
Abstract
Interleukin-37 is a newly discovered cytokine that plays a pivotal role in suppressing innate inflammation and acquired immunity. We have recently expressed both the mature(mat-) and pro-forms of human IL-37b in plants and demonstrated that while both forms of the plant-made hIL-37b are functional, pmat-hIL37b exhibited significantly greater activity than ppro-IL-37b. Compared to ppro-hIL-37b, on the other hand, the expression level of pmat-hIL-37b was substantially lower (100.5 µg versus 1.05 µg/g fresh leaf mass or 1% versus 0.01% TSP). Since the difference between ppro-hIL-37b and pmat-hIL-37b is that ppro-hIL-37b contains a signal sequence not cleavable by plant cells, we reasoned that this signal sequence would play a key role in stabilizing the ppro-hIL-37b protein. Here, we describe a novel approach to enhancing pmat-hIL-37b production in plants based on incorporation of a gene sequence encoding tobacco etch virus (TEV) protease between the signal peptide and the mature hIL-37b, including a TEV cleavage site at the C-termini of TEV protease. The rationale is that when expressed as a sp-TEV-matIL-37b fusion protein, the stabilizing properties of the signal peptide of pro-hIL-37b will be awarded to its fusion partners, resulting in increased yield of target proteins. The fusion protein is then expected to cleave itself in vivo to yield a mature pmat-hIL-37b. Indeed, when a sp-TEV-matIL-37b fusion gene was expressed in stable-transformed plants, a prominent band corresponding to dimeric pmat-hIL-37b was detected, with expression yields reaching 42.5 µg/g fresh leaf mass in the best expression lines. Bioassays demonstrated that plant-made mature pmat-hIL-37b is functional.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interleukin-37 (IL-37) is a newly discovered member of the IL-1 family cytokines and functions as a natural inhibitor of innate inflammation and acquired immune responses. Human IL-37 exists as five splice transcript variants, namely IL-37a–e, with IL-37b being the longest and most studied isoform (Boraschi et al. 2011). IL-37 gene in mice is missing, thus mice do not express IL-37, but human IL-37 is active on mouse cells (Dinarello et al. 2016). There has been a growing interest in exploring IL-37 as a new therapeutic agent in the treatment of inflammation-related diseases. For instance, administration of recombinant IL-37 to mice was found to ameliorate experimental psoriasis (Teng et al. 2014), alleviate rheumatoid arthritis (Ye et al. 2015) and bleomycin induced experimental lung injury/fibrosis (Li et al. 2018) and atherosclerosis (Ji et al. 2017), decrease renal ischemia–reperfusion injury (Yang et al. 2015) and inhibit the growth of cancer cells (Deng et al. 2018). Moreover, in mice with transgenic overexpression of IL-37, inflammation is reduced in models of LPS shock (Nold et al. 2010), chemical colitis (McNamee et al. 2011), cardiac ischemia (Xiao et al. 2018), cerebral ischemia (Patel et al. 2014), obesity-induced type 2 diabetes (Ballak et al. 2014) and contact dermatitis (Fujita et al. 2013). In a more recent study, Tsilioni et al (2019) have found that in brains of children with autism spectrum disorder (ASD), a neurodevelopmental disability characterized by impaired social skills, communication problems, and repetitive behaviors that is majorly caused by widespread inflammation in the brain due to elevated levels of pro-inflammatory cytokines, IL-37 expression is increased and inhibits the secretion of pro-inflammatory cytokines and chemokines such as IL-1β and IL-18, highlighting the potential role of IL-37 in treating autism. Another new study by Lonnemann et al. (2022) showed that transgenic expression of IL-37 is able to limit inflammation in the brain after acute inflammatory events and prevent loss of cognitive abilities in a mouse model of Alzheimer’s disease. Taken together, these findings strongly support the potential utility of hIL-37 as a new therapeutic agent in human inflammatory diseases.
Presently, recombinant IL-37 produced from E. coli is available only in limited quantities. While E. coli provides a useful expression system for production of foreign proteins, it is limited by expression level and protein solubility. Moreover, scaling up protein production using this system is often difficult and expensive. To overcome these limitations, we have recently developed the use of plants as an alternative expression platform for production of hIL-37 (Alqazlan et al. 2019), taking unique advantages of plant-based expression systems: low production cost, virtually unlimited scalability, the ability of being able to produce complex proteins that are properly glycosylated, folded and assembled, and a significantly lower chance of contamination with prion or mammalian viruses as compared to microbial and mammalian cell-based expression systems (Tremblay et al. 2010, 2011). In our previous work, we reported the production of transgenic plants synthesizing both the mature and pro-forms of hIL-37b and demonstrated that while plant-made both forms of hIL-37b are biologically active, pmat-hIL-37b exhibited significantly greater activity than the pro-form of hIL-37b. Compared to ppro-hIL-37b, on the other hand, the expression level of pmat-hIL-37b in plants was considerably lower (100.5 µg vs. 1.05 µg/g fresh leaf mass or roughly 1% vs. 0.01% TSP). Given that the difference between ppro-hIL-37b and pmat-hIL-37b is that ppro-hIL-37b contains a native 45-aa non-classical signal peptide at its N-terminus that was not recognized and cleaved by plant cellular machinery (Alqazlan et al. 2019), we reasoned that this uncleavable signal peptide would most likely play a key role in stabilizing ppro-hIL-37b protein, although other factors also may play a role, such as subcellular localization of ppro-hIL-37b in plant cells. It should be mentioned that in our previous work, the approach we used to achieve the expression of mature hIL-37b protein in plants involves replacing the native signal peptide of pro hIL-37b protein with one from a well-characterized secretory protein of plant origin, such as barley α-amylase or tobacco pathogenesis-related PR1b protein. While this commonly used approach is effective in allowing for production of the desired mature form of hIL-37b, the protein yield obtained is poor accounting only for 1.05 µg/g fresh leaf mass or 0.01% TSP (Alqazlan et al. 2019).
Here, we describe a novel approach to enhancing the expression yield of mature hIL-37b protein in plants based on genetic modification of the junction between the native signal peptide and the mature hIL-37b protein by in-frame insertion of a gene sequence encoding the catalytic domain of TEV protease, including a TEV cleavage site engineered at the C-terminus of TEV protease. The approach takes advantage of the proposed stabilizing properties of the native N-terminal signal peptide of pro-hIL-37b to improve the stability and thus increase the level of expression of recombinant proteins when fused to it. As the TEV protease is highly used for the cleavage of fusion proteins and removal of tags from recombinant proteins in vitro and in vivo, the sp-TEV-mathIL-37b fusion protein is then expected to be able to self-cleave in vivo via its TEV protease activity at the desired cleavage site to remove the sp-TEV portion, yielding a tag-free pmat-hIL-37b with original N-termini. Indeed, when a sp-TEV-IL-37b fusion gene was constructed and expressed in transgenic tobacco plants, a prominent band corresponding to dimeric pmat-hIL-37b in size was detected, with expression yields reaching up to 42.5 µg/g fresh leaf mass in the highest transgene expression lines. The new expression yield of mature hIL-37b protein represents a roughly 40-fold increase compared to the previously reported protein yield in plants (Alqazlan et al. 2019). Functional assays using a cell-based in vitro assay demonstrated that plant-made pmat-hIL-37b is biologically active.
Materials and methods
Plasmid vector construction
A chimeric fusion gene, consisting of nucleotide sequences encoding the N-terminal signal peptide of 45 amino acid (aa) residues of pro-hIL-37b followed by a 241-aa, 27 kDa catalytic domain of the nuclear inclusion a (NIa) protein encoded by the tobacco etch virus (TEV), a 7-aa cleavage recognition site (ENLYFQG) for TEV protease and the mature form of hIL-37b, was commercially synthesized by BGI Genomics Co. Ltd (Shenzhen, China), with codon optimization based on its expression in tobacco plants. The nucleotide sequence encoding the 27‐kDa catalytic domain of TEV was according to NCBI sequence database (accession number: M15239 and NP_734212). To facilitate subsequent cloning steps, a PciI site that overlaps the translation start site of the fusion gene and a 3′ BamH1 site immediately after the stop codon were added. The single Pcil/BamH1 fragment of the fusion gene was subcloned into the Nco1/BamH1 sites of pTRL-GUS in replacement of the GUS gene (Carrington and Freed 1990). The fusion gene expression cassette, consisting of cauliflower mosaic virus (CaMV) 35S promoter and 5′untranslated region from tobacco etch virus (TEV), sp-TEV-matIL-37b and 3′ untranslated region from Agrobacterium nopaline synthase gene, was released from pTRL-sp-TEV-matIL-37b as a single HindIII fragment and cloned into binary plant transformation vector pBI101.1 (Brandsma et al. 2010), generating vector pBI-sp-TEV-matIL-37b. To allow for easy downstream purification, a second vector that incorporated a 6xHis-tag at the C terminus of sp-TEV-mathIL-37b, pBI-sp-TEV-matIL-37bx6His, was additionally constructed.
Genetic transformation of tobacco plants
For tobacco genetic transformation, plasmid constructs were first introduced into Agrobacterium tumefaciens strain LBA4404 by a method of triparental mating previously described by Ma et al (2005). Agrobacterium harbouring either pBI-sp-TEV-matIL-37b or pBI-sp-TEV-matIL-37bx6His was then grown in liquid YEP medium (10 g/L yeast extract, 10 g/L bacto peptone, 5 g/L NaCl, pH 7.0) supplemented with rifampicin (25 mg/L) and kanamycin (50 mg/L) at 280C with shaking until OD600 of 1.0–1.5 was reached. The cells were collected by centrifugation at 13 000 g for 5 min at 4 °C. The pellet was resuspended in MS liquid basal medium (Murashige and Skoog 1962) supplemented with 100 µM acetosyringone (Sigma Chemical Co., St Louis, MO). Leaf disk explants (1 × 1 cm2) of 4-week-old in vitro-obtained plantlets of tobacco (Nicotiana tabacum cv. 81V9) were then infected with the above-prepared agrobacterium suspension for 1–2 min in Petri dishes. After infection, leaf discs were blotted with sterile filter paper to remove the excess of bacterial cells and medium, and then cultured on antibiotic-free MS medium (MS mineral salts, 3% (w/v) sucrose, 1 × Gamborg’s vitamin solution, 0.8% (w/v) agar, pH 5.8) supplemented with 0.1 mg/L indole-3-acetic acid (IAA), 1 mg/L 6-benzylaminopurine (6-BAP) and 100 µM acetosyringone in a growth chamber at 25 °C with 60 to 70% relative humidity for 48 h (in dark). Leaf discs were then transferred to the regeneration and selection medium (MS mineral salts, 3% (w/v) sucrose, 1 × Gamborg’s vitamin solution, 0.1 mg/L indole-3-acetic acid (IAA), 1 mg/L 6-benzylaminopurine (6-BAP), carbenicillin 500 mg/L, kanamycin 100 mg/L, 0.8% agar) and incubated in a 25 °C growth chamber with 60–70% relative humidity under a 16-h/8-h (light/dark) photoperiod. Regular subcultures of leaf discs were performed in the same regeneration and selection medium. The emerging shoots were then transferred to the rooting medium for root growth and development. The rooting medium is identical to regeneration and selection medium but omitting plant hormones. After rooting, plantlets were transferred to plastic pots containing autoclaved perlite:peat moss (1:1) and were grown in a greenhouse under controlled environment conditions. Young tobacco plants with fully expanded leaves were then subjected to further analysis.
Protein extraction from transgenic tobacco leaves
Total soluble protein (TSP) was extracted from young leaves of independent transgenic tobacco lines, including wild-type tobacco leaves as a negative control. Briefly, leaf samples were ground using a mortar and pestle in liquid nitrogen until a fine powder was obtained. TSP was extracted by using extraction buffer (50 mM Tris pH 8.0, 100 mM NaCl, 200 mM sucrose, 10 mM EDTA, 14 mM b-mercaptoethanol, 0.05% Tween-20, 1 mM phenylmethylsulfonyl fluoride, 2 µg/mL leupeptin, 2 µg/mL aprotinin) with a ration of 2 ml of extraction buffer for 1 g of leaf material. Cell debris was removed by two rounds of centrifugation at 13,000 g for 10 min at 4 °C. Supernatant was collected and TSP concentration was determined with the Biorad Protein Assay Kit (BioRad Laboratories) before proceeding with hIL-37b expression analysis and purification.
Western blotting analysis of pmat-hIL-37b expression
The expression of pmat-hIL-37b in leaf extracts was analysed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting as described previously (Alqazlan et al. 2019). Briefly, samples of crude leaf extracts were mixed with Laemmli Protein Loading Buffer (VWR Life Science) and were boiled for 10 min at 950C. Samples were then separated on a 12.5% (w/v) SDS–PAGE gel and were blotted to a polyvinylidene difluoride (PVDF) membrane (Millipore, Burlington, MA, USA) by using a semi-dry transfer system (Thermo Fisher Scientific, CA, USA). Blots were blocked with 5% (w/v) skim milk in TBS-T (20 mM Tris, 150 mM NaCl, 0.02% (v/v) Tween 20, pH 7.6) at 370C for 1 h, and then incubated overnight at 4 °C with a 1:2000 dilution (v/v) of rabbit anti-human IL-37 primary antibody (ab116282, Abcam), followed by goat anti-rabbit secondary antibody conjugated with peroxidase (074-1506, KLP) at a dilution of 1:5000. For detection of sp-TEV moiety, a by-product arisen from cleavage of the fusion protein, blots were incubated with rabbit anti-TEV protease primary antibody (200-401-B91, Rockland Immunochemicals, Gilbertsville, PA, USA) at 1:500 dilution. All blots were incubated with enhanced chemiluminescence (ECL) plus detection reagent (Merck Millipore) according to manufacturer’s instructions prior to imaging with the FluorChem Q imaging system (ProteinSimple, San Jose, CA, USA).
Quantification of plant‑derived pmat-hIL‑37b
The yield of plant-derived pmat-IL-37b in crude leave extracts was determined by ELISA as described previously (Alqazlan et al. 2019). Briefly, 96-well microtiter plate (Thermo Fisher Scientific) was coated in triplicate with crude leaf extracts and incubated overnight at 4 °C. After removing the coating solution, plates were blocked with 3% BSA (w/v) in PBS-T (phosphate buffered saline containing 0.05% Tween-20) for 2 h at room temperature. After blocking, a 1:3000 dilution of rabbit anti-hIL-37 polyclonal antibody (Abcam) was added into the wells and incubated for 2 h at room temperature. Then, horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Abcam ab205718) was added with the dilution of 1:2000 and incubated at room temperature for 1 h. Between each step, the plates were washed three times with 1xPBS-T. The colorimetric signal was developed by adding TMB (tetramethylbenzidine) that acts as a substrate for HRP (R&D Systems) and the enzymatic reaction was terminated using 2 N H2SO4. The optical density (OD) was measured at 450 nm using a 96-well microplate reader. A standard curve was created by plating E. coli-derived hIL-37b standard (46-218aa, R&D Systems) at various concentrations ranging from 3 to 30 ng per well in PBS buffer to calculate the amount of plant-made recombinant pmat-hIL-37b. Crude leaf extracts were diluted to fit in the linear range of the E. coli-derived hIL-37b standard curve.
Purification of plant-produced pmat-hIL-37b
Small-scale purification of histidine-tagged pmat-hIL-37b for functional characterization was achieved by using immobilized-metal affinity chromatography (IMAC). In brief, the supernatant containing TSP, obtained from crude leaf extracts following two rounds of centrifugation at 13,000 g for 10 min at 4 °C as described above, was filtered by 0.45 µM membrane filter (MilliporeSigma, United States). After equilibration with the binding buffer (50 mM Tris, pH 7.5, 50 mM NaCl and 20 mM imidazole), pre-treated samples were loaded onto a 1-ml HisTrap™ HP column (GE Healthcare Life Sciences, USA), prepacked with Ni sepharose affinity resin equilibrated with the same binding buffer, at a flow rate of 1 ml/per min. The column was then washed with the same binding buffer at the same flow rate until the absorbance reaches a steady baseline. The immobilized protein was eluted from the column with elution buffer (50 mM Tris, pH 7.5, 50 mM NaCl, 450 mM imidazole). Protein elution was followed by monitoring the absorbance at 280 nm. Fractions containing the protein of interest were combined and dialyzed extensively against PBS to remove the excess of imidazole. Eluted protein was then concentrated using a speed vacuum at 4 °C. The integrity and identity of purified protein was verified by western blotting, and its concentration was determined by using an ELISA assay as described above. The percent recovery of pmat-hIL-37b following IMAC procedures was estimated based on the collected amount of purified pmat-hIL-37b divided by the starting amount of pmat-hIL-37b in crude leaf extracts that were loaded onto a 1-ml HisTrap™ HP column, then multiplied by 100.
Determination of biological activity of plant‑made pmat-hIL-37b protein
The biological activity of plant‑derived pmat-hIL-37b protein was determined using a cell-based bioassay as described previously (Alqazlan et al. 2019). Briefly, primary mouse renal cells isolated from the kidney cortex of B6 mice were grown in K1 medium (50:50 DMEM and Ham’s F12; Invitrogen) supplemented with 10% (v/v) FBS, hormone mix (5 μg/mL insulin, 34 pg/mL triiodothyronine, 5 μg/mL transferrin, 1.73 ng/mL sodium selenite, 8 ng hydrocortisone and 25 ng/mL epidermal growth factor), 100 U/mL penicillin and 0.1 mg/mL streptomycin at 37 °C in 5% CO2. To perform the assay, cells were seeded in triplicate in a 96-well flat-bottom tissue culture plate at a density of 1–5 × 104 cells/well in the same cell culture medium and grown overnight at 37 °C. After incubation, the cells were treated with different concentrations of purified plant-made pmat-hIL-37b or commercial recombinant hIL-37b standard for 24 h. The supernatant was then removed, and cells were treated with 1 μg/mL lipopolysaccharide (LPS), an endotoxin from Gram-negative bacteria that acts as a potent inducer of proinflammatory cytokines, for 24 h at 37 °C. Cells treated with LPS only or with LPS plus the IMAC elution fractions obtained from leave crude extracts of untransformed control tobacco plants (Elu-C) were used as a negative control. After treatment, cell culture supernatants were collected, and the TNF-α levels were assessed using a commercial TNF-α-specific ELISA kit according to the manufacturer’s instructions (R&D Systems).
Statistical analysis
Unless otherwise noted, each sample was assayed in triplicate. The in vitro cell-based assays for assessing the TNF-alpha content were repeated two times. Data were presented as the mean ± SE, and two tailed Student’s t test was used to compare two groups. The differences were considered statistically significant when the p values were < 0.05 at the alpha level of 0.05. Normality of the data was checked by Shapiro–Wilk test whereas equal variances were established by F-test in MS Excel before applying the t-test.
Results
Construction of expression vector
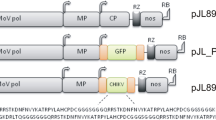
The plasmid vectors designed specifically for production of a mature form of recombinant hIL-37b in plants are illustrated in Fig. 1. The procedures used for construction of pBI-sp-TEV-matIL-37b and pBI-sp-TEV-matIL-37bx6His was given in detail in the Methods section. Expression of the fusion gene was under the transcriptional control of an enhanced 35S promoter with a translational enhancer sequence of TEV. A flexible polypeptide linker of 3xGGGGS repeats was placed between the signal sequence of pro-IL-37b and the TEV protease to minimize the negative impacts of the N-terminal flanking region on the enzymatic activity of the TEV moiety when fused to hIL-37b by promoting its independent folding. The constructed vectors were transformed into A. tumefaciens LBA4404 and used for stable transformations of tobacco plants.

Schematic diagrams of T-DNA regions of plant transformation vectors. The right and left borders of the T-DNA are indicated with RB and LB, respectively. PNOS, Promoter of nopaline synthase; NPT II, Neomycin phosphotransferase gene; TNOS, Nopaline synthase terminator; e35S, Enhanced CaMV 35S promoter including a tobacco etch virus 5′nontranslated leader sequence; SP, Signal peptide sequence of proIL-37b; TEV, The 27 kDa catalytic domain of the Nuclear Inclusion a (NIa) protein encoded by the tobacco etch virus; matIL-37b, mature form of IL-37b coding sequence; 3xGGGGS, a flexible linker consisting of three copies of GGGGS; ENLYFQ↓G, the TEV cutting site (where “↓ “ represents the cleavage position)
Expression of pmat-h37b in transgenic tobacco plants
Transgenic tobacco plants (N. tabacum cv. 81V9) harbouring pBI-sp-TEV-matIL-37b or pBI-sp-TEV-matIL-37bx6His were obtained by Agrobacterium-mediated transformation. The expression of pmat-hIL-37bx6His protein in individual regenerated plant lines was evaluated by western blot analysis of total crude leaf extracts. As shown in Fig. 2a, western blot analysis showed that the anti-human IL-37 antibody (ab116282, Abcam) reacted, with high intensity, against a specific band at around 37 kDa, which most likely represents the homodimeric form of plant-made pmat-hIL-37b. A weak anti-IL-37 reactive band at around 20 kDa was also seen, representing the monomeric form of pmat-hIL-37b. In addition, as observed in those higher pmat-hIL-37b-expressing transgenic lines such as T13, T14 and T15, there was a minor fraction of plant-made pmat-hIL-37b that appears to be present in even higher-order multimers as trimers, tetramers and pentamers (see supplementary Fig. S1). These results, however, appear to be inconsistent with the findings of our previous studies. Shown in Fig. 3, when one of our old hIL-37b expression constructs, PBI-sp(pr1b)-IL-37bxHis6, was used for pmat-hIL-37b expression, the above-mentioned dominant band for homodimeric pmat-hIL-37b was not observed on western blot probed with anti-hIL-37b antibody even after a prolonged film exposure time (5 min), with only a protein band having the predicted size for monomeric pmat-hIL-37b being seen. A slight difference in mobility between the monomeric form of plant-made pmat-hIL-37b and E. coli-derived mature hIL-37b standard is likely due to the addition of extra amino acid residues such as 6xHis to the C-terminus of plant-made pmat-hIL-37b. As demonstrated below, those dark-coloured (same color as the background) bands found within the white-colored bands for dimeric pmat-hIL-37b are of anti-IL-37 antibody-nonreactive proteins representing the sp-TEV portion, a cleavage by-product of the fusion protein that co-migrated (overlapped) with dimeric pmat-hIL-37b.

Immunoblot analysis of the expression of pmat-hIL37b in transgenic tobacco plants. Protein samples (40 μg/per well) were separated by 12.5% SDS–PAGE, transferred onto PVDF membranes and then incubated with A anti-human IL-37 (ab116282, Abcam) or B anti-TEV protease (200-401-B91, Rockland Immunochemicals) antibodies overnight at 4 °C. Blot signals were detected using a FluorChem Q system. The solid double arrow indicates the position of dimeric pmat-hIL-37b and the solid single arrow indicates the location of monomeric pmat-hIL-37b. The sp-TEV moiety arisen from cleavage of the fusion protein was indicated by an empty arrow. The triangle indicates non-specific bands recognized by the antibody. WT, wild-type tobacco; ST, recombinant human IL-37b mature protein (aa 46–218; MW, 17–19 kDa; R&D Systems) standard (20 ng loaded). T8–T19, representative independent transgenic tobacco lines. M, protein molecular mass markers with size (kDa) indicated by the numbers at left

Immunoblot analysis of mature pmat-hIL-37b expression in transgenic tobacco plants using an old IL-37b expression construct, PBI- sp(pr1b)-IL-37bxHis6, as previously described by us (Alqazlan et al. 2019). Protein samples (50 μg/per well) were separated by 12.5% SDS–PAGE, separated by 12.5% SDS–PAGE, transferred onto PVDF membranes and then probed with the same anti-human IL-37 antibody (ab116282, Abcam). Blot signals were detected using a FluorChem Q system after a prolonged exposure time (5 min). The triangle indicates the position of pmat-hIL-37b. Nonspecific bands of around 25 kDa that are recognized by the anti-hIL-37 antibody are indicated by an asterisk. A22–A28 representative independent transgenic tobacco lines; WT, wild-type tobacco; ST, recombinant human IL-37b mature protein standard (aa 46-218); M, protein molecular mass markers with size (kDa) indicated by the numbers at left
Site-specific cleavage of sp-TEV-mathIL-37b fusion protein via its expressed TEV protease activity is expected to result in two un-related smaller proteins, i.e., the target protein pmat-hIL-37b and the sp-TEV tag moiety. To demonstrate the expression of the sp-TEV part, we additionally analyzed total crude leaf extracts by western blotting using anti-TEV antibodies. As shown in Fig. 2b, the anti-TEV antibody specifically recognized those of the above-mentioned anti-IL-37 antibody-nonreactive protein bands (dark in colour) found within the anti-IL-37 reactive bands for homodimeric pmathIL-37b protein (Fig. 2a), indicating that they truly represent the sp-TEV portion. The immune reactivity with anti-TEV antibody was highly specific, as no bands of the similar size was detected in crude extracts from untransformed control plants using same anti-TEV antibody under the same experimental conditions. Sp-TEV moiety, consisting of the 45-aa signal peptide of hIL-37b, a linker of 3 GGGGS repeats, the 27 kDa the catalytic domain of TEV protease and 6 extra amino acid residues (Glu-Asn-Leu-Tyr-Phe-Qln) due to the introduction of a TEV protease-specific recognition site, has a predicted molecular weight of approximately 36 kDa. Therefore, sp-TEV portion and plant-made dimeric pmat-hIL-37b protein are similar in size. To ensure the reproducibility of these results, we also conducted western blot analysis in the use of reduced sample volumes as the signal derived from the protein bands on a western blot may vary with the amount of sample loaded onto the protein gel and moreover, reduction in sample volume may also help improve the signal-to-noise ratio. As shown in Fig. 4, similar band patterns were obtained in western blot when a reduced volume of total leaf extract (20 μg/per lane in Fig. 4 vs. 40 μg/per lane in Fig. 2) was tested. Moreover, we investigated the impact of different concentrations of both primary and secondary antibodies on size, shape or colour of the bands in western blot and found that reducing antibody concentration yielded no difference in band appearance and patterns (data not shown). Similar results were obtained when total extracts from transgenic plants containing pBI-sp-TEV-mathIL-37b (His-tag free) were analyzed by western blotting with both anti-human IL-37 and anti-TEV antibodies (data not shown). The expression level of pmat-hIL-37b in individual transgenic lines was measured by using ELISA. While the expression levels of pmat-hIL-37b were shown to be highly variable among different primary transgenic lines (T0) probably due to random sites of transgene integration (position effect), several lines (T9, T13, T14, T15, T28, T30 and T33) showed pmat-hIL-37b expression at the yield of greater than 35 µg/g fresh leaf mass, with transgenic lines T15 and T33 being the highest reaching up to 41 and 42.5 µg/g fresh leaf mass, respectively. Therefore, the fusion protein-based method reported here led to a roughly 40-fold increase in production yield for mature hIL-37b protein when compared with that obtained previously using our old hIL-37b expression constructs (Alqazlan et al. 2019).

Western blot analysis with reduced amount of leaf crude extract samples. One-half of the original amount of protein samples (20 μg/per well) were tested. Samples were separated by 12.5% SDS–PAGE, transferred onto PVDF membranes and then incubated with A anti-human IL-37 or B anti-TEV protease antibodies overnight at 4 °C. The solid double arrow indicates the position of dimeric pmat-hIL-37b and the solid single arrow indicates the location of monomeric pmat-hIL-37b. The sp-TEV moiety arisen from cleavage of the fusion protein was indicated by an empty arrow. The triangle indicates non-specific bands picked up by the anti-hIL-37 antibody used. WT, wild-type tobacco; ST, recombinant human IL-37b mature protein (aa 46-218; MW, 17–19 kDa; R&D Systems) standard (20 ng loaded). T26–T37, representative transgenic tobacco lines. M, protein molecular mass markers with size (kDa) indicated by the numbers at left
Purification of plant-made pmathIL-37b and its characterization
For characterization of protein function, we performed small-scale affinity purification (in microgram quantities) of the expressed pmat-hIL-37b from leaf extracts of transgenic plants harbouring sp-TEV-matIL-37bx6His by IMAC as described in detail in the Methods section. The identity and integrity of the purified protein was verified by western blot analysis using both anti-IL-37 and anti-TEV protease antibodies. As shown in Fig. 5, while both antibodies were able to react with total protein extracts or the flow-through (FT) fraction as expected, only anti-hIL-37 antibody, but not the anti-TEV protease antibody, showed reactivity with the protein of interest in collected elution fractions at the expected size. These results indicated that: (1) purified pmat-hIL-37b obtained through IMAC structurally remains intact; (2) the fusion protein was efficiently self-cleaved in vivo via its expressed TEV protease activity resulting into two separate proteins, pmat-hIL-37bx6His and its by-product sp-TEV; and (3) small-scale preparative purification of plant-made pmat-hIL-37b can be achieved by using a simple one-step IMAC method. Based on the relative amount of target protein that is retrieved after purification compared with amount loaded on the column, the recovery yield of purified pmat-hIL-37b was approximately 50%.

Immunoblot analysis of purified plant-made pmat-hIL-37b. His-tagged pmat-hIL-37b was partially purified using an IMAC method. Purified protein was electrophoresed on a 12.5% SDS–PAGE, transferred onto PVDF membranes and then incubated with A anti-human IL-37 or B anti-TEV protease antibodies. T, Total leaf extracts; FT, Flow through; E1 to E5, Different elution fractions collected; M, Protein molecular mass markers with size (kDa) indicated by the numbers at left
Characterization of the biological activity of plant-made pmathIL-37b
In our previous study, we only obtained low production yield of pmat-hIL-37b in plants (1.05 µg/g fresh leaf mass or 0.01%TSP), with plant-made pmat-hIL-37b being detected as a monomeric protein (Alqazlan et al. 2019). Using the fusion protein strategy, in the present study we were able to significantly increase the yield of the target protein (up to 42.5 µg/g fresh leaf mass) and at current expression levels plant-made pmat-hIL-37b is present predominantly as a dimeric protein. To determine the functionality of the recombinant pmat-hIL-37b, we performed an in-vitro assay in mouse primary renal tubular epithelial cells. As shown in Fig. 6, treatment of mouse renal tubular epithelial cells with purified dimer-dominant pmat-hIL-37b or E. coli- derived IL-37b mature protein standard (aa 46–218) at all test concentrations significantly reduced LPS-induced TNF-α secretion in a concentration-dependent manner compared with control cells treated with LPS alone or LPS plus IMAC purified elution samples derived from leave crude extracts of untransformed control tobacco plants (LPS + Elu-C). Inclusion of IMAC purified elution samples from untransformed control plants was to eliminate possible effects of plant endogenous protein contaminants present in purified pmat-hIL37bx6His preparations. The inhibitory effect of plant-made dimer-dominant pmat-hIL-37b on TNF-α release in mouse renal tubular epithelial cells was comparable to E. coli-derived mature IL-37b protein standard (aa 46–218). There were no significant differences in TNF-α concentrations in the supernatants of cells treated with either LPS or LPS + Elu-C.

Functional analysis of plant-derived dimeric pmat-hIL-37b. Functional characterization of plant-derived dimeric pmat-hIL-37b was carried out using a cell-based in vitro assay. Mouse primary renal tubular epithelial cells were treated with different concentrations of partially purified plant-derived dimeric pmat-hIL-37b or E. coli-derived recombinant IL-37b standard for 24 h and then treated with 1 μg/ml of LPS for 24 h. Control cells were treated with LPS alone or LPS plus the IMAC elution samples from untransformed control plants (LPS + Elu-C). TNF-α levels in the supernatants were assayed by ELISA. The assay was performed in triplicate and repeated twice. Representative data are shown with standard error bars. **Statistically significant difference (p < 0.0035) in TNF- α levels released from LPS-induced cells treated with either plant-made pmat-hIL-37b or recombinant IL-37 protein standard versus cells treated with LPS alone or LPS + Elu-C. *Statistically not significant difference (p > 0.25) in TNF- α levels in supernatants from plant matIL-37b treated cells versus IL-37 standard treated cells. IL-37b(S), recombinant IL-37 mature protein (aa 46-218) standard; pmat-hIL-37b, plant-made mature IL-37b
Discussion
In the present study, we present a novel approach to improving the production yield of pmathIL-37 in plants. The new approach is based on genetic incorporation of a DNA sequence encoding the catalytic domain of TEV protease between the native signal peptide and the mature form of hIL-37b, including a TEV recognition site at the C-terminus of TEV enzyme. Such a fusion protein-based approach allows us to explore the stabilizing property of the atypical signal peptide sequence of the pro form of hIL-37b to improve the yield of the protein of interest. On the other hand, TEV protease is a highly sequence-specific cysteine protease, and this exquisite specificity is frequently used for the controlled cleavage of fusion proteins and removal of tags from recombinant proteins in vitro and in vivo (Nallamsetty et al. 2004). Therefore, the plant-made sp-TEV-matIL-37b fusion protein is expected to be able to cleave itself in vivo to release a recombinant native pmat-hIL-37b, except for one extra amino acid at its N-terminus. Indeed, western blot analysis of crude leaf extracts of tobacco plants containing sp-TEV-IL-37bx6His fusion gene showed a dominant band at a size corresponding to the dimeric pmat-hIL-37b, with transgenic lines T15 and T33 being the highest reaching up to 41 and 42.5 µg/g fresh leaf mass, respectively. These expression yields are substantially higher than the previously reported yield of 1.05 µg/g fresh leaf mass by us, having important implications. Firstly, our new results demonstrate that the fusion protein-based strategy represents an effective innovative method for enhancing the production yield of recombinant pmatIL-37b and is likely applicable to the high-level expression in plants of many other mammalian therapeutic proteins in their native states. Secondly, the atypical signal peptide of pro form of hIL-37b has the potential to become a new versatile stabilization tag for recombinant protein production. Thirdly, the TEV protein retains its in vivo enzyme activity even when expressed as a fusion protein, evidenced by production of a desired tag-free native target protein.
The results of present study demonstrate that plant-made pmat-hIL-37b is present predominantly as a dimer. In our previous studies, however, we showed that plant-made pmat-hIL-37b was only expressed as a monomeric protein (Alqazlan et al. 2019). The difference in recombinant protein forms may be related to differences in protein yields in plants. It is known that native IL-37 exists in both monomeric and dimeric forms, and that the monomer–dimer equilibrium appears to be concentration-dependent with dimer form dominating at higher protein concentration (Eisenmesse et al. 2019). This view is also supported by our previous results showing that at an expression level of 100.5 µg/g fresh leaf mass or 1% TSP, plant-made pro form of hIL-37b, ppro-hIL-37b, is present predominantly as a dimeric protein (Alqazlan et al. 2019). It was reported that the dimer form of hIL-37, which is held together by noncovalent bonds, is more stable and such a structure is not affected by the reducing agent such as dithiothreitol (DTT) (Kumar et al. 2002). It should be pointed out that cytokine dimerization or multimerization is not uncommon. Cytokines are cell signaling proteins mediating communication among immune and non-immune cells. They are important in both health and disease, and therefore their expression and effector functions must be tightly regulated. Multimerization is regarded as one of mechanisms to regulate their functional roles (Nold-Petry and Nold 2022).
Mature pmat-hIL-37b protein in its monomeric form has a theoretical molecular weight of 20 kDa deduced from its amino acid sequence. However, the dimeric form of plant-made pmat-hIL-37b was shown in western blot to be around 37 kDa that is a slightly smaller than 40 kDa, while the monomeric form of pmat-hIL-37b showed an expected size of 20 kDa. This difference may reflect some conformational changes induced by dimerization, resulting in a more compact structure. It is not unusual to notice that a protein displaying a size on the western blot different from its predicted size, as several factors can contribute the difference between observed and calculated molecular weights such as net charge and protein shape (Quan et al. 2015). It has been reported that mature hIL-37b protein (aa 46-218) undergoes large conformational changes that are linked to greater biological activity (Cavalli and Dinarello 2018).
In the present study we presented additional data to demonstrate the removal and separation of the sp-TEV tag from recombinant target protein through analysis of crude leaf extracts as well as collected IMAC elution fractions by western blot using an anti-TEV antibody. As expected, the data are consistent with the results obtained from western blot analysis of the same plant samples using anti-IL-37 antibodies. While a commercial TEV proteinase protein was not included as a western blot loading control, it would not undermine the reliability of the results, since the anti-TEV antibody we used is highly specific as shown in Figs. 2b, 4b and 5. No bands of similar size were detected in samples from untransformed control tobacco leaves in western blot probed with same anti-TEV antibody under the same experimental conditions. These data also provide evidence that directly links those anti-IL-37 antibody-nonreactive bands (dark in colour) found within the bands (white in colour) for dimeric pmathIL-37b, revealed by western blot analysis of crude extracts using an anti-IL-37 antibody as shown in Fig. 2a, to the cleaved sp-TEV portion that comigrated (overlapped) with dimeric pmathIL-37b. The findings that no anti-TEV antibody reactive bands were detected in samples of collected pmatIL-37b elution fractions (Fig. 5b) provide further support for our conclusion above. Taken together, our findings suggest that sp-TEV-mathIL-37b fusion protein was efficiently self-cleaved in vivo to release pmat-hIL-37b lacking sp-TEV moiety.
The main objective of this study was to investigate whether a fusion protein-based approach is effective in increasing the expression of mature hIL-37b protein in plants and as such, we, in the present work, made a major effort in the new plant expression vector construction, protein expression analysis and functional characterization of the expressed target protein. In addition, we performed small-scale affinity purification of plant produced pmat-hIL-37b by IMAC for use in functional assays. Our preliminary results suggest that IMAC can be utilized as a simple method for rapid pmat-hIL-37b purification at least on a small scale, with the recovery yield accounting for approximately 50%. These results are encouraging and will allow us to move forward with more focus on optimization and upscaling of pmat-hIL-37b purification using IMAC method and preclinical application of the plant-made cytokine as future direction.
Results from functional analysis using an in vitro cell-based bioassay indicate that plant-made dimer-dominant pmat-hIL-37b is biologically active, with its activity comparable to E. coli-derived mature hIL-37b standard at all tested concentrations (Fig. 6). These results offer further support for efficient and site-specific self-cleavage of the fusion protein in vivo. The results also suggest that plant-made dimeric pmatIL-37b with a molecular mass slightly smaller than expected could not be resulted from undesired cleavage within the target protein but rather a consequence of the conformational change after dimerization.
In conclusion, we have developed a novel and effective strategy to increase the yield of a mature form of hIL-37b in plants. Using this approach, we achieved the expression level of pmathIL-37b up to 42.5 µg/g fresh leaf mass in transgenic tobacco, which represents 40-fold greater than previously reported (Alqazlan et al. 2019). We have recently used this approach to express several other high-value therapeutic proteins including interferon alpha-2a and IL-10, and obtained encouraging preliminary results (unpublished data). We believe that this approach holds great promise for enhancing the production yield of many recombinant proteins in plants.
References
Alqazlan N, Diao H, Jevnikar AM, Ma S (2019) Production of functional human interleukin 37 plants. Plant Cell Rep 38:391–401
Ballak DB, van Diepen JA, Moschen AR, Jansen HJ, Hijmans A, Groenhof GJ, Leenders F, Bufler P, Boekschoten MV, Müller M, Kersten S, Li S, Kim S, Eini H, Lewis EC, Joosten LA, Tilg H, Netea MG, Tack CJ, Dinarello CA, Stienstra R (2014) IL-37 protects against obesity-induced inflammation and insulin resistance. Nat Commun 5:4711–4722
Boraschi D, Lucchesi D, Hainzl S, Leitner M, Maier E, Mangelberger D, Oostingh GJ, Pfaller T, Pixner C, Posselt G, Italiani P, Nold MF, Nold-Petry CA, Bufler P, Dinarello CA (2011) IL-37: a new anti-inflammatory cytokine of the IL-1 family. Eur Cytokine Netw 22:127–147
Brandsma ME, Diao H, Wang X, Kohalmi SE, Jevnikar AM, Ma S (2010) Plant-derived recombinant human serum transferrin demonstrates multiple functions. Plant Biotechnol J 8:489–505
Carrington JC, Free DD (1990) Cap-independent enhancement of translation by a plant potyvirus 5′ nontranslated region. J Virol 64:1590–1597
Cavalli G, Dinarello CA (2018) Suppression of inflammation and acquired immunity by IL-37. Immunol Rev 281:179–190
Deng HB, Zhang H, Liang JM, Xian HB, Chen ZC, Tang YC, Yang S, Feng WN (2018) IL-37 mediates the anti-tumor activity in non-small cell lung cancer through IL-6/STAT3 pathway. J Appl Biomed 16:15–21
Dinarello CA, Nold-Petry C, Nold M, Fujita M, Li S, Kim S, Bufler P (2016) Suppression of innate inflammation and immunity by interleukin-37. Eur J Immunol 46:1067–1081
Eisenmesser EZ, Gottschlich A, Redzic JS, Paukovich N, Nix JC, Azam T, Zhang L, Zhao R, Kieft JS, The E, Meng X, Dinarello CA (2019) Interleukin-37 monomer is the active form for reducing innate immunity. Proc Natl Acad Sci USA 116:5514–5522
Fujita H, Inoue Y, Seto K, Komitsu N, Aihara M (2013) Interleukin-37 is elevated in subjects with atopic dermatitis. J Dermatol Sci 69:173–175
Ji Q, Meng K, Yu K, Huang S, Huang Y, Min X, Zhong Y, Wu B, Liu Y, Nie S, Zhang J, Zhou Y, Zeng Q (2017) Exogenous interleukin 37 ameliorates atherosclerosis via inducing the Treg response in Apo E deficient mice. Sci Rep 7:3310
Kumar S, Hanning CR, Brigham-Burke MR, Rieman DJ, Lehr R, Khandekar S, Kirkpatrick RB, Scott GF, Lee JC, Lynch FJ, Gao W, Gambotto A, Lotze MT (2002) Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 18:61–71
Li Y, Gao Q, Xu K, Peng X, Yuan X, Jiang W, Li M (2018) Interleukin-37 attenuates bleomycin-induced pulmonary inflammation and fibrosis in mice. Inflammation 41:1772–1779
Lonnemann N, Hosseini S, Ohm M, Geffers R, Hiller K, Dinarello CA, Korte M (2022) IL-37 expression reduces acute and chronic neuroinflammation and rescues cognitive impairment in an Alzheimer’s disease mouse model. Elife 11:e75889
Ma S, Huang Y, Davis A, Yin Z, Mi Q, Menassa R, Brandle JE, Jevnikar AM (2005) Production of biologically active human interleukin-4 in transgenic tobacco and potato. Plant Biotechnol J 3:309–318
McNamee EN, Masterson JC, Jedlicka P, McManus M, Grenz A, Collins CB, Nold MF, Nold-Petry C, Bufler P, Dinarello CA, Rivera-Nieves J (2011) Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci USA 108:16711–16716
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nallamsetty S, Kapust RB, Tözsér J, Cherry S, Tropea JE, Copeland TD, Waugh DS (2004) Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif 38:108–115
Nold MF, Nold-Petry CA, Zepp JA, Palmer BE, Bufler P, Dinarello CA (2010) IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol 11:1014–1022
Nold-Petry CA, Nold MF (2022) Rationale for IL-37 as a novel therapeutic agent in inflammation. Expert Rev Clin Immunol 18:1203–1206
Patel FJ, Volkmann DT, Taylor GW, Hansson MA, Anderson JF, Zhou Y, Scoazec LM, Hartford CV, Hainz DL (2014) IL-37 reduces inflammatory response after cerebral ischemia and reperfusion injury through down-regulation of pro-inflammatory cytokines. Cytokine 69:234–239
Quan YH, Zhu QF, Huang DL, Zhao SY, Lo LJ, Peng JR (2015) An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Sci Rep 5:13370
Teng X, Hu ZL, Wei XQ, Wang Z, Guan T, Liu N, Liu X, Ye N, Deng G, Luo C, Huang N, Sun C, Xu M, Zhou X, Deng H, Edwards CK III, Chen X, Wang X, Cui K, Wei Y, Li J (2014) IL-37 ameliorates the inflammatory process in psoriasis by suppressing proinflammatory cytokine production. J Immunol 192:1815–1823
Tremblay R, Feng M, Menassa R, Huner NP, Jevnikar AM, Ma S (2011) High-yield expression of recombinant soybean agglutinin in plants using transient and stable systems. Transgenic Res 20:345–356
Tremblay R, Wang D, Jevnikar AM, Ma S (2010) Tobacco, a highly efficient green bioreactor for production of therapeutic proteins. Biotechnol Adv 28:214–221
Tsilioni I, Patel AB, Pantazopoulos H, Berretta S, Conti P, Leeman SE, Theoharides TC (2019) IL-37 is increased in brains of children with autism spectrum disorder and inhibits human microglia stimulated by neurotensin. Proc Natl Acad Sci USA 116:21659–21665
Xiao HP, Li BD, Yang XM, Yin QL (2018) IL-37 protects myocardial ischemia reperfusion injury in mice through mediating inflammation response. Biomed Res 29:663–666
Yang Y, Zhang ZX, Lian D, Haig A, Bhattacharjee RN, Jevnikar AM (2015) IL-37 inhibits IL-18-induced tubular epithelial cell expression of pro-inflammatory cytokines and renal ischemia-reperfusion injury. Kidney Int 87:396–408
Ye L, Jiang B, Deng J, Du J, Xiong W, Guan Y, Wen Z, Huang K, Huang Z (2015) IL-37 alleviates rheumatoid arthritis by suppressing IL-17 and IL-17-triggering cytokine production and limiting Th17 cell proliferation. J Immunol 194:5110–5119
Acknowledgements
This research was supported by funding from the National Natural Science Foundation of China (Grant number 32072588, U22A20495) to Aoxue Wang.
Author information
Authors and Affiliations
Contributions
YZ and NA participated in project planning and experimental design and performed majority experiments. ZM, JZ, NL performed tobacco genetic transformation, immunoblotting, ELISA and protein affinity purification. YZ and MF performed vector construction and statistical analysis of experimental data. AW and SM wrote the manuscript. AW and SM designed and guided intellectually all processes of the work. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
Cite this article
Zhang, Y., Alqazlan, N., Meng, Z. et al. A novel approach to achieving more efficient production of the mature form of human IL-37 in plants. Transgenic Res 32, 279–291 (2023). https://doi.org/10.1007/s11248-023-00351-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-023-00351-z