemia. Cureus 5(3):3110. doi:10.17759/cureus.110
Katz MA (1973) Hyperglycemia-induced hyponatremia: calculation of the expected serum sodium depression. N Engl J Med 289(16):843–844
Al-Kudzi RR, Daugirdas JT, Ing TS et al (1982) Extreme hyperglycemia in dialysis patients. Clin Nephrol 17(5):228–231
Liamis G, Liberopoulos E, Barkas F, Elisaf M (2013) Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol 38(1):50–57
Agarwal R, Emmett M (1994) The post-transurethral resection of prostate syndrome: therapeutic proposals. Am J Kidney Dis 24(1):108–111
Ayus JC, Achinger SG, Arieff A (2008) Brain cell volume regulation in hyponatremia: role of sex, age, vasopressin, and hypoxia. Am J Physiol Renal Physiol 295(3):F619–F624
Edelman IS, Leibman J, O’Meara MP, Birkenfeld LW (1958) Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest 37(9):1236–1256
Rose BD (1986) New approach to disturbances in plasma sodium concentration. Am J Med 81(6):1033–1040
Ayus JC, Moritz ML (2010) Bone disease as a new complication of hyponatremia: moving beyond brain injury. Clin J Am Soc Nephrol 5(2):167–168
Pham PC, Pham PM, Pham PT (2006) Vasopressin excess and hyponatremia. Am J Kidney Dis 47(5):727–737
Thaler SM, Teitelbaum I, Berl T (1998) “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis 31(6):1028–1031
Subramanian D, Ayus JC (1992) Severe symptomatic hyponatremia associated with lisinopril therapy. Am J Med Sci 303(3):177–179
Olesen ET, Fenton RA (2013) Is there a role for PGE2 in urinary concentration? J Am Soc Nephrol 24(2):169–178
Leaf A, Bartter FC, Santos RF, Wrong O (1953) Evidence in man that urinary electrolyte loss induced by pitressin is a function of water retention. J Clin Invest 32(9):868–878
Warms PC, Michelis MF, Brafdon RW et al (1974) Micropuncture study of tubule sodium reabsorption in dilutional hyponatremia. Metabolism 23:203–208
Verbalis JG (2006) Whole-body volume regulation and escape from antidiuresis. Am J Med 119(7 Suppl 1):S21–S29
Bourque CW (2008) Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci 9(7):519–531
Verbalis JG (2010) Brain volume regulation in response to changes in osmolality. Neuroscience 168(4):862–870
Ayus JC, Arieff AI (1995) Pulmonary complications of hyponatremic encephalopathy: noncardiogenic pulmonary edema and hypercapnic respiratory failure. Chest 107(2):517–521
Decaux G (2006) Is asymptomatic hyponatremia really asymptomatic? Am J Med 119(7 Suppl. 1):S79–S82
Lee DS, Austin PC, Rouleau JL et al (2003) Predicting mortality among patients hospitalized for heart failure: derivation of and validation of a clinical model. JAMA 290(19):2581–2587
Ayus JC, Negri AL, Kalantar-Zadeh K, Moritz ML (2012) Is chronic hyponatremia a novel risk factor for hip fracture in the elderly? Nephrol Dial Transplant 27(10):3725–3731
Verbalis JG, Barsony J, Sugimura Y et al (2010) Hyponatremia-induced osteoporosis. J Bone Miner Res 25(3):554–563
Adrogué HJ, Madias NE (2000) Hyponatremia. N Engl J Med 341(21):1581–1589
Lien YH, Shapiro JI (2007) Hyponatremia: clinical diagnosis and management. Am J Med 120(8):653–658
Tzamaloukas AH, Malhotra D, Rosen BH et al (2013) Principles of management of severe hyponatremia. J Am Heart Assoc 2:3005199. doi:10.1161/JAHA.112.005199
Mellits ED, Cheek DB (1970) The assessment of body water and fatness from infancy to adulthood. Monogr Soc Res Child Dev 45(7):12–16
Watson PE, Watson ID, Batt RD (1980) Total body water volumes for adult males and females estimated from simple anthropometric measurements. Am J Clin Nutr 33(1):27–39
Chumlea WC, Guo SS, Zeller CM et al (2002) Total body water reference values and prediction equations for adults. Kidney Int 59(6):2250–2258
Ellison DH, Berl T (2007) The syndrome of inappropriate antidiuresis. N Engl J Med 356(20):2064–2072
Furst H, Hallows KR, Post J et al (2000) The urine/plasma electrolyte ratio: a predictive guide to water restriction. Am J Med Sci 319(4):240–244
Gennari FJ, Weise WJ (2008) Acid–base disturbances in gastrointestinal disease. Clin J Am Soc Nephrol 3(6):1861–1868
Oster JR, Singer I, Thatte L et al (1997) The polyuria of solute diuresis. Arch Intern Med 157(7):721–729
Berl T (2008) Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol 19(6):1076–1078
Soupart A, Silver S, Schroeder B et al (2002) Rapid (2-hour) reaccumulation of brain organic osmolytes (particularly myo-inositol) in azotemic rats after correction of chronic hyponatremia. J Am Soc Nephrol 13(6):1433–1441
Jovanovich AJ, Berl T (2013) Where vaptans do and do not fit in the treatment of hyponatremia. Kidney Int 83(4):563–567
Sood L, Sterns RH, Hix JK et al (2013) Hypertonic saline and desmopressin: a simple strategy for safe correction of severe hyponatremia. Am J Kidney Dis 61(4):571–578
Tzamaloukas AH, Shapiro JI, Raj DS et al. Management of severe hyponatremia: infusion of hypertonic saline and desmopressin or infusion of vasopressin inhibitors. Am J Med Sci (in press)
Sterns RH, Hix JK, Silver S (2010) Treating profound hyponatremia: a strategy for controlled correction. Am J Kidney Dis 56(4):774–779
Norenberg MD (2010) Central pontine myelinolysis: historical and mechanistic considerations. Metab Brain Dis 25(1):97–106
King JD, Rosner MH (2010) Osmotic demyelination syndrome. Am J Med Sci 339(6):561–567
Gangam Kengne F, Nicaise C, Soupart A et al (2011) Astrocytes are an early target in osmotic demyelination syndrome. J Am Soc Nephrol 22(10):1834–1845
Berl T, Rastegar A (2010) A patient with severe hyponatremia and hypokalemia: osmotic demyelination following potassium repletion. Am J Kidney Dis 55(4):742–748
Soupart A, Penninckx R, Crenier L et al (1994) Prevention of brain demyelination in rats after excessive correction of chronic hyponatremia by serum sodium lowering. Kidney Int 45(1):193–200
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rondon-Berrios, H., Agaba, E.I. & Tzamaloukas, A.H. Hyponatremia: pathophysiology, classification, manifestations and management. Int Urol Nephrol 46, 2153–2165 (2014). https://doi.org/10.1007/s11255-014-0839-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-014-0839-2