
Abstract
To further understand the genetic structure that is associated with insulin (INS) and thyroid hormones (TH), including triiodothyronine (T3) and thyroxine (T4), in Chinese Holstein cows, we conducted a genome-wide association study (GWAS) of thyroid hormones and insulin in cows. We conducted GWAS analysis on 1217 Chinese Holstein cows raised in southern China and found 19 significant single nucleotide polymorphisms (SNPs) in this study: 10 SNPs were associated with INS, 5 SNPs were associated with T3, and 4 SNPs were associated with T4. In our study, the GWAS method was used for preliminary screening on related genes of traits, and due to insufficient relevant literature, a functional analysis of genes could only be based on human studies. We observed that DGKB from Bos taurus chromosome (BTA)4 is strongly associated with insulin secretion. We found that EXOC4 gene was significantly correlated with T3 and T4 traits. Another significant SNP was located in the CYP7A1 gene, which has been confirmed to be affected by thyroid hormones.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hormones are used to communicate between organs and tissues for physiological regulation and behavioral activities. In this study, we mainly investigated INS, T3, and T4. Some studies have shown that insulin has the same function as growth hormone and oxytocin, which can promote cell proliferation and hormone secretion (Choi et al. 2004; Bossaert et al. 2010). Triiodothyronine, also known as T3, is a thyroid hormone. It affects almost every physiological pr socess in the body, including growth and development, metabolism, body temperature, and heart rate. T4 is a tyrosine-based hormone produced by the thyroid gland which is primarily responsible for the regulation of metabolism.
When investigating the effects of hormones on various physiological activities, an increasing number of studies have found that genes influence hormones. With the advent of high-throughput gene typing and sequencing technologies, genome-wide association study (GWAS) has become a popular research approach to analyze gene variation. However, in the studies on cows to date, there is little correlation between hormones and genetic variation, and most studies address economic traits (Daetwyler et al. 2008; Schopen et al. 2011). The results of a GWAS study on cows, which was first reported in 2008, found 133 significant SNP sites associated with milk yield (Daetwyler et al. 2008). Huang et al. (2010) used the selective DNA pooling method to study the breeding traits of Holstein cattle in 2010 and found 22 significant SNP sites related to the fertilization rate and 5 SNPs related to the embryo bubble rate.
GWAS analysis showed that FAM13A and POM121C were candidate genes for insulin secretion (Lundbäck et al. 2018). In the existing genome-wide association analysis of insulin, 19 loci were found that are associated with thyroid-stimulating hormone (TSH) circulation, and 6 loci were associated with free T4 (fT4) (Eriksson et al. 2012; Wilson. 2013; Malinowski et al. 2014).
To further understand the genetic structures that are related to insulin, triiodothyronine, and thyroxine secretion in Chinese Holstein cows, insulin and thyroid hormones in serum samples of cows were studied.
Materials and methods
Animal resource
The animals used in this study comprised 1217 Holstein cows that were inter-crossed with domestic Holstein cows and imported semen from high-yield bulls. These 1217 cows came from 48 half-sib lines, and each line contained 5 to 50 individuals. The Holstein cows were housed on two commercial farms in the southeast of China.
Blood sample collection and phenotypes
Blood samples of 10 mL were obtained from coccygeal vessels of the cows. Half of each blood sample (another 5 mL of blood was used to extract genomic DNA for genotyping) was immediately injected into a tube containing 30 μL of 20% EDTA anticoagulant. All samples were stored at 4 °C and sent to the hospital of Fuzhou within 24 h for analysis. Three hormones (INS, T3, and T4) were examined in this hospital, and these hormone concentrations were used as phenotype data for performing GWAS.
Genotypes
We used a cost-effective method to genotype all 1217 cows. For each line, two cows were randomly selected to be genotyped using 50K chips (Illumina BovineSNP), and the remaining cows were genotyped using 26K chips (GeneSeek). The imputation for the 26K chips as well as sporadic missing genotypes was performed using Beagle version 3.3.1 (Browning and Browning, 2009). Quality control was conducted using PLINK 1.07 (Purcell et al. 2007) to remove SNPs with call rates of ≤ 95%, minor allele frequencies of ≤ 0.05, and significant deviation from Hardy-Weinberg equilibrium (p < 10−5). After these criteria were applied, 47,396 SNPs from 1217 animals were retained for further analyses.
Statistical model
The FarmCPU algorithm (Liu et al. 2016) was used to solve a linear mixed model for three hormone traits in Chinese Holstein cows. The model iteratively uses a fixed-effect model (FEM) and a random-effect model (REM). The FEM tests markers, one at a time, and uses a set of pseudo-quantitative trait nucleotides (QTNs) as covariates. The model can be written as follows:
where yi is the observation of the ith individual; Mi1, Mi2……Mitare the genotypes of t pseudo QTNs, initiated as an empty set; b1, b2, ……bt are the corresponding effects of the pseudo QTNs; Sij is the genotype of the ith individual and jth genetic marker; dj is the corresponding effect of the jth genetic marker; and ei is the residuals having a distribution with zero mean and variance of \( {\sigma}_e^2 \).
The REM, using the SUPER algorithm, optimized the possible correlation sites of different combinations by using the p values and position information of genetic markers. The model can be written as follows:
where yi and ei are the same as in Eq. (1), and ui is the total genetic effect of the ith individual. The expectation of the individuals’ total genetic effects is 0.
The variance and covariance matrix of the individuals’ total genetic effects is\( G=2K{\sigma}_{\mathrm{a}}^2 \), where \( {\sigma}_{\mathrm{a}}^2 \) is an unknown genetic variance, and K is the kinship derived from the pseudo QTNs.
Equations (1) and (2) are used interchangeably in the FarmCPU model.
This algorithm solves a linear mixed model by performing marker tests with associated markers as covariates in a fixed-effect model and separately optimizing based on the associated covariate markers in a random-effect model.
After the association testing, the Bonferroni method was used to conduct a multiple-test correction. An SNP was considered significant when its p value < 0.05/N (N = number of markers).
Gene annotation
The UMD3.1 assembly of bovine genome sequences in the Ensembl database (http://asia.ensembl.org/index.html) was used to locate genes that contained the significant SNPs or were within 400 kb from the significant SNPs.
Results
The serum samples of 1217 cows involved in the experiment were counted, and the relevant concentrations of hormones in the serum were calculated: the minimum (Min), maximum (Max), mean, standard error (SE), standard deviation (SD), variance (Var), and coefficient of variation (CV) (Table 1).
The heritability of insulin, T3, and T4 was 0.0635, 0.1059, and 0.1948, respectively; the traits we chose all had low heritability.
In this study, a total of 47,396 SNP sites were detected by whole-gene scanning, among which 19 were significantly correlated with hormone concentration (Table 2).
There were 10 significant SNPs associated with insulin concentrations.
Among these 10 SNPs, one SNP was located within CLCN3 on Bos taurus chromosome 8 (BTA8), and one SNP on BTA22 was RBMS3. One SNP was located near ZFP36L2 on BTA11, and one SNP on BTA2 was 21 kb away from ACTL8. One SNP was 6 kb away from the B3GN76 on BTA15. Another SNP was 143 kb away from SHOX on BTA2. There were 4 SNPs located within known genes: DGKB on BTA4, BAZ1A on BTA21, SMOC2 on BTA9, and FAM65B on BTA23.
Five SNPs located in different chromosomes showed significant associations with triiodothyronine concentrations. The most significant SNP was located within EXOC4 on BTA4. One SNP on BTA14 was from CYP7A1, and one SNP was located within SH3GLB2 on BTA11. Two SNPs were located near SATB1 on BTA1 and TSHZ2 on BTA13.
For thyroid hormone concentrations, there were 4 SNPs located in different chromosomes that showed significant associations. The most significant SNP was located on BTA14 133,805 bp away from PAG1. Two SNPs were located within CTNND2 on BTA20, and another was 160 kb away from ANO6 on BTA5.
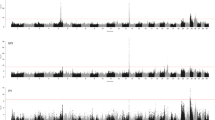
These results are visualized via Manhattan plots (Fig. 1).

Manhattan plots of −log10 (p values) for three hematological hormone traits. In GWAS Manhattan plots, genomic coordinates are displayed along the x-axis, with the negative logarithm of the association p value for each single nucleotide polymorphism (SNP) displayed on the y-axis, with each dot on the Manhattan plot signifying an SNP
Figure 2 contains the corresponding Quantile-Quantile plots, which were used to assess the goodness of fit of the model. If there is no systematic error in the correlation analysis results, most points should be attached to the straight line of y = x, and a small number of the significant correlation points deviate from the straight line upwards. There are no systematic errors such as false-positives or false-negatives

Q-Q plots of −log10 (p values) for three hematological hormone traits. In the Q-Q diagram of GWAS, the expected p value is the value of each point when the null hypothesis is true. With the x-axis as the expected p value and the y-axis as the observed p value
We found the locations of these SNPs through the website http://asia.ensembl.org/index.html and observed that some of the SNPs were located not in the genes but near the coding regions (Table 2).
Discussion
To our knowledge, the present work is the first GWAS to analyze the genetic variants related to hormone concentrations in Holstein cattle. We preliminarily identified genes associated with insulin, triiodothyronine, and thyroxine concentrations and considered their putative functions. RBMS3 on BTA22, DGKB on BTA4, SHOX on BTA2, CYP7A1 on BTA14, and SCN5A on BTA22 were identified as being involved in hormone balance in cattle. However, there is a lack of evidence for these genes in cow studies, and we can only speculate from the existing literature.
Based on previous studies, RBMS3 is a gene encoding a glycine-rich RNA-binding protein and belongs to the family of c-Myc gene single-strand binding proteins (MSSP). Experimental results indicate that RBMS3 also plays an important role in maintaining the physiological function of mature pancreatic exocrine cells (Lu et al. 2012). We speculate that this gene might have an association with insulin secretion.
The SNP within DGKB likely influences signal transduction, cell proliferation, development, glucose-sensing, and circadian regulation. Dupuis et al. (2010) and collaborators found in a 2010 study that the relationship between DGKB and type 2 diabetes may affect fasting insulin levels. We therefore hypothesize that the DGKB gene might affect insulin levels to some extent. Research by Wagner et al. (2011) showed that the diabetogenic alleles of DGKB were nominally associated with reduced insulin secretion. Although little research has been conducted on the regulation of insulin by DGKB, we speculate that this gene might have an association with insulin levels.
SHOX belongs to the paired homeobox family and is located in the pseudo-autosomal region 1 (PAR1) of X and Y chromosomes. It is not certain that SHOX directly regulates insulin secretion, but the available literature suggests that this gene has a potential association with insulin. Kim et al. (2012) showed the expression of SHOX could be the origin of human labia minora dermis–derived fibroblasts (hLMDFs). This result indicates that hLMDFs have the capacity to differentiate into IPCs (insulin-producing cells).
In our study, five valuable SNPS were associated with thyroxine and triiodothyronine. CYP7A1 encodes a member of the cytochrome P450 superfamily of enzymes. Kuipers et al. (2014) included CYP7A1 as a research candidate because it may indirectly affect insulin resistance. Song et al. (2015) showed that thyroid-stimulating hormone (TSH) inhibits CYP7A1 activity.
In the analysis of T3 and T4, we found a common SNP within EXOC4. The protein encoded by this gene is a component of the exocyst complex, a multiple protein complex essential for targeting exocytic vesicles to specific docking sites on the plasma membrane. Unfortunately, we have not found reported evidence that the EXOC4 gene is related to thyroid hormones, but because of the function of this gene, we hope to be able to explore its relationship with thyroid hormones in future studies.
Conclusion
From this study, it was possible to conclude that the GWAS analysis using the FarmCPU model was accurate, effectively avoiding the emergence of false-positives, and it can make the initial screening sites more valuable.
Through this study, genes potentially related to hormone secretion in dairy cows were identified, which provided gene sources and driving forces for the subsequent development of molecular marker–assisted selection and other breeding measures to cultivate new dairy cow strains.
Data availability
All the data used in this article were uploaded at https://figshare.com (https:// 10.6084/m9.figshare.7865810).
References
Bossaert, P., De, Cock H., Leroy, JL, De, Campeneere S., Bols, P.E., Filliers, M., Opsomer, G., 2010. Immunohistochemical visualization of insulin receptors in formalin-fixed bovine ovaries post mortem and in granulosa cells collected in vivo. Theriogenology. 73, 1210-1219.
Browning, B.L., Browning, S.R., 2009. A Unified Approach to Genotype Imputation and Haplotype-Phase Inference for Large Data Sets of Trios and Unrelated Individuals. The American Journal of Human Genetics. 84, 210-223.
Choi, K.M., Barash, I., Rhoads, R.E., 2004. Insulin and prolactin synergistically stimulate beta-casein messenger ribonucleic acid translation by cytoplasmic polyaden. Molecular Endocrinology. 18, 1670-1686.
Daetwyler, H.D., Schenkel, F.S., Sargolzaei, M., Robinson, J.A., 2008. A Genome Scan to Detect Quantitative Trait Loci for Economically Important Traits in Holstein Cattle Using Two Methods and a Dense Single Nucleotide Polymorphism Map. Journal of Dairy Science. 91, 3225-3236.
Eriksson, N., Tung, J.Y., Kiefer, A.K., Hinds, D.A., Francke, U., Mountain, J.L., Do, C.B., 2012. Novel associations for hypothyroidism include known autoimmune risk loci. PLoS One. 7, e34442.
Huang, W., Kirkpatrick, B.W., Rosa, G.J.M., Khatib, H., 2010. A genome-wide association study using selective DNA pooling identifies candidate markers for fertility in Holstein cattle. Animal Genetics. 41, 570-578.
Dupuis, J., Langenberg, C., Prokopenko, I., Saxena, R., Soranzo, N., Jackson, A.U., Wheeler, E., Glazer, N.L., Bouatia-Naji, N., Gloyn, A.L., et al, 2010. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature Genetics. 42, 105-116.
Kim, B., Yoon, B.S., Moon, J.H., Kim, J., Jun, E.K., Lee, J.H., Kim, J.S., Baik, C.S., Kim, A., Whang, K.Y., You, S., 2012. Differentiation of human labia minora dermis-derived fibroblasts into insulin-producing cells. Experimental and Molecular Medicine, 44, 26-35.
Kuipers, F., Bloks, V.W., Groen, A.K., 2014. Beyond intestinal soap—bile acids in metabolic control. Nature Reviews Endocrinology. 10, 488-498.
Liu, X., Huang, M., Fan, B., Buckler, E.S., Zhang, Z., 2016. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 12, e1005767.
Lu, C. K., Lai, Y. C., Chen, H. R., Chiang, M. K., 2012. Rbms3, an RNA-binding protein, mediates the expression of Ptf1a by binding to its 3'UTR during mouse pancreas development. DNA & Cell Biology. 31, 1245-1251.
Lundbäck, V., Kulyte, A., Strawbridge, R.J., Ryden, M., Arner, P., Marcus, C., Dahlman, I., 2018. FAM13A and POM121C are candidate genes for fasting insulin: functional follow-up analysis of a genome-wide association study. Diabetologia. 61, 1112-1123.
Malinowski, J.R., Denny, J.C., Bielinski, S.J., Basford, M.A., Bradford, Y., Peissig, P.L., et al, 2014. Genetic Variants Associated with Serum Thyroid Stimulating Hormone (TSH) Levels in European Americans and African Americans from the eMERGE Network. PLoS One. 9, e111301.
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., de Bakker, PI., Daly, M.J., Sham, P.C., 2007. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human Genetics. 81, 559-575.
Wagner, R., Dudziak, K., Herzberg-Schäfer, S.A., Machicao, F., Stefan, N., Staiger, H., Häring, H.U., Fritsche, A., 2011. Glucose-raising genetic variants in MADD and ADCY5 impair conversion of proinsulin to insulin. Plos One, 6, e23639.
Schopen, G.C., Visker, M.H., Koks, P.D., Mullaart, E., Arendonk, J.A., Bovenhuis, H., 2011. Whole-genome association study for milk protein composition in dairy cattle. Journal of Dairy Science. 94, 3148-3158.
Song, Y.F., Xu, C., Shao, S., Liu, J., Xing, W., Xu, J., Qin, C., Li, C., Hu, B., Yi, S., et al, 2015. Thyroid-stimulating hormone regulates hepatic bile acid homeostasis via SREBP-2/HNF-4α/CYP7A1 axis. Journal of Hepatology, 62, 1171-1179.
Acknowledgments
The authors are thankful to the ChangFu ranch in Fujian for the sample support for this experiment. We are particularly grateful to Mr. Zhang Yuelei and Mr. Miao Jian for their support and help in the process of data collection and analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
The manuscript does not contain clinical studies or patient data.
All procedures involving animals followed the guidelines for the care and use of experimental animals approved by the State Council of the People’s Republic of China.
Additional information
This article belongs to the topical Collection: Camelids
Guest Editor: Bernard Faye
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gan, Q., Li, Y., Liu, Q. et al. Genome-wide association studies for the concentrations of insulin, triiodothyronine, and thyroxine in Chinese Holstein cattle. Trop Anim Health Prod 52, 1655–1660 (2020). https://doi.org/10.1007/s11250-019-02170-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-019-02170-z